RASopathies are a family of developmental disorders that share germline mutations in the components of the RAS-MAPK pathway leading to dysregulated signalling. A high prevalence of hypertrophic cardiomyopathy (HCM) in these patients suggested genetic involvement. Whether RASopathies are an aetiology of HCM or a comorbidity with ominous prognostic implications remained a debatable issue for sometimes in the past. However, recent evidence demonstrates RASopathies can result in a specific form of cardiomyopathy (CM), particularly in the paediatric population. Although this type of CM shares many pathologic cardiac manifestations with HCM, it is a more severe form with significantly higher mortality rates. Same to other genetic diseases, RASopathies have a very low prevalence, and hence, neglected in pathophysiological research. Consequently, pathogenic mechanisms of RASopathy-associated CM remains unclear and lacks aetiological treatment. Clinical overlap between RASopathy phenotypes, extensive cardiac variability within each phenotype and within families, and between unrelated individuals with the sane genetic mutation has complicated diagnosis and treatment. Thus, the understanding of RASopathy-associated CM and accurate diagnosis is essential to guide the choice of the most appropriate treatment. This paper reviews published evidence on RASopathy-associated CM with a focus on its phenotypic expression, pathogenesis, clinical evaluation and management.
rasopathy-associated cardiomyopathy, hypertrophic biventricular obstructive cardiomyopathy, leopard syndrome, noonan syndrome, costello syndrome
RASopathies (previously known as neuro-cardio-facio-cutaneous syndromes or RAS-MAPK syndromes) are a clinically defined group of medical genetic syndromes caused by pathogenic variants in the RAS-mitogen-activated protein kinase (MAPK) pathway. Typical examples of this group of syndromes includes neurofibromatosis type 1 (NF1), Noonan syndrome (NS), NS with multiple lentigines (NSML), capillary malformation–arteriovenous malformation (CM-AVM), Costello syndrome (CS), cardiofaciocutaneous syndrome (CFCS), and Legius syndrome (LS). A frequent manifestation of cardiomyopathy (CM) in these patients suggests RASopathies may be a potential aetiologic agent of CM. However, current supporting evidence is sporadic and unclear. In the CM literature, RASopathies lack a unifying terminology, variously termed cardiofacial syndromes or syndromic diseases, which undermines comparative studies and a general understanding. The 2006 and 2007 consensus statements by the American Heart Association (AHA) [1] and the European Society of Cardiology (ESC) [2] respectively on the definitions and classifications of CMs recognize RASopathies as potential secondary causes of CMs. The AHA lists cardiofacial disorders (Noonan syndrome and Lentiginosis) as a secondary cause of CM whereas the ESC lists Noonan Syndrome as a cause of syndromic hypertrophic CM (HCM).
Recent evidence also suggest the involvement of RASopathies in the development of CM. Farag et al. [1] described a difficult case of a 26-year old male diagnosed with Noonan syndrome associated hypertrophic biventricular obstructive cardiomyopathy (HOCM). In a subsequent commentary of the Farag case report, Spartalis et al. [2] report that it was not the seminal case report. Four other case reports had already described an association between hypertrophic cardiomyopathy (HCM) and left ventricular (LV) and right ventricular (RV) outflow tract obstruction [5-8]. HCM occurs in 20% of patients with RASopathies [5]. Whether RASopathies are a cause of a specific form of CM or is a comorbidity in HCM with prognostic implications remained a conundrum that warrants additional prospective clinical trials for clarification. This paper reviews the current clinical understanding of CMs associated with RASopathies as well as identifies areas of limited knowledge that could benefit from addition research.
Genetic disorders in humans fall into four different categories based on the type of mutation (genotype) and environmental involvement or expression (phenotype). The four categories are monogenic (Mendelian), complex (multifactorial), chromosomal and mitochondrial disorders. RASopathies belong to the group of monogenic disorders that result from mutation in one single gene or its regulatory sequences. However, allelic heterogeneity is often present – when different mutations in the same gene cause the same disorder. For example, locus heterogeneity (mutations at different loci causing the same phenotype) or clinical heterogeneity (mutations in the same gene causing different disorders) known as allelic disorders [9,10]. The pattern of inheritance can be autosomal dominant, autosomal recessive, x-linked dominant and x-linked recessive. Inheritance in RASopathies is through the autosomal, dominant pattern. The affected individuals are heterozygous for the mutated allele, located on one of the autosomes. A disorder with autosomal dominant inheritance pattern can affect both male and female and transmitted by either sex [9].
Over the past decade, there has been an increased recognition of human genetic syndromes due to germline mutations in the genes encoding components or regulators of the RAS/MAPK pathway. The RAS genes constitute a multi-gene family that includes HRAS, NRAS, KRAS, PTPN11, SOS1, RAF1, SHOC2, and CBL. RAS proteins are small guanosine nucleotide-bound GTPases that function as a critical hub within the cell [9-11]. Extra cellular input in the form of growth factors binding to receptor tyrosine kinases, G-protein-coupled receptors, cytokine receptors, and extracellular matrix receptors activate Ras proteins. The MAPK pathway is among the several critical downstream signalling cascades of RAS [12]. Taken together, the RAS-MPAK pathway plays a critical role in the regulation of cell cycle and cellular growth, differentiation and senescence, all of which are vital processes to normal human development [9]. Thus, the dysregulation of the RAS-MPAK pathway by pathogenic genetic variants has profound consequences for normal human development. Myocardial involvement may lead to the development of CM. RASopathy-associated CM has been defined as a heterogeneous genetic condition characterized by the early onset ventricular hypertrophy and a high prevalence of co-existing congenital heart disease related to mutations in the genes encoding components of the RAS-MAPK pathway [13].
Each RASopathy exhibits a unique phenotype but because they share a common underlying mechanism – the dysregulation of the RAS-MAPK pathway – they share many overlapping characteristics. They include craniofacial dysmorphology, cardiac malformations, cutaneous, musculoskeletal and ocular abnormalities, neurocognitive impairment, hypotonia, and an increased cancer risk [9-11]. However, distinguishing one RASopathy phenotype from another remains a clinical challenge. Fortunately, animal models have improved our understanding of the molecular causes of autosomal dominant traits and missense mutations altering genes whose proteins are responsible for RAS-mediated intracellular signalling, has reinforced the existing nosology. The identification of the causative mutation has also allowed the detection of several genotype-phenotype associations including those responsible for HCM. It has also allowed elucidation of the pathogenesis of CM and provided valuable insights that suggest RASopathies-associated CM may be treatable. A genetic mutation previously known to cause NS has been recently discovered to cause non-syndromic dilated CM (DCM) with a pathogenesis that also suggests therapy [10].
In addition to NS, there is a group of other less prevalent autosomal dominant disorders clinically and genetically related to NS. This group of disorders share dysregulation of the RAS signalling as the common pathogenic mechanism and collectively termed RASopathies. They include NS with multiple Lentigines (NSML), Costello syndrome, Neurofibromatosis type 1 (NF1), cardiofaciocutaneous syndrome (CFCS), Legius syndrome and NS wit loose Anagen hair
Noonan syndrome (NS: (MIM 163950) is the most common RASopathy affecting about 1 in 1000 to 1 in 2500 newborns [10]. It is a clinically variable developmental disorder inherited in an autosomal dominant trait. Its cardinal clinical feature is dysmorphic facial features, most commonly most commonly hypertelorism, ptosis, low-set ears, and short webbed neck. Other typical phenotypic features include congenital cardiac defects, short stature, ectodermal and skeletal anomalies (particularly, sternal deformities and cubitus valgus), and variable cognitive and developmental defects [14]. Individuals with NS also have an elevated risk of developing cancer [9,10]. Although NS is a rare disease, it is among the leading non-chromosomal disorders affecting development and growth.
Seven genes have been associated with the development of NS: PTPN11 [15], SOS1 [16,17], RAF1 [18,19], KRAS [20], NRAS [21], SHOC2 [22], and CBL [23,24]. All these genes harbour heterozygous germline mutation and encode various components or proteins associated with the RAS/MAPK pathway. The most common gene associated with NS is PTPN11 accounting for about half of all NS cases followed by SOS1 accounting for 13% of all NS cases [11]. A greater majority of NS patients (about 80-90%) exhibit cardiovascular involvement. This involvement includes a broad range of congenital heart defects most commonly valvar pulmonic stenosis and/or early onset of HCM. The prevalence of HCM – classified by the AHA as a secondary form of myocardial disease [1] – is 20% in NS patients. Half of these HCM-NS patients have congenital cardiac defects. Histological evidence of the myocardium in NS-associated HCM is indistinguishable from that seen in sarcomeric HCM [25].
The natural history of the NS-associated HCM is distinct in several ways from the primary HCM due to mutations in the sarcomeric proteins or other paediatric forms of HCM [26,27]. NS-associated CM has an early onset. The median age of presentation is five months, with more than half diagnosed by the sixth month of life. The onset of NS-associated CM is earlier than other paediatric forms of HCM, which present at an average age of eight years. At diagnosis, patients with NS-associated HCM are more likely to have congestive HF than other children with HCM at 24% and 9% respectively. NS also manifests with significant LVOT with a mean gradient of 32 mm Hg [11].
The presence of HCM in NS patients portends a worse prognosis with significant differences in survival compared to NS children without HCM. In the absence of HCM, children with NS have nearly a complete survival 15 years after diagnosis, whereas in the presence of HCM survival at 15 years after diagnosis is 70%. The primary cause of death for NS-CM is HF [26-28]. The presence of HCM affects survival in paediatric patients with and without NS. Children with NS are more likely to die early, with most deaths occurring during infancy. Comparatively, other forms of paediatric HCM exhibit a steady, slower mortality rate and survival declines to NS levels 13 years after diagnosis. Long-term follow-up however suggests NS-HCM has higher late mortality compared to sarcomeric HCM [26]. Other important prognostic factors in NS-HCM children include age of presentation and the presence or absence of HF at presentation. The presence of HF in infants with NS presenting before six months of age have an ominous prognosis with 2-year survival of ~30%. In contrast, infants with NS presenting after 6 months of age in the absence of HF have a favourable prognosis with survival of ~90% after 2 years. Infants with NS presenting before six months in the absence of HF have ~75% survival at two years [27].
NS with multiple lentigines (NSML: MIM 151100) is a rare autosomal dominant disorder. NSML is a similar phenotype to NS because it presents with craniofacial features mimicking NS but with multiple lentigines [9-11]. Formerly, NSML was termed LEOPARD, which is an acronym for the characteristic abnormalities associated with the disorders. Lentigines – multiple black or dark brown spots on the skin. Electrocardiographic (ECG) conduction abnormalities reflecting derangements in electrical activity and the coordination of proper contractions. Ocular hypertelorism (widely spaced eyes. Pulmonary stenosis (obstruction of the normal outflow of blood from the RV. Abnormalities of the genitals. Retarded growth leading to short stature. Deafness or hearing loss due to malfunction of the inner ear (sensorineural deafness) [10]. Some affected individuals may present with mild intellectual disability, speech difficulty and/or additional physical disabilities. However, NSML exhibits wide individual variations in the range and severity of symptoms and physical characteristics vary from one individual to another [11].
NSML and NS result from mutations in the PTPN11 and RAF1 genes. Both are allelic disorders resulting from different heterozygous missense mutations in the same genes, PTPN11 [29,30] and RAF1 [18]. The most common NSML-associated PTPN11 mutation affect amino acids in the catalytic PTP domain, resulting in reduced SHP2 catalytic activity in vitro resulting in a loss of function [29,31]. In contrast, in an in vivo Drosophila model, residual catalytic activity in the NSML mutant SHP2 protein is sufficient to produce a gain-of-function phenotype because of dysregulation of the protein causing continuous MAPK pathway activity during development [32].
In NSML patients, HCM occurs in ~80%, which is the highest rate of HCM among the RASopathies [33]. However, due to its relative rarity, outcome data for HCM in NSML patients is not currently available. Approximately 50% of NSML patients with HCM have LVOT from the hypertrophy. The clinical course of HCM in most individuals with NSML appears relatively benign although a rare case of HCM may present in early infancy and progress rapidly. However, published data about outcomes of HCM in NS came from clinical diagnoses. Since distinguishing NS from NSML remains a clinical challenge in infants and toddlers who often do not exhibit lentigines even in the presence of NSML, some proportion of infants with poor outcomes described in the NS literature may be patients with NSML. Distinguishing NSML from NS is not only a classification issue but also has implications in the choice of therapy [11].
Costello syndrome (CS: MIM 218040) is among the rarer RASopathies. It is a multiple congenital syndrome having many overlapping features with other RASopathy syndromes. It is a more severe phenotype relative to NS. The cause of CS is heterozygous activation of germline mutation in HRAS. The distribution of mutation shows > 80% of CS patients have a p.G12S substitution followed by p.G12A substitution. These substitutions disrupt guanine nucleotide binding and reduces intrinsic and GAP-induced GTPase activity leading to RAS remaining in the active state [9].
Typical clinical features of CS include dysmorphic craniofacial features, failure to thrive especially in the newborn period, cardiac musculoskeletal, ectodermal and ocular anomalies, hypotonia, and variable neurocognitive delay [34]. Phenotypic features manifest in the perinatal period with polyhydramnios in utero, and many births are premature with low-birth weight. Typical facial features include macrocephaly with prominent forehead, epicanthal folds, down-slanting palpebral fissures, a short nose with depressed nasal bridge and broad base, and posteriorly rotated ears with thickened helices and lobes [9]. Dermatologic manifestations that support the diagnosis of CS include soft skin with excessive wrinkling [35].
The majority of CS individuals present with cardiac abnormalities including HCM, valve anomalies, septal defects and arrhythmias. The prevalence of HCM in CS ranged from 20% to 61%, notably higher than in NS depending on the series [36,37]. In the series of Swiwik et al. [38], the age of discovery of HCH varied between 5 months to 20 years. In another case report, Lin et al. [36] reported that diagnosis of HCM is not always performed at birth and some patients develop HCM later possibly due to the accumulation of metabolites over time [39]. In individuals with CS and HCM, the pattern is asymmetric in more than 60%. The natural history of CS-HCM is variable but includes severe or progressive disease in 40% of the cases and nearly 25% of patients undergo septal myectomy. The natural history of CS-HCM is indistinguishable from that of sarcomeric HCM with myocardial fibre disarray [36,40]. Regular echocardiographic monitoring is necessary for the possibility of rapidly evolving forms of HCM.
Neurofibromatosis Type 1 (NF1: MIM 162200) was the first multiple congenital anomaly to be associated with germline mutation in the RAS-MAPK pathway. NF1 affects about 1 in 3,000 newborns [41]. NFI results from mutations in the NF1 gene, with about 50% of the mutations being de novo [42-44]. The NF1 gene encodes neurofibromin, which is a RAS-GAP (GTPase-activating protein that is a negative regulator of RAS. NF1 mutations cause neurofibromin loss of function leading to haploinsufficiency with the cell, reduced RAS-GTPase activity and eventually overall increase in active GTP-bound RAS. Due to these effects, NF1 is a cancer predisposition syndrome and thus, individuals with NF1 are at a greater risk for developing malignancies [9]. NF1 has a complex array of clinical signs and symptoms. Its clinical diagnosis relies on the presence of café-au-lait maculae, intertriginous freckling, neurofibromas and plexiform neurofibromas, iris Lisch nodules, osseous dysplasia, optic pathway glioma, and/or a first-degree NF1 relative. Other clinical manifestations of NF1 include cardiac malformation, cardiovascular disease, vasculopathy, hypertension, vitamin D deficiency, brain malformations and seizure. Some may manifest dysmorphic craniofacial features similar to those observed in NS [45,46].
Many reports in literature have described cardiac involvement in NF1 patients. They include valvular pulmonary stenosis, branch peripheral pulmonary stenosis, atrial and ventricular septal defects, and coarctation of the aorta, and association with HCM [47-50]. Analysis of 2,322 cases of NF1 in the National Neurofibromatosis Foundation International Database from 1991-98 reported a low prevalence rate of cardiovascular abnormalities 2.3% [49]. Related studies reported higher prevalence (18.8%) [50] and 27% [48] of cardiac abnormalities in 69 and 48 NF1 patients respectively. Cardiac abnormalities included mitral/aortic regurgitation, mitral valve prolapse, mild left pulmonary artery stenosis and coarctation of the aorta [48,50]. Sutton et al. [51] suggested a strong association between NF1 and HCM. In published series, the frequency of congenital heart defects range between 0.4% and 6.4%. More recently, Incecik et al. [52] evaluated 65 NF1 patients for cardiac abnormalities based on the findings of standard ECG and echocardiography. Eleven of the 65 patients (15.3%) had cardiac abnormalities: secundum atrial septal defect (ASD), pulmonary valvular stenosis, ventricular septal defect, tricuspid/aortic valve regurgitation. There were no reports of CM.
In 1988, Fitzpatrick and Emanuel [53] were the first to describe a co-existent of NF and idiopathic HCM in two siblings – first degree relative. They suggested HCM occurred by chance or NF and HCM were manifestations of a common hereditary defect of neural crest tissue, or abnormalities of the catecholamine metabolism and nerve growth factor in NGF patients can cause secondary ventricular hypertrophy with septal involvement. Since then, two case reports have reported an association between NF1 and HCM [54,55]. Jurko et al. [54] presented a case of 18-year old male with NF1 and HCM with systolic anteward movement of the anterior leaflet of the mitral valve. Gradient in the LVOT was 85 mm Hg secondary to sub valvular aortic stenosis with LV diastolic dysfunction. Abnormalities in catecholamine metabolism and nerve factor growth suggests an aetiological link between NF1 and HCM. These abnormalities can cause secondary valvular hypertrophy with septal involvement [54]. More recently, Ritter et al. [55] was the first to report foetal CM as the presenting feature of NF1 and a review of NF1-related LV hypertrophy. The case report suggests that differential diagnosis for foetuses with CM is essential even in the absence of a known family history of the condition. Since cardiac abnormalities in NF1 patients have potential long-term effect hemodynamic consequences to justify early diagnosis, cardiologic assessment should be mandatory at the time of diagnosis with regular follow-up [54,55].
Recently, a rare case of co-existing DCM and NF1 has been reported in a 44-year-old male [56]. Chest x-ray showed cardiomegaly and pulmonic congestion while laboratory results were unremarkable. Two-dimensional (2D) echocardiographic examination revealed dilated and diffusely hypokinetic LV (end-diastolic/end-systolic dimension 70/62 mm and LV ejection fraction 24%). Colour Doppler examination demonstrated severe mitral regurgitation, moderate tricuspid regurgitation with increased pulmonary artery pressure. The right atrium and the right ventricle were also enlarged Coronary angiography revealed normal coronary arteries. This is the first case showing the co-occurring NF1 and DCM in an adult patient. Despite substantial improvement in the understanding of the natural history, pathology and pathogenies of cardiovascular disease over the past few decades, it is still inadequate to explain the pathogenesis of DCM in RASopathies.
Cardiofaciocutaneous syndrome (CFCS: MIM 115150) is among the rarer RASopathies with overlapping phenotypic features with NS and CS. Individuals with CFCS have NS-like craniofacial features [31]. Cardiac abnormalities have a similar frequency with those of NS and CS. The most prevalent cardiac abnormalities include pulmonic stenosis, septal defects and HCM. Musculoskeletal and ocular abnormalities are common. Failure to thrive is a typical characteristic in infancy. Neurologic abnormalities are universally present to varying degrees and include hypotonia, motor delay, seizures, speech delay, and/or learning disability [57]. FCFCS results from mutation in four genes that encode proteins in the RAS-MAPK pathway downstream of RAS. These genes are BRAF [58,59], MAP2K1 and MAP2K2 [59] and KRAS [58]. The role of KRAS in CFCS is unclear since its mutations occur in NS [22]. Heterozygous BRAF mutations occur in about 75% of mutation positive CFCS patients [60]. The pattern of CFCS transmission is autosomal dominant [59]. Since it is rare for CFCS individuals to reproduce, the greater majority of CFCS patients result from de novo dominant mutation. Although CFCS-associated mutations are in an oncogenic pathway, it is still not clear whether CFCS individuals are more likely to develop malignancies. CFCS does not appear to have the malignancy risk associated with NF1, NS and CS [61,62].
Similar to NS, cardiovascular involvement in CFCS is prevalent in ~75% of the patients with pulmonary valve stenosis being the most common cardiac manifestation [63]. Published and unpublished data including three autopsies on cardiac involvement in RASopathies report 53 individuals with CFCS [64-69]. Cardiac ultrasonography revealed no evidence of cardiac abnormality in 24.5% of CFCS patients. The remaining 75.5% had one or multiple cardiac abnormalities. ASD (isolated or associated with pulmonary valve stenosis) occurred in 22.5% (n=9) of those with cardiac anomalies, five of them had mild pulmonary stenosis and two had severe HCM. Other cardiac lesions included small ventricular septal defects in two patients and partial AV canal in another two patients. Sixteen (40%) patients had some form of myocardial disease as the primary or secondary diagnosis. Eight has mild HCM with a bulge in subaortic area or with moderate diastolic dysfunction [63]. Overall, diagnosis of HCM is common in ~40 of CFCS patients. Severity varies widely from localized sub-aortic involvement to severe global hypertrophy with obstruction. However, exact data on outcome of HCM in the context of CFCS are not available [11].
Legius syndrome (LS: MIM: 611431) is a recently discovered rare autosomal dominant genetic skin pigmentation disorder. Brems et al [70] first described LS in 2007 after they identified a heterozygous mutation in SPRED1 gene was responsible for this mild NF phenotype. Previously, LS was called NF1-like syndrome because LS shares many phenotypic features with NF1 as well as with other RASopathies but LS is a less severe phenotype [9]. The typical phenotypic characteristic of LS is multiple café-au-lait maculae without neurofibromas or other tumour manifestations of NF1. Additional commonly reported clinical manifestations include intertriginous freckling, mild neurocognitive impairment, and macrocephaly, with some having dysmorphic craniofacial features resembling those of NS. On the other hand, typical NF1 neoplastic features such as neurofibromas, plexiform neurofibromas, iris Lisch nodules, and central nervous system tumours appear dissociated with LS. The LS results from heterozygous inactivating mutations in SPRED1 [10]. Definitive diagnosis of LS remains a clinical challenge. It is difficult to diagnose LC based solely on clinical grounds. The detection of the pathogenic variant in SPRED1, the only gene known to cause LS, is necessary to confirm diagnosis [10]. Due to the rarity of the syndrome, there are very few reports in literature. The current knowledge of the natural history of LF relies on the clinical manifestations of fewer than 200 individuals with molecularly confirmed diagnosis. Cardiovascular involvement remains undocumented in LS patients. Thus, better delineation of the clinical manifestation, natural history and cardiac involvement of this rare syndrome will likely occur following identification of more affected individuals.
Noonan syndrome with loose Anagen hair is a relatively recently described RASopathy in 2003. Typical characteristic include ectodermal features including darkly pigmented skin with eczema or ichthyosis and loose Anagen hair [62]. A very limited number of patients with this RASopathy have been described in published literature, a high percentage of affected individuals have cardiovascular involvement [71-74]. HCM prevalence in patients with NS with loose Anagen hair is about 25% but its natural history has not be delineated [11].
Reports of other rarer RASopathies without documented involvement of the myocardium are available. Whether these rarer RASopathies case CM or not is not clear because of inadequate description in literature. These rarer RASopathies are capillary malformation-arteriovenous malformation (CM-AVM), hereditary gingival fibromatosis (HGF), and autoimmune lymphoproliferative syndrome (ALPS). Knowledge of these rarer RASopathies are important to create a holistic understanding of this potentially life threatening syndrome.
CM-AVM is a rarer RASopathy inherited in an autosomal dominant pattern. Its cardinal clinical characteristics are multifocal capillary malformations, sometimes associated with AVMs and fistulas [75]. The genetic cause of CM-AVM syndrome is heterozygous inactivating mutation in the gene RASA1, which like NF1, encodes a RAS-GAP [76]. The cardinal feature of CM-AVM syndrome is the multifocality of the malformations. AVMs can manifest in many tissues including the skin, muscle bone and in various internal organs including the heart and the brain. RASA1 gene mutation have also been associated with Parkes Weber syndrome and vein of Galen malformations [77]. Haploinsufficiency of p120-RAS-GAP (the protein product of RASA1 gene) causes a reduction in the hydrolysis of RAS-GTP causing an increasing in RAS-MAPK pathway signalling. Same to NS and NF1 syndromes, patients with CM-AVM may be at an elevated risk of developing tumours [10].
HGF is a gradually progressive, benign, fibrous overgrowth of the keratinized gingiva [78]. It is a genetically heterogeneous syndrome inherited in both autosomal dominant and recessive pattern. A rare autosomal dominant form of HGF, HGF Type 1 results from an insertion mutation in the SOS1 gene. The insertion mutation causes a frameshift that generated 22 novel amino acids prior to a premature stop codon that abolished four proline-rich SH3 binding domains required for GRB2 binding in the C terminus [79]. Ectopic expression of the SOS1 protein mutation reveals the truncated protein localizes to the plasma membrane without growth factor binding, causing RAS hyperactivation and sustained signalling in the RAS-MAPK pathway [80]. In HGF Type 1 has no observable development effects due to SOS1 gene mutation, whereas in NS activating SOS1 mutation is associated with development effects [10].
ALPS is a rare form of RASopathy that manifests with defective lymphocyte apoptosis, an accumulation of non-malignant lymphocytes and increased risk of developing haematological malignancies [81]. In a majority of cases, ALPS results from impaired extrinsic Fas-receptor mediated apoptosis due to mutation associated with the CD95 pathway [82]. A germline mutation in NRAS may also cause ALPS independent of CD95 pathway. NRAS mutation causes ALPS by activating G13D amino acid residue substitution resulting in the stabilization of the active GTP-bound form of NRAS and increased signalling of the RAS-MAPK pathway. Increased ERK phosphorylation due to NRAS activation, inhibits the expression of the apoptosis promoting protein BCL-2-interacting mediator of cell death in lymphocytes, in turn inhibits intrinsic mitochondrial apoptosis [81]. The effects of activating mutations in HRAS and KRAS genes have developmental effects but the activating NRAS mutation associated with ALPS does not affect development [11].
Considerable work has been performed to understand the pathogenic mechanisms underlying cardiac defects in RASopathies. These efforts have focused on HCM in RASopathies although cases of DCM have also been reported. The main mechanisms postulated to cause HCM in RASopathies are germline hyperactivation of the RAS-MAPK pathway and RAS-MAPK independent mechanisms. In the rarer DCM form of RASopathy-associated CM, the underlying mechanisms remain unclear, although the overlap between genes causing HCM and DCM and the roles of RAS pathway signalling in myocardial biology may be useful in providing an explanation.
Hypertrophic cardiomyopathy variant
Germline hyperactivation of the RAS/MAPK pathway: RASopathy-CM has been associated with HCM, and the ESC classification of CMs lists it as a secondary aetiology of HCM, rather than a specific and a distinct form of CM [2]. Thus, most literature describe the pathogenesis of RASopathy-CM is terms of HCM. Functional analyses of a wide variety of mutations both in vitro and in vivo and in animal models of RASopathies have led to the general concept of RAS-MAPK hyperactivation as the primary pathogenic mechanism of CM in these patients. Thus, sustained RAS-MAPK activation has a causal link to the abnormal development of several tissues resulting in some congenital symptoms such as cardiac and craniofacial abnormalities as well as alterations in response to different hormones leading to endocrine dysfunction. Several mouse models have demonstrated the contribution of germline hyperactivation of different RAS-MAPK signalling pathways, which has greatly contributed to the current understanding of the differences between RASopathy syndromes.
A mouse model of NS bearing mutation in the RAF1 gene generated using homologous recombination in embryonic stem cell recapitulated the major phenotypic features of NS including HCM, observed as early as two weeks. The mice model demonstrated increased signalling through the RAS-MAPK cascade in multiple cell types, including cardiac fibroblasts and neonatal cardiomyocytes. Consistent with this evidence, treatment with a MEK inhibitor reversed the HCM in RAF1 mice [83].
A mouse model of NS bearing mutation in the SOS1 generated using homologous recombination in embryonic stem cells recapitulated phenotypic features of RASopathies including aortic valve leaflet thickening observed after 8.5 months of age in 40% of the mice and 20% exhibited HCM with increased signalling through the RAS-MAPK and Rho-GTPase Rac [84]. Since HCM has a negative association with SOS1 mutations in humans, these latter findings in this mouse model are less informative.
A mouse model of NSML with mutation in the PTPN11 gene generated using homologous recombination in embryonic stem cells recapitulated several features of the NSML phenotype including HCM with postnatal onset. The mouse model demonstrated increased signal flow through the PI3K-AKT-mTOR pathway in cardiomyocytes, whereas ligand-evoked ERK phosphorylation was observed to be impaired in these mice. Treatment of the PTPN11 mice with rapamycin (an mTOR inhibitor) or with the AKT inhibitor reversed HCM [85,86]. These findings support previous data demonstrating a positive role of NSML-associated PTPN11 mutation on AKT activation [87].
A mouse model with mutation in the HRAS gene generated through homologous recombination in the embryonic stem cells recapitulated some features of the CS but the cardiovascular features were not easily interpretable. These mouse models exhibited cardiac hypertrophy with fibrosis by four months of age. However, systematic hypertension was absent. Treating the mice with angiotensin converting enzyme inhibitor (ACE-I) captopril reversed the cardiac hypertrophy [88]. A conditional HRAS allele may be needed to dissect the direct effects on the myocardium from the indirect effects.
RAS/MAPK independent mechanisms: In addition to RAS-MAPK hyperactivation, several signalling pathways appear dysregulated in RASopathies and may be involved in their pathophysiology. Several mouse models have shown hyperactivation of P13K/AKT signalling in NSML [84-86,89], SOS1-associated and HRAS-associated CS/CFCS [87-89], and KRAS-related NS/CFCS [90,91,93-96]. From a functional standpoint, several cellular and animal models of NSML associate cardiomyocyte hypertrophy with P13K/AKT/mTOR hyperactivation. Treatment with rapamycin or AKT inhibitor and AKTI genetic invalidation reverts HCM [84-86,97,98]. In a recent study, a 12-week everolimus (a rapamycin analogue) treatment improved cardiac function in an infant with NSML and rapidly progressive HCM. Although cardiac hypertrophy did not reverse within the 12-week period, the report provides important insight into new therapies to alleviate HCM [8]. The observation that hyperactivation of both RAS-MAPK and P13K/AKT pathways leads to similar HCM-inducing effect remains unclear. However, the possibility that dysregulation of both pathways contributes jointly to HCM pathophysiology has not been addressed in the same model although crosstalk between the two is well-established [99]. In addition to P13K dysregulation, CS-associated CM have been casually linked to upregulation of the renin-angiotensin II system, driving a hypertensive phenotype and pointing to angiotensin convertase inhibitors as potent strategies [100].
Nearly all clinical forms of RASopathy-associated CM present with hypertrophied ventricular myocardium. However, some case reports have suggested in some RASopathies, a variant of DCM may develop, although a rarity [56,101]. The observation of DCM in some RASopathy patients has led to the exploration of an aetiological link between mutations in the RAS-MAPK pathway genes and the development of DCM [102]. The basis of this exploration were the known roles of the RAS pathway signalling in myocardial biology and the overlap between genes causing HCM and DCM in general. Out of nine RAS-MAPK genes screened, missense mutation occurred in only one RAF1 gene in five out of 218 individuals of South Asia ancestry with DCM. In subsequent screening of RAF1 in individual cohorts of variable ancestry revealed small numbers of additional missense and one frameshift mutation in South/North Indian and Japanese cohorts but none in an Italian cohort. No individual with a RAF1 mutation had any extra-cardiac involvement to suggest RASopathy syndrome. Although limited families were available for analysis, one individual with RAF1 gene variant arose de novo. Available clinical information reveals only one difference among individuals with RAF1 gene mutation in comparison to others – the mean age of presentation was younger (12.6 versus 20 years) [102].
Overall, 9% of DCM cohort presenting in childhood or adolescent have RAF1 mutations. The exploration of the functional impact of the RAF1 gene mutations through transient expression in human embryonic cells revealed the effects of MAPK activation after ligand stimulation was inconsistent among the various mutations and different to those linked with RASopathy-associated mutations causing HCM [102]. Notably, the over-expression of the DCM-associated RAF1 mutations resulted in a consistent activation of AKT and its downstream target tuberin. Examination of the effects of DCM-associated RAF1 mutations in vivo, zebrafish models generated by injecting mRNAs into 1-cell embryos. Over expression of two RAF1 mutations resulted in a cardiac phenotype with elongation of the atrial and ventricular chambers, marked pericardial oedema and reduced heart rates, whereas the over-expression of the wild-type RAF1 did not perturb heart development. In the HEK293 cells, ERK activation remained unaltered in the zebrafish models but AKT was hyper-activated. The blocking of AKT hyper-activation using rapamycin partially reversed cardiac abnormalities. Overall, this study reveals the existence of a non-syndromic RASopathy due to RAF1 mutation that can cause DCM presenting in childhood or in the adolescent. The biochemical profile of DCM-RASopathy, same to NSML, caused AKT activation, suggesting possible therapeutic efficacy using rapamycin or related drugs that block signalling through mTOR [102]. Short-term therapy with mTOR inhibitor has recently been used to prevent congestive HF in NSML patients with a severe form of HCM although no reversal of cardiac hypertrophy was observed [8,85].
Meta-analysis of cardiac features and management
The seminal description of a RASopathy (NF1) was in 1882 but the initial genetic association of a RASopathy (the NF1 gene) was as recent as 1990. A decade later, the involvement of a second gene was discovered, PTPN11 gene, in NS [103]. Initially, the syndrome was termed neuro-cardio-facio-cutaneous syndrome after its most frequent clinical features. Subsequent identification of genetic defects in the RAS-MAPK pathway led to their classification as RASopathies. Despite a clinical overlap between RASopathies, each individual syndrome exhibits extensive clinical variability as well as within families and between unrelated individuals having the same genetic mutation [9-11]. These clinical overlap and variability poses significant challenges for r definitive diagnosis as well as renders the development of generalized diagnostic criteria clinically infeasible. Correct diagnosis is clinically relevant for future follow-up because different syndromes have different prognoses concerning certain features. In addition, studies on the diagnosis of RASopathy-associated CM have focused on genetic features while others have centred research on the prevalence of CM and other cardiac manifestations in patients with RASopathies. With clinical variability, RASopathy-associated CM lacks pathognomonic and current diagnostic work-up depend on detecting genetic mutations and accompanied cardiac manifestations. The present meta-analysis aggregates published data on patients with RASopathies to determine the common genetic mutations and pathologic cardiac features. Such a determination is useful to refine therapeutic approaches that is currently tailored to specific phenotype including symptoms, arrhythmias, the degree of hypertrophy and LV outflow tract obstruction (LVOTO).
A systematic search was performed on online databases PubMed, Cochrane and Google Scholar for relevant studies on RASopathies. Reference list of the included studies were screened to identify additional studies missed in the online search. The key terms used in the online search were cardiomyopathy, cardiac defects, cardiovascular abnormalities, RASopathies and each of the following individual diseases – Noonan syndrome, Noonan syndrome with multiple lentigines, Costello syndrome, Neurofibromatosis Type 1, cardiofaciocutaneous syndrome, Legius syndrome and Noonan syndrome with loose Anagen hair. The eligibility criteria were studies that enrolled patients with RASopathies, evaluated genetic and/or cardiac defects, and reported numeric outcomes of the genetic and cardiac defects in these patients. The excluded studies were case reports, conference papers and review articles.
Eleven studies met the inclusion criteria and formed the final data set for a pooled analysis [5,13,26,48,50,52,68,104-107]. These studies were relatively recent, published between 1999 [68] and 2019 [13]. All the eleven studies adopted a retrospective observational design. The primary objectives of the 11 studies were to determine prevalence of cardiac defects and genetic mutation in patients with RASopathies. Most of the studies [3,104,106,107] evaluated patients with RASopathies in general, three each evaluated NS [5,26,68] and NF1 [48,50,52], and one evaluated NSML [105]. The total number of patients enrolled in the 11 studies was 1,572 at a mean age of 10.2 years (range 7 to 14). In five studies [5,13,50,52,105], the male to female representation showed a slight male majority (58.6%).
Cardiac defects are common in patients with RASopathies. In five studies enrolling 895 had a cardiac defect, translating into an event rate (ER) of 59.2% (95% CI: 42.5% to 74.0%; Figure 1) [5,13,48,50,52,104-107]. HCM was also a common occurrence. In eight studies enrolling 980 patients, 155 had HCM with an event rare of 17.5% (95% CI: 8.9% to 31.5%; Figure 2) [5,13,26,48,50,68,104,105]. Analysis of the total percentage of individual cardiac defects reveals the most common defect is pulmonary stenosis (46.8%) followed by LVOTO (44.7%), secundum atrial septal defect (13.4%) and ventricular septal defect 11.1%). Other common cardiac defects occurring with > 10% of RASopathy patients are patent ductus arteries (8.9%), mitral valve regurgitation (5.6%), mitral valve prolapse (5.1), aortic root dilation (4.8%), aortic coarctation (3.8%), septal aneurysm (3.5%), Tetralogy of Fallot (2.4%), valvar aortic stenosis (2.4%), aortic valve regurgitation (2.0%), and tricuspid valve regurgitation (1.5%; Table 1).
Table 1. Summary of Included Studies
1st Author |
Year |
No. |
Male (n) |
Mean Age |
Type |
Cardiac Defects (n) |
HCM (n) |
Primary Objective |
Key Findings |
Marino [68] |
1999 |
136 |
NR |
NR |
NS |
NR |
14 |
To report the prevalence of CHD in NS children |
Left-sided lesions (aortic coarctation and anomalies of the mitral valve) are frequent NS-CHD patients |
Tedesco [48] |
2002 |
48 |
NR |
10 |
NF1 |
13 |
2 |
To evaluate the prevalence of CVD abnormalities in NF1 patients |
A high prevalence of CVD abnormalities. Congenital lesions have potential long-term hemodynamic consequences to justify an early diagnosis. |
Lama [50] |
2004 |
69 |
36 |
11 |
NF1 |
13 |
2 |
To evaluate blood pressure and CVD involvement in NF1 children |
Cardiac defects are common in BF1 patients. No significant clinical and cardiac differences between the normotensive and hypertensive NF1 patients |
Hickey [26] |
2011 |
138 |
NR |
NR |
NS |
NR |
30 |
To examine links between relationships and survival implications between structural heart disease and NS-HCM |
NS-HCM frequently coexists with structural cardiac malformations. Late survival is significantly worse for NS-HCM than non-syndromic HCM |
Ezquieta [104] |
2012 |
230 |
NR |
NR |
All |
156 |
18 |
To investigate the genotypic profile and the diagnostic yield of molecular analysis in NS patients |
Most patients (94%) with a positive genotype had known congenital heart disease, 79% pulmonary stenosis and 12% hypertrophic cardiomyopathy. |
Carcavilla [105] |
2013 |
19 |
13 |
7.4 |
NSML |
17 |
12 |
To characterize the clinical and molecular features of LEOPARD syndrome patients |
NSML display a higher prevalence of HCM. Given its clinical implications, active search for HCM is warranted in NS patients. |
Prendiville [5] |
2014 |
293 |
176 |
14 |
NS |
237 |
47 |
To define the spectrum of cardiac morphology and clinical course of NS |
NS patients have a distinct spectrum of cardiac phenotypes with a natural history and response to therapy atypical to non-syndromic heart disease |
Incecik [52] |
2015 |
65 |
37 |
9 |
NF1 |
11 |
NR |
To evaluate cardiac abnormalities in patients with NF1 |
Cardiac abnormalities have potential long-term hemodynamic consequences that justify an early diagnosis. |
Calcagni [106] |
2016 |
371 |
NR |
NR |
All |
298 |
NR |
To analyze cardiac morbidity and mortality in patients with RASopathy |
In a large cohort of RASopathy patients, cardiac abnormalities are a frequent observation |
Jhang [107] |
2016 |
155 |
NR |
NR |
All |
118 |
NR |
To review the clinical manifestations and genotype-phenotype associations on cardiac lesions of RASopathies |
Variety of clinical presentations and their progression of severity, proper management with regular long-term follow-up of these patients is essential |
Chen [13] |
2019 |
47 |
27 |
NR |
All |
32 |
30 |
To review the mutation spectrum and clinical outcome of RASopathy patients |
RASopathy-associated HCM is characterized by early-onset cardiac hypertrophy and a high prevalence of co-existing CHD |
CHD: Congenital Heart Diseases; CVD: Cardiovascular; HCM: Hypertrophic Cardiomyopathy; NF1: Neurofibromatosis Type 1; NS: Noonan Syndrome; NR: Not Reported
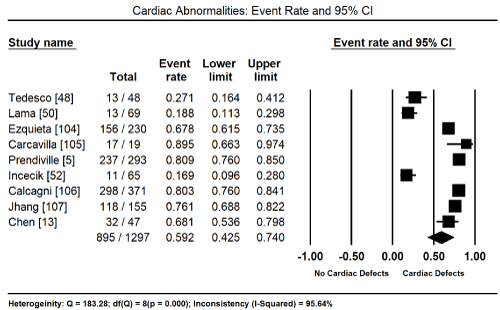
Figure 1. Event Rate and 95% CI for Cardiac Abnormalities in RASopath
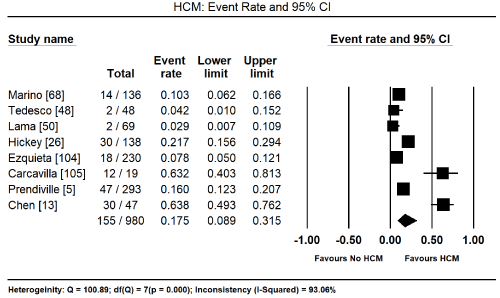
Figure 2. Event Rate and 95% CI for HCM in RASopathies
The present analysis finds cardiac defects are a common manifestation in all patients with RASopathies. They occur in approximately 60% and HCM in 17.5% of all patients with RASopathies. Pulmonary stenosis, LVOTO and secundum atrial septa defect were also common with an event rate of 44.7%, 13.4% and 11.1%. However, due to a smaller number of studies and a wide variation in baseline characteristics of patients warrants additional studies to clarify the most frequent cardiac features. Additional studies are also essential to determine cardiac manifestations by type of RASopathy. These findings are consistent with current reviews and diagnostic guidelines that recommend the diagnosis of the underlying genetic mutation (type of RASopathy) and cardiac manifestation to support the diagnosis of CM. Cardiac defects in RASopathies are clinically relevant to guide the choice of an optimal therapeutic intervention.
The clinical diagnosis for RASopathy-associated CM rests determination of RASopathy and evidence of associated pathologic cardiac defects. The initial diagnosis relies on clinical recognition of the phenotypic features and molecular testing used to confirm the clinical diagnosis [9]. The correlation between clinic and molecular diagnosis often relies on the clinical diagnostic criteria. Although not all causative genes for RASopathy have been identified, the progressive growth of genotype-phenotype correlations will increase the importance of molecular diagnosis and assist in overcoming intrinsic limitation of clinical diagnosis, improve patient management and aid in the design of clinical trials to develop potential treatment options. At present, clinical diagnosis rests on demonstrating the presence of cardiac defects, which the present meta-analysis suggest are prevalent in patients with RASopathies [9].
The cardinal cardiac characteristic of RASopathy-associated CM supporting clinical diagnosis is asymmetrical hypertrophy with major involvement of basal interventricular septum and sometimes the apex, the mid-point and the posterior wall of the LV. The 2014 ESC guidelines on the diagnosis and management of HCM recommends LV wall thickness > 15 mm as the gold standard for the diagnosis of HCM in adults [108]. Since minor degrees of LV wall thickening (13-14 mm) may occur in genetic and non-genetic disorders, diagnosis of HCM may require additional evaluation of supporting features such as family history, extra-cardiac symptoms and signs, ECG abnormalities and laboratory examination and multimodal cardiac imaging. In children, diagnosis requires segmental LV wall thickness > 2 standard deviations greater than the predicted mean for age, sex and height-weight ratio [108]. Hypertrophy in children is often non-specific and asymmetrical with the ratio of septal thickness to LV free wall > 1.3 to 1.0. In comparison with other types of HCM, RASopathy-associated HCM exhibits more ventricular hypertrophy and increased prevalence of LVOTO as well as a higher grade of RV hypertrophy [7] (Table 2).
Table 2. Cardiac Defects (%) In RASopathies
Cardiac Defect |
Event Rate (%) |
95% CI |
Studies (References) |
Pulmonary Valvular Stenosis |
46.8 |
38.9-55.0 |
5,13,26,104,105 |
LV Outflow Tract Obstruction |
44.7 |
31.2-58.9 |
13 |
Secundum Atrial Septal Defect |
13.4 |
6.4-26.0 |
5,13,26,48,50,52,68 |
Ventricular Septal Defect |
11.1 |
8.6-14.2 |
5,13,26,52 |
Patent Ductus Arterios |
8.9 |
6.1-12.7 |
5 |
Mitral valve Regurgitation |
5.6 |
3.5-8.8 |
48,50,52,68 |
Mitral Valve Prolapse |
5.1 |
3.307.9 |
5,48,50 |
Aortic Root Dilation |
4.8 |
3.0-7.7 |
5,48 |
Aortic Coarctation |
3.8 |
1.8-7.9 |
5,26,50,68,105 |
Septal aneurysm |
3.5 |
1.3-8.9 |
48,50 |
Tetralogy of Fallot |
2.4 |
1.4-4.1 |
5,26,68 |
Valvar Aortic Stenosis |
2.4 |
1.1-4.9 |
5 |
Aortic Valve Regurgitation |
2.0 |
0.9-4.5 |
5,48,50,52 |
Tricuspid Valve Regurgitation |
1.5 |
0.2 -10.1 |
52 |
Other morphological cardiac defects that can support clinical diagnosis RASopathy-associated CM include structural abnormalities such as elongation of the mitral valve with anomalous insertion (associated with mitral valve prolapse and regurgitation as reported in the present review) and abnormal displacement of papillary muscles [109,110]. Same to the present findings, congenital abnormalities of the pulmonary valve are frequent in RASopathies. Particularly, in NS, severe HCM has an early onset associated with an increased risk of long-term morbidities to suggest a tailored management based on the aetiology of HCM [111]. In this patient cohort, severe PVS is often associated with HCM. Dilated coronary arteries manifest in the context of HCM due to RAS-MAPK mutations irrespective of the extent of HCM. Due to the higher degree of hypertrophy and the associated valvular defects, several pathophysiologic features need consideration during clinical evaluation such as LV outflow tract obstruction, diastolic dysfunction, myocardial ischemia and cardiac arrhythmias [106].
The presence of diastolic dysfunction in patients with RASopathy-associated CM in the setting of abnormal relaxation and impaired ventricular compliance may also support diagnosis. RASopathy patients with diastolic dysfunction often present with are exertional dyspnoea and exercise intolerance since infancy. Infants have pronounced symptoms of feeding difficulties and the failure to thrive [7]. The NS phenotype manifests with marked HCM and diastolic dysfunction, which may explain the heterogeneous symptoms and clinical events, and define appropriate therapeutic options. During clinical evaluation, clinicians should also consider LVOTO diagnosis due to systolic anterior movement of the mitral valve or due to mid-cavitary LV obstruction [33]. Exertional activities appear to worsen LVOTO, which is more severe in NS children [7]. Myocardial ischemia is a common occurrence in HCM patients including adolescents with RASopathies, which manifest because of impaired myocardial perfusion and increased myocardial mass.
Abnormalities of the coronary arteries (the dilation of the main left coronary artery, anterior descending artery or right coronary artery) may also occur in patients RASopathy-associated HCM [33]. Abnormalities in coronary arteries could be a consequence of CHD independent from the specific genetic change could help use explain the increased risk of myocardial ischemia in NS-HCM patients. Frequent symptomatic manifestations in RASopathy-associated CM are arrhythmias (supraventricular and ventricular ectopic beats, and non-sustained ventricular tachycardia). The current paediatric HCM guidelines including those with NS provide markers for assessing the risk of sudden cardiac death. LV wall thickness > 6 standard deviation and /or a previous history of life-threatening arrhythmias are indication for implantable cardioverter defibrillator (ICD) implantation [7]. Children with HCM have other specific risk factors such as congestive HF, increased LV posterior wall thickness and the presence of coronary myocardial bridging [112]. There is need for a specific risk score tool for the estimation of sudden cardiac death in NS children.
Pathological cardiac features in RASopathies support the clinical diagnosis of CM and guide the choice of the most appropriate treatment. However, clinical management of RASopathy-associated CM remains a challenge for cardiologists. Generally, since genetic diseases including RASopathies have a very low prevalence, they remain neglected in pathophysiological research. Consequently, they lack aetiological treatment. The sole option for the clinical management of RASopathies is symptomatic therapies for CMs developmental delay but treatment efficacy and safety of this option remain debatable [102]. Tailored treatment approach based on specific phenotype includes cardiac symptoms, arrhythmias, the extent of hypertrophy and the presence of LVOTO [106].
Symptomatic management: Symptomatic management of patients with RASopathy-associated CM as applies treatment strategies developed for HCM and other non-syndromic congenital heart defects. Infants with NS-associated CM have worse prognosis compared with older children, and a more severe cardiac involvement correlates with a lower survival rate due to significant diastolic dysfunction [27]. Patients with CS-associated CM require periodic cardiac follow-up in the first two years of life, and annual evaluation and risk stratification at puberty. The severity of hypertrophy should guide the frequency of cardiac evaluation. Clinical management should also include periodic examination in asymptomatic patients [36].
Medical therapy is the first-line therapy in the treatment of the majority of patients with RASopathies. General guidelines for medical treatment of HCM includes drugs such as beta-blockers, dysopiramide and L-type calcium channel blockers to supress symptoms and the degree of LVOTO [113]. The ESC guidelines on HCM diagnosis and management recommends beta-blockers as the first-line medicine for management of HCM using non-vasodilating beta-blockers titrated to the maximum tolerated dose. Beta-blocker therapy improves diastolic dysfunction and ventricular remodelling [113]. Indication for dysopiramide is often an addition to beta-blocker to reduce the degree of obstruction. Calcium channel blockers are effective in patients non-responsive to beta-blockers. In RASopathy patients who have developed congestive HF, diuretics are useful to counterbalance the effect on hypotension and LVOTO [108].
Surgical myectomy is the preferred treatment for patients with RASopathy-associated CM who appear symptomatic for severe LVOTO despite optimal medical therapy [114,115]. The presence of LVOTO in children with RASopathy-associated CM exhibit earlier cardiac symptoms compared with non-syndromic HCM as well as suffer from greater mortality. Some clinical setting advocate an early cardiac transplantation for selected patients ineligible for surgery [27]. However, in specialized centres experienced in paediatric surgical myectomy, surgical relief of LVOTO is a practical alternative to transplantation [116].
Orthotropic cardiac transplantation is a rare intervention in RASopathy-associated CM [28]. However, it may be considered in eligible patients with LVEF < 50% and New York Heart Association (NYHA) functional class III to IV despite optimal medical treatment, intractable ventricular arrhythmias or severe diastolic dysfunction (even in the case of preserved LVEF) [108]. Orthotropic cardiac transplantation can also be a life-saving strategy for some HCM patients but the risk factors and outcomes lack scientific scrutiny but rejection is a potential cause of death in these patients [117].
Specific data with respect to genotype-phenotype RAS-MAPK cascade versus eligibility for cardiac transplantation is lacking. An accurate analysis of all additional risk factors should be considered carefully to define better the timing of listing and determine patient subgroup may derive optimal benefit from a cardiac transplant [118]. Cumulative wait list mortality based on the Paediatric CM Registry and Australian Registry is significantly higher within 2 to 3 months after listing. The mortality rate is much higher in infants with HCM suggesting a more malignant natural history for the infantile form in comparison to HCM with a late onset [119,120]. The relatively higher mortality rate following orthotropic cardiac transplantation may be also related to high-priority and critically ill RASopathy infants. Thus, orthotropic cardiac transplantation in children with RASopathies should be considered as early as possible and prior to clinical deterioration because of favourable outcomes following success cardiac transplantation [118].
In addition to HF therapies, symptomatic treatment for NS-associated growth retardation with recombinant human growth hormone (rhGH) has had debatable efficacy on both growth, and cardiac function and symptoms. Most studies report enhanced growth during the first years of treatment but the benefit of long-term outcomes remain uncertain. In RASopathy patients with CM, rhGH treatment might potentially affect ventricular development in NS-patients with cardiac defects although two studies did not find any cause for concern [121,122]. Evidence for the effect of rhGH on cardiac defects in patients with RASopathy is sporadic and comparison across studies is difficult due to different protocols used (variable enrolment, ages, treatment durations and doses) and outcome criteria. None of the studies evaluation the effect of rhGH on cardiac morphology and function are randomized clinical trial, and thus, major biases may affect all the studies [123].
Targeted molecular therapies: The identification of specific gene mutations and pathogenic mechanisms now provide new insights into specific molecular therapies for RASopathies. Shared genetic mutations altering the RAS-MAPK and phosphinositide-3-kinase/AKT pathways are promising targets for developing specific molecular therapies for RASopathy-associated CM [11]. However, because of the variations in the pathogenetic mechanisms of RASopathy-associated CMs, no single drug is likely to be effective for all the RASopathies. In addition, a better understanding of long-term systemic consequences of RASopathy-associated mutations will also provide insight into additional processes to target alleviating specific traits [102].
In NSML caused by mutation in the PTPN11 gene, CM appears to result from increased activation of AKT via mTOR. Already, the Federal Drug Administration in the U.S. has approved several mTOR inhibitors such as rapamycin and related compounds (also known as rapalogues). Rapalogues are immunosuppressive and widely used to prevent post-transplantation rejection and coronary re-stenosis after stent placement. Serious side effect from the use of rapalogues include interstitial pneumonitis, elevated risk for cancer and diabetes.
Phase III trial is ongoing on the efficacy of rapalogues (everolimus) on tuberous sclerosis (a genetic disease due to increased signalling via the AKT) and Phase II trial with everolimus in affected children for neurocognition. The everolimus trial is establishing the safety of the drug in children, thereby providing an opportunity to use the drug and other rapalogues for NSML-associated CM. The two trials inspired an ongoing trial on the use of rapamycin therapy of HCM in patients with LEOPARD syndrome” (https://ncats.nih.gov/trnd/projects/active/rapamycin-hypertrophic-cardiomyopathy-leopard). The trial first evaluates the use of rapalogues on existing NSML-mouse models, and if successful advance to early phase clinical trials in NMSL individuals. Other RASopathy-associated CM appear to result from gain-of-function mutation causing an increase in signalling via the ERK although HRAS gene mutations appear to cause hyperactivation of both AKT and ERK.
Preclinical work on mouse model with RAF1L613V inhibition of MEK could prevent the development of HCM but no information is available on the reversal of HCM, which is clinically relevant. Novartis is sponsoring a Phase II open-label trial examining the safety, pharmacokinetic and tolerability for their MEK inhibitor in adults with NS-associated CM (ClinicalTrials.gov identifier: NCT01556568). The primary end-points for the clinical trial are changes in LV mass at the third and sixth month of treatment relative to baseline.
The use of molecular inhibitors of the central RAS-MAPK pathway promises safety and efficacy for RASopathies patients, concerns exist on the safety of long-term therapy (relative to the duration of the typical chemotherapy courses for cancers) especially for infants and growing children. In addition, RASopathy children may respond differently to these new drugs because their of a broader increased RAS pathway signalling compared to cancer patients where abnormal signalling is restricted to the somatically mutated tumour.
Another promising approach to the treatment of RASopathies is drugs that alter the RAS pathway signalling in a less dramatic pattern. Instead of the potent inhibitor of the central RAS-mediated signalling pathways, small molecules that alter the signalling more subtly including altering cross talk or feedback mechanisms could also be effective. Already, Lee and colleagues [124] demonstrated mouse model of NS with PTPN11 D61G missense mutation exhibiting deficiency in learning and memory that HMG-Co reductase-inhibitor lovastatin therapy could rescue. Lovastatin, approved by the Food and Drug Administration in the U.S. appears to decrease rather than inhibit RAS signaling. However, the efficacy of Lovastatin for the treatment of RASopathy-associated CM remained untested although the beneficial outcomes on neurocognition suggests promising results [11].
RASopathies are a group of syndromes that share germline mutations in the genes encoding components of the RAS-MAPK pathway. The RAS genes is a multi-gene family that includes HRAS, NRAS, KRAS, PTPN11, SOS1, RAF1, SHOC2, and CBL. Noonan syndrome (NS) is the most prevalent and its phenotypic variants are NS with multiple lentigines, neurofibromatosis type 1, Costello syndrome, Legius syndrome, and cardiofaciocutaneous syndrome. The primary pathogenic mechanism is germline hyperactivation of the different RAS-MAPK signalling pathways leading to abnormal development of several tissues leading to the development of cardiac and craniofacial abnormalities. P13K/AKT/mTOR hyperactivation is another possible pathogenetic mechanism resulting in cardiomyocyte hypertrophy. The most prevalent variant of CM in RASopathy is HCM although there are also sporadic cases of DCM. Diagnosis relies on genetic testing to establish genetic mutation and phenotypic expression, and evidence of myocardial involvement to demonstrate CM. Clinical overlap between RASopathies, extensive cardiac variability within each phenotype, as well as within families and between unrelated individuals with the same mutation has complicated diagnosis. Treatment targets the relief of cardiac symptoms or altering the pathophysiologic mechanism of RASopathies. Symptomatic management follows guidelines for HCM and consists of beta-blocker (first-line therapy), dysopiramide and calcium channel blockers for those non-responsive to beta-blockers. Surgery (cardiac transplantation) for those non-responsive to medical therapy. Research is ongoing for molecular therapies that prevent or reduce RAS-MAPK pathway signalling with promising results in mouse models but untested in humans.
- Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, et al. (2006) Contemporary definitions and classification of the cardiomyopathies: an American Heart Association scientific statement from the council on clinical cardiology, heart failure and transplantation committee; quality of care and outcomes research and functional genomics and translational biology interdisciplinary working groups; and council on epidemiology and prevention. Circulation 113: 1807-1816. [Crossref]
- Farag ES, Planken RN, Boekholdt SM, Kluin J (2017) Systolic anterior motion of the tricuspid valve in a patient with hypertrophic obstructive cardiomyopathy Interact Cardiovasc Thorac Surg 25: 496-497. [Crossref]
- Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, et al. (2007) Classification of the cardiomyopathies: a position statement from the european society of cardiology working group on myocardial and pericardial diseases. Eur Heart J 29: 270-276. [Crossref]
- Spartalis M, Tzatzaki E, Athanasiou A, Spartalis E (2017) Comment. noonan syndrome and biventricular hypertrophic obstructive cardiomyopathy. Interact Cardiovasc Thorac Surg 25: 498. [Crossref]
- Prendiville TW, Gauvreau K, Tworog-Dube E, Patkin L, Kucherlapati RS, et al. (2014) Cardiovascular disease in noonan syndrome. Arch Dis Child 99: 629-634. [Crossref]
- Schinkel AF, Vos J (2007) Biventricular hypertrophic obstructive cardiomyopathy in noonan syndrome. Int J Cardiol 115: e22-e23. [Crossref]
- Cerrato F, Pacileo G, Limongelli G, Gagliardi MG, Santoro G, et al. (2008) A standard echocardiographic and tissue doppler study of morphological and functional findings in children with hypertrophic cardiomyopathy compared to those with left ventricular hypertrophy in the setting of noonan and leopard syndromes. Cardiol Young 18: 575. [Crossref]
- Hahn A, Lauriol J, Thul J, Behnke-Hall K, Logeswaran T, et al. (2015) Rapidly progressive hypertrophic cardiomyopathy in an infant with Noonan syndrome with multiple lentigines: palliative treatment with a rapamycin analog. Am J Med Genet A 167A: 744-751. [Crossref]
- Rauen KA (2013) The rasopathies. Annu Rev Genomics Hum Genet 14: 355-369. [Crossref]
- Tidyman WE, Rauen KA (2009) The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev 19: 230-236. [Crossref]
- Gelb BD, Roberts AE, Tartaglia M (2015) Cardiomyopathies in noonan syndrome and the other RASopathies. Prog Pediatr Cardiol 39: 13-19. [Crossref]
- Yoon S, Seger R (2006) The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors 24: 21-44. [Crossref]
- Chen H, Li X, Liu X, Wang J, Zhang Z, et al. (2019) Clinical and mutation profile of pediatric patients with RASopathy-associated hypertrophic cardiomyopathy: results from a Chinese cohort. Orphanet J Rare Dis 14: 29. [Crossref]
- Roberts AE, Allanson JE, Tartaglia M, Gelb BD (2013) noonan syndrome. Lancet 381: 333-342. [Crossref]
- Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, et al. (2001) Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause noonan syndrome. Nat Genet 29: 465-468. [Crossref]
- Tartaglia M, Pennacchio LA, Zhao C, Yadav KK, Fodale, et al. (2007) Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat Genet 39: 75-79. [Crossref]
- Roberts AE, Araki T, Swanson KD, Montgomery KT, Schiripo T, et al. (2007) Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat Genet 39: 70-74. [Crossref]
- Pandit B, Sarkozy A, Pennacchio LA, Carta C, Oishi K, et al. (2007) Gain-of-function RAF1 mutations cause Noonan and leopard syndromes with hypertrophic cardiomyopathy. Nat Genet 39: 1007-1012. [Crossref]
- Razzaque MA, Nishizawa T, Komoike Y, Yagi H, Furutani M, et al. (2007) Germline gain-of-function mutations in RAF1 cause Noonan syndrome. Nat Genet 39: 1013-1017. [Crossref]
- Schubbert S, Zenker M, Rowe SL, Boll S, Klein C, et al. (2006) Germline KRAS mutations cause Noonan syndrome. Nat Genet 38: 331-336. [Crossref]
- Cirstea IC, Kutsche K, Dvorsky R, Gremer L, Carta C, et al. (2010) A restricted spectrum of NRAS mutations causes noonan syndrome. Nat Genet 42: 27-29. [Crossref]
- Cordeddu V, Di Schiavi E, Pennacchio LA, Ma’ayan A, Sarkozy A, et al. (2009) Mutation of SHOC2 promotes aberrant protein N-myristoylation and causes noonan-like syndrome with loose anagen hair. Nat Genet 41: 1022-1026. [Crossref]
- Martinelli S, De Luca A, Stellacci E, Rossi C, Checquolo S, et al. (2010) Heterozygous germline mutations in the CBL tumor-suppressor gene cause a Noonan syndrome-like phenotype. Am J Hum Genet 87:250-257. [Crossref]
- Niemeyer CM, Kang MW, Shin DH, Furlan I, Erlacher M, et al. (2010) Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukaemia. Nat Genet 42: 794-800. [Crossref]
- Burch M, Mann JM, Sharland M, Shinebourne EA, Patton MA, et al. (1992) Myocardial disarray in Noonan syndrome. Br Heart J 68: 586-588. [Crossref]
- Hickey EJ, Mehta R, Elmi M, Asoh K, McCrindle BW, et al. (2011) Survival implications: hypertrophic cardiomyopathy in noonan syndrome. Congenit Heart Dis 6: 41-47. [Crossref]
- Wilkinson JD, Lowe AM, Salbert BA, Sleeper LA, Colan SD, et al. (2012) Outcomes in children with noonan syndrome and hypertrophic cardiomyopathy: A study from the Paediatric Cardiomyopathy Registry. Am Heart J 164: 442-448. [Crossref]
- Shaw AC, Kalidas K, Crosby AH, Jeffery S, Patton MA (2007) The natural history of Noonan syndrome: a long-term follow-up study. Arch Dis Child 92: 128-132. [Crossref]
- Digilio MC, Conti E, Sarkozy A, Mingarelli R, Dottorini T, et al. (2002) Grouping of multiple-lentigines/LEOPARD and Noonan syndromes on the PTPN11 gene. Am J Hum Genet 71: 389-394. [Crossref]
- Legius E, Schrander-Stumpel C, Schollen E, Pulles-Heintzberger C, Gewillig M, et al. (2002) PTPN11 mutations in LEOPARD syndrome. J Med Genet 39: 571-574. [Crossref]
- Kontaridis MI, Swanson KD, David FS, Barford D, Neel BG (2006) PTPN11 (Shp2) mutations in LEOPARD syndrome have dominant negative, not activating, effects. J Biol Chem 281: 6785-6792. [Crossref]
- Oishi K, Zhang H, Gault WJ, Wang CJ, Tan CC, et al. (2009) Phosphatase-defective LEOPARD syndrome mutations in PTPN11 gene have gain-of-function effects during Drosophila development. Hum Mol Genet 18: 193-201. [Crossref]
- Limongelli G, Pacileo G, Marino B, Digilio MC, Sarkozy A, et al. (2007) Prevalence and clinical significance of cardiovascular abnormalities in patients with the leopard syndrome. Am J Cardiol 100: 736-741. [Crossref]
- Rauen KA (2007) HRAS and the costello syndrome. Clin Genet 71: 101-118. [Crossref]
- Siegel DH, Mann JA, Krol AL, Rauen KA (2011) Dermatological phenotype in Costello syndrome: consequences of Ras dysregulation in development. Br J Dermatol 164: 521-529. [Crossref]
- Lin AE, Alexander ME, Colan SD, Kerr B, Rauen KA, et al. (2011) Clinical, pathological, and molecular analyses of cardiovascular abnormalities in costello syndrome: a Ras/MAPK pathway syndrome. Am J Med Genet A 155A: 486-507. [Crossref]
- Hakim K, Boussaada R, Hamdi I, Msaad H (2014) Cardiac events in costello syndrome: One case and a review of the literature. J Saudi Heart Assoc 26: 105-109. [Crossref]
- Siwik ES, Zahka KG, Wiesner GL, Limwongse C (1998) Cardiac disease in Costello syndrome. Pediatrics 101: 706-709. [Crossref]
- Hinek A, Teitell MA, Schoyer L, Allen W, Gripp KW, et al. (2005) Myocardial storage of chondroitin sulfate‐containing moieties in costello syndrome patients with severe hypertrophic cardiomyopathy. Am J Med Genet A 133: 1-2. [Crossref]
- Tomita H, Fuse S, Ikeda K, Matsuda K, Chiba S (1998) An infant with costello syndrome complicated with fatal hypertrophic obstructive cardiomyopathy. Acta Paediatr Jpn 40: 608-611. [Crossref]
- Williams VC, Lucas J, Babcock MA, Gutmann DH, Korf B, et al. (2009) Neurofibromatosis type 1 revisited. Pediatrics 123: 124-133. [Crossref]
- Cawthon RM, Weiss R, Xu GF, Viskochil D, Culver M, et al. (1990) A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations. Cell 62: 193-201. [Crossref]
- Viskochil D, Buchberg AM, Xu G, Cawthon RM, Stevens J, et al. (1990) Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell 62: 187-192. [Crossref]
- Wallace MR, Marchuk DA, Andersen LB, Letcher R, Odeh HM, et al. (1990) Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. Science 249: 181-186. [Crossref]
- Huffmeier U, Zenker M, Hoyer J, Fahsold R, Rauch A (2006) A variable combination of features of Noonan syndrome and neurofibromatosis type I are caused by mutations in the NF1 gene. Am J Med Genet A 140A: 2749-2756. [Crossref]
- Stevenson DA, Viskochil DH, Rope AF, Carey JC (2006) Clinical and molecular aspects of an informative family with neurofibromatosis type 1 and Noonan phenotype. Clin Genet 69: 246-253. [Crossref]
- Kurien A, John PR, Milford DV (1997) Hypertension secondary to progressive vascular neurofibromatosis. Arch Dis Child 76: 454-455. [Crossref]
- Tedesco MA, Di Salvo G, Natale F, Pergola V, Calabrese E, et al. (2002) The heart in neurofibromatosis type 1: an echocardiographic study. Am Heart J 143: 883-888. [Crossref]
- Lin AE, Birch PH, Korf BR, Tenconi R, Niimura M, et al. (2000) Cardiovascular malformations and other cardiovascular abnormalities in neurofibromatosis 1. Am J Med Genet 95: 108-117. [Crossref]
- Lama G, Graziano L, Calabrese E, Grassia C, Rambaldi PF, et al. (2004) Blood pressure and cardiovascular involvement in children with neurofibromatosis type1. Pediatr Nephrol 19: 413-418. [Crossref]
- Sutton MG, Tajik AJ, Giuliani ER, Gordon H, Su WD (1981) Hypertrophic obstructive cardiomyopathy and lentiginosis: a little known neural ectodermal syndrome. Am J Cardiol 47: 214-217. [Crossref]
- Incecik F, Herguner OM, Erdem SA, Altunbaşak S (2015) Neurofibromatosis type 1 and cardiac manifestations. Turk Kardiyol Dern Ars 43: 714-716. [Crossref]
- Fitzpatrick AP, Emanuel RW (1988) Familial neurofibromatosis and hypertrophic cardiomyopathy. Heart 60: 247-251. [Crossref]
- Jurko Jr A, Minarik M, Minarikova E, Nagi AS (2010) Neurofibromatosis associated with hypertrophic cardiomyopathy. Saudi Med J 31: 935-936. [Crossref]
- Ritter A, Cuddapah S, Degenhardt K, Kasperski S, Johnson MP, et al. (2019) Fetal cardiomyopathy in neurofibromatosis type I: Novel phenotype and review of the literature. Am J Med Genet A 179: 1042-1046. [Crossref]
- Dogan A, Aksoy H, Okutucu S, Karakulak UN, Evranos B, et al. (2012) PP-343 Dilated cardiomyopathy in a patient with neurofibromatosis type 1. Int J Cardiol 155: S212.
- Yoon G, Rosenberg J, Blaser S, Rauen KA (2007) Neurological complications of cardio-facio-cutaneous syndrome. Dev Med Child Neurol 49: 894-899. [Crossref]
- Niihori T, Aoki Y, Narumi Y, Neri G, Cave H, et al. (2006) Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat Genet 38: 294-296. [Crossref]
- Rodriguez-Viciana P, Tetsu O, Tidyman WE, Estep AL, Conger BA, et al. (2006) Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science 311: 1287-1290. [Crossref]
- Tidyman, WE, Rauen, KA (2009) Molecular cause of cardio-facio-cutaneous syndrome. Noonan Syndrome and Related Disorders: A Matter of Deregulated Ras Signaling. In: Zenker, M., Eds. Monogr Hum Genet. 17: 73-82.
- Rauen KA, Tidyman WE, Estep AL, Sampath S, Peltier HM, et al. (2010) Molecular and functional analysis of a novel MEK2 mutation in cardio-facio-cutaneous syndrome: transmission through four generations. Am J Med Genet A 152A: 807-814. [Crossref]
- Mazzanti L, Cacciari E, Cicognani A, Bergamaschi R, Scarano E, et al. (2003) Noonan-like syndrome with loose anagen hair: a new syndrome? Am J Med Genet A 118A: 279-286. [Crossref]
- Roberts A, Allanson J, Jadico SK, Kavamura MI, Noonan J, et al. (2006) The cardiofaciocutaneous syndrome. J Med Genet 43: 833-842. [Crossref]
- Tartaglia M, Kalidas K, Shaw A, Song X, Musat DL, et al. (2002) PTPN11 mutations in Noonan syndrome: molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. Am J Hum Genet 70: 1555-1563. [Crossref]
- Lin AE, Grossfeld PD, Hamilton RM, Smoot L, Gripp KW, et al. (2002) Further delineation of cardiac abnormalities in Costello syndrome. Am J Med Genet 111: 115-129. [Crossref]
- Sarkozy A, Conti E, Deripa D, Digilio MC, Grifone M, et al. (2003) Correlation between PTPN11 gene mutations and congenital heart defects in Noonan and LEOPARD syndromes. J Med Genet 40: 704-708. [Crossref]
- Bertola DR, Kim CA, Sugayama SM, Albano LM, Wagenfuhr J, et al. (2000) Cardiac findings in 31 patients with Noonan’s syndrome. Arq Bras Cardiol 75: 409-412. [Crossref]
- Marino B, Digilio MC, Toscano A, Gianotti A, Dallapiccola B (1999) Congenital heart disease in children with Noonan syndrome: an expanded cardiac spectrum with high prevalence of atrioventricular canal. J Pediatr135:703-706. [Crossref]
- Zenker M, Buheitel G, Rauch R, Koenig R, Bosse K, et al. (2004) Genotype-phenotype correlations in Noonan syndrome. J Pediatr 144: 368-374. [Crossref]
- Brems H, Chmara M, Sahbatou M, Denayer E, Taniguchi K, et al. (2007) Germline loss‑of‑function mutations in SPRED1 cause a neurofibromatosis 1‑like phenotype. Nat Genet 39: 1120‑1126. [Crossref]
- Kratz CP, Rapisuwon S, Reed H, Hasle H, Rosenberg PS (2011) Cancer in Noonan, Costello, cardiofaciocutaneous and LEOPARD syndromes. Am J Med Genet C 157C: 83-89. [Crossref]
- Komatsuzaki S, Aoki Y, Niihori T, Okamoto N, Hennekam RC, et al. (2010) Mutation analysis of the SHOC2 gene in Noonan-like syndrome and in hematologic malignancies. J Hum Genet 55: 801-809. [Crossref]
- Mazzanti L, Tamburrino F, Scarano E, Perri A, Vestrucci B, et al. (2013) GH Therapy and first final height data in Noonan-like syndrome with loose anagen hair (Mazzanti syndrome). Am J Med Genet A 161A: 2756-2761. [Crossref]
- Cordeddu V, Di Schiavi E, Pennacchio LA, Ma’ayan A, Sarkozy A, et al. (2009) Mutation of SHOC2 promotes aberrant protein N-myristoylation and causes Noonan-like syndrome with loose anagen hair. Nat Genet 41: 1022-1026. [Crossref]
- Boon LM, Mulliken JB, Vikkula M (2005) RASA1: variable phenotype with capillary and arteriovenous malformations. Curr Opin Genet Dev 15: 265-269. [Crossref]
- Eerola I, Boon LM, Mulliken JB, Burrows PE, Dompmartin A, et al. (2003) Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet 73: 1240-1249. [Crossref]
- Revencu N, Boon LM, Mulliken JB, Enjolras O, Cordisco MR, et al. (2008) Parkes Weber syndrome, vein of Galen aneurysmal malformation, and other fast-flow vascular anomalies are caused by RASA1 mutations. Hum Mutat 29: 959-965. [Crossref]
- Hart TC, Pallos D, Bowden DW, Bolyard J, Pettenati MJ, et al. (1998) Genetic linkage of hereditary gingival fibromatosis to chromosome 2p21. Am J Hum Genet 62: 876-883. [Crossref]
- Hart TC, Zhang Y, Gorry MC, Hart PS, Cooper M, et al. (2002) A mutation in the SOS1 gene causes hereditary gingival fibromatosis type 1. Am J Hum Genet 70: 943-954. [Crossref]
- Jang SI, Lee EJ, Hart PS, Ramaswami M, Pallos D, et al. (2007) Germ line gain of function with SOS1 mutation in hereditary gingival fibromatosis. J Biol Chem 282: 20245-20255. [Crossref]
- Oliveira JB, Fleisher T (2004) Autoimmune lymphoproliferative syndrome. Curr Opin Allergy Clin Immunol 497-503. [Crossref]
- Bidere N, Su HC, Lenardo MJ (2006) Genetic disorders of programmed cell death in the immune system. Annu Rev Immunol 24: 321-352. [Crossref]
- Wu X, Simpson J, Hong JH, Kim KH, Thavarajah NK, et al. (2011) MEK-ERK pathway modulation ameliorates disease phenotypes in a mouse model of Noonan syndrome associated with the Raf1(L613V) mutation. J Clin Invest 121: 1009-1025. [Crossref]
- Chen PC, Wakimoto H, Conner D, Araki T, Yuan T, et al. (2010) Activation of multiple signaling pathways causes developmental defects in mice with a Noonan syndrome-associated Sos1 mutation. J Clin Invest 120: 4353-4365. [Crossref]
- Marin TM, Keith K, Davies B, Conner DA, Guha P, et al. (2011) Rapamycin reverses hypertrophic cardiomyopathy in a mouse model of LEOPARD syndrome-associated PTPN11 mutation. J Clin Invest 121: 1026-1043. [Crossref]
- Wang J, Chandrasekhar V, Abbadessa G, Yu Y, Schwartz B, et al. (2017) In vivo efficacy of the AKT inhibitor ARQ 092 in Noonan syndrome with multiple lentigines-associated hypertrophic cardiomyopathy. PLoS One 12: e0178905. [Crossref]
- Edouard T, Combier JP, Nédélec A, Bel-Vialar S, Métrich M, et al. (2010) Functional effects of PTPN11 (SHP2) mutations causing LEOPARD syndrome on epidermal growth factor-induced phosphoinositide 3-kinase/AKT/glycogen synthase kinase 3beta signalling. Mol Cell Biol 30: 2498-2507. [Crossref]
- Schuhmacher AJ, Guerra C, Sauzeau V, Canamero M, Bustelo XR, et al. (2009). A mouse model for Costello syndrome reveals an Ang II-mediated hypertensive condition. J Clin Invest 118: 2169-2179. [Crossref]
- Hanna N, Montagner A, Lee WH, Miteva M, Vidal M, et al. (2006) Reduced phosphatase activity of SHP-2 in LEOPARD syndrome: consequences for PI3K binding on Gab1. FEBS Lett 580: 2477-2482. [Crossref]
- Tumurkhuu M, Saitoh M, Takita J, Mizuno Y, Mizuguchi M (2013) A novel SOS1 mutation in Costello/CFC syndrome affects signaling in both RAS and PI3K pathways. J Recept Signal Transduct Res 33: 124-128. [Crossref]
- Rosenberger G, Meien S, Kutsche K (2009) Oncogenic HRAS mutations cause prolonged PI3K signalling in response to epidermal growth factor in fibroblasts of patients with Costello syndrome. Hum Mutat 30: 352-362. [Crossref]
- Gremer L, De Luca A, Merbitz-Zahradnik T, Dallapiccola B, Morlot S, et al. (2010) Duplication of Glu37 in the switch I region of HRAS impairs effector/GAP binding and underlies Costello syndrome by promoting enhanced growth factor dependent MAPK and AKT activation. Hum Mol Genet 19: 790-802. [Crossref]
- Tajan M, Batut A, Cadoudal T, Deleruyelle S, Le Gonidec S, et al. (2014) LEOPARD syndrome-associated SHP2 mutation confers leanness and protection from diet-induced obesity. Proc Natl Acad Sci USA 111: E4494–E4503. [Crossref]
- Yin JC, Platt MJ, Tian X, Wu X, Backx PH, et al. (2017) Cellular interplay via cytokine hierarchy causes pathological cardiac hypertrophy in RAF1-mutant Noonan syndrome. Nat Commun 8: 15518. [Crossref]
- Schubbert S, Bollag G, Lyubynska N, Nguyen H, Kratz CP, et al. (2007) Biochemical and functional characterization of germ line KRAS mutations. Mol Cell Biol 27: 7765-7770. [Crossref]
- Dasgupta B, Yi Y, Chen DY, Weber JD, Gutmann DH (2005) Proteomic analysis reveals hyperactivation of the mammalian target of rapamycin pathway in neurofibromatosis 1-associated human and mouse brain tumours. Cancer Res 2755-2760. [Crossref]
- Schramm C, Fine DM, Edwards MA, Reeb AN, Krenz M (2012) The PTPN11 loss-of-function mutation Q510E-Shp2 causes hypertrophic cardiomyopathy by dysregulating mTOR signalling. Am J Physiol Heart Circ Physiol 302: H231-H243. [Crossref]
- Roy R, Krenz M (2017) Heterozygous deletion of AKT1 rescues cardiac contractility, but not hypertrophy, in a mouse model of Noonan syndrome with multiple lentigines. J Mol Cell Cardiol 112: 83-90. [Crossref]
- De Luca A, Maiello MR, D’Alessio APergameno M, Normanno N (2012) The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin Ther Targets16: S17-S27. [Crossref]
- Limal JM, Parfait B, Cabrol S, Bonnet D, Leheup B, et al. (2006) Noonan syndrome: relationships between genotype, growth, and growth factors. J Clin Endocrinol Metab 91: 300-306. [Crossref]
- Dhandapany PS, Razzaque MA, Muthusami U, Kunnoth S, Edwards JJ, et al. (2014) RAF1 mutations in childhood-onset dilated cardiomyopathy. Nat Genet 46: 635-639. [Crossref]
- Tajan M, Paccoud R, Branka S, Edouard T, Yart A (2018) The RASopathy family: consequences of germline activation of the Ras/MAPK pathway. Endocr Rev 39: 676-700. [Crossref]
- Vyas KS (2013) Pulmonary Hypertension in the RASopathies. J Cardio Vasc Med 1: 1-6.
- Ezquieta B, Santomé JL, Carcavilla A, Guillén-Navarro E, Pérez-Aytés A, et al. (2012) Alterations in RAS-MAPK genes in 200 Spanish patients with Noonan and other neuro-cardio-facio-cutaneous syndromes. Genotype and cardiopathy. Rev Esp Cardiol (Engl Ed) 65: 447-455. [Crossref]
- Carcavilla A, Santomé JL, Pinto I, Sánchez-Pozo J, Guillén-Navarro E, et al. (2013) LEOPARD syndrome: a variant of Noonan syndrome strongly associated with hypertrophic cardiomyopathy. Rev Esp Cardiol (Engl Ed) 66: 350-356. [Crossref]
- Calcagni G, Limongelli G, d’Ambrosio A, Gesualdo F, Digilio MC, et al. (2016) Clinical Impact of Cardiovascular Diseases in Rasopathies. Circulation 134: A14870.
- Jhang WK, Choi JH, Lee BH, Kim GH, Yoo HW (2016) Cardiac manifestations and associations with gene mutations in patients diagnosed with RASopathies. Pediatr Cardiol 37: 1539-1547. [Crossref]
- Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, et al. (2014) 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 35: 2733-2779. [Crossref]
- Marino B, Gagliardi MG, Digilio MC, Polletta B, Grazioli S, et al. (1995) Noonan syndrome: structural abnormalities of the mitral valve causing subaortic obstruction. Eur J Pediatr 154: 949-952. [Crossref]
- Hirsch HD, Gelband HE, Garcia OT, Gottlieb ST, Tamer DM (1975) Rapidly progressive obstructive cardiomyopathy in infants with Noonan’s syndrome. Report of two cases. Circulation 52: 1161-1165. [Crossref]
- Colquitt JL, Noonan JA (2014) Cardiac findings in Noonan syndrome on long-term follow-up. Congenit Heart Dis 9: 144-150. [Crossref]
- Suda K, Kohl T, Kovalchin JP, Silverman NH (1997) Echocardiographic predictors of poor outcome in infants with hypertrophic cardiomyopathy. Am J Cardiol 80: 595-600. [Crossref]
- Ostman-Smith I (2014) Beta-blockers in paediatric hypertrophic cardiomyopathies. Rev Recent Clin Trials 9: 82-85. [Crossref]
- Minakata K, Dearani JA, O’Leary PW, Danielson GK, et al. (2005) Septal myectomy for obstructive hypertrophic cardiomyopathy in paediatric patients: early and late results. Ann Thorac Surg 80: 1424-1429. [Crossref]
- Hickey EJ, McCrindle BW, Larsen SH, Benson L, Manlhiot C, et al. (2012) Hypertrophic cardiomyopathy in childhood: disease natural history, impact of obstruction, and its influence on survival. Ann Thorac Surg 93: 840-848. [Crossref]
- Poterucha JT, Johnson JN, O'Leary PW, Connolly HM, Niaz T, et al. (2015) Surgical ventricular septal myectomy for patients with Noonan syndrome and symptomatic left ventricular outflow tract obstruction. Am J Cardiol 116: 1116-1121. [Crossref]
- Calcagni G, Limongelli G, D'Ambrosio A, Gesualdo F, Digilio MC, et al. (2017) Cardiac defects, morbidity and mortality in patients affected by RASopathies. CARNET study results. Int J Cardiol 245: 92-88. [Crossref]
- Gajarski R, Naftel DC, Pahl E, Alejos J, Pearce FB, et al. (2009) Outcomes of pediatric patients with hypertrophic cardiomyopathy listed for transplant. J Heart Lung Transplant 28: 1329-1334. [Crossref]
- Colan SD, Lipshultz SE, Lowe AM, et al. (2007) Epidemiology and cause-specific outcome of hypertrophic cardiomyopathy in children: findings from the pediatric cardiomyopathy registry. Circulation 115: 773-781. [Crossref]
- Nugent AW, Daubeney PE, Chondros P, Carlin JB, Colan SD, et al. (2005) Clinical features and outcomes of childhood hypertrophic cardiomyopathy: results from a national population-based study. Circulation 112: 1332-1338. [Crossref]
- Urosevic J, Sauzeau V, Soto-Montenegro ML, Reig S, Desco M, et al. (2011) Constitutive activation of B-Raf in the mouse germ line provides a model for human cardio-facio-cutaneous syndrome. Proc Natl Acad Sci USA 108: 5015-5020. [Crossref]
- Andreadi C, Cheung LK, Giblett S, Patel B, Jin H, et al. (2012) The intermediate-activity (L597V)BRAF mutant acts as an epistatic modifier of oncogenic RAS by enhancing signaling through the RAF/MEK/ERK pathway. Genes Dev 26: 1945-1958. [Crossref]
- Cotterill AM, McKenna WJ, Brady AF, Sharland M, Elsawi M, et al. (1996) The short-term effects of growth hormone therapy on height velocity and cardiac ventricular wall thickness in children with Noonan’s syndrome. J Clin Endocrinol Metab 81: 2291-2297. [Crossref]
- Lee YS, Ehninger D, Zhou M, Oh JY, Kang M, et al. (2014) Mechanism and treatment for learning and memory deficits in mouse models of Noonan syndrome. Nat Neurosci 417: 1736-1743. [Crossref]