Alzheimer’s disease (AD) is a chronic progressive, fatal neurodegenerative disease which is the most common form of dementia among the elderly affecting over 5.8 million patients in the United States (US) and 36 million worldwide. Currently there is no effective treatment. Based on the observation that radiation therapy (RT) has been used to successfully treat systemic amyloidosis for over 2 decades our group hypothesized that RT could be used to treat AD. We have published preclinical data which supports this hypothesis using a transgenic murine model of AD. The mechanisms by which RT improves cognition in this model may include a combination of amyloid and tau reduction, alteration of immune responses, reduction of maladaptive neuroplasticity and other effects. Further preclinical work is underway and an FDA approved phase 1 human trial, examining the safety of RT in AD subjects is open at our institution.
alzheimer’s disease, radiation therapy, treatment, amyloid, neuroplasticity
Alzheimer’s disease (AD) is a chronic progressive neurodegenerative disease. It is the most common form of dementia among the elderly affecting over 5.8 million patients in the United States (US) and 36 million worldwide. The US incidence is expected to double by 2050 with the cost of caring for these patients expected to exceed 1.2 trillion dollars [1]. AD is a fatal disease. Within 12 years of initial diagnosis virtually 100% of AD patients will die [2].
The U.S. Food and Drug Administration (FDA) has approved five medications to treat the symptoms of Alzheimer's disease. donepezil, rivastigmine, galantamine, memantine and the combination of donepezil and memantine. The first three are acetylcholinesterase inhibitors that enhance the neurotransmission of acetylcholine in the brain, which is thought to be essential for cognition. There are many other strategies that have been studied for AD treatment including immunologic, nutraceutical, mitochondrial, phosphodiesterase, 5-HT6 receptor agonists and stem cell-based treatments [3]. The results from large-scale randomized clinical trials of pharmacological agents have not identified an effective and safe treatment to date: “Although treatment can help manage symptoms for a limited period of time in some people, no intervention is currently available to slow or prevent the underlying disease process.” [4]
The widespread distribution of amyloid plaques in post- mortem brains of patients with AD was one of the initial hallmarks of the disease as described by Alzheimer. Further studies in human and animal models of AD provide good evidence that an imbalance of β amyloid, over production, under clearance or both, is an early, critical step in the disease [5]. This understanding has provided the rationale for developing treatments aimed at lowering β amyloid as evidenced by reduction in size and quantity of amyloid plaques [6].
Eliminating amyloid-β has been advocated as a beneficial treatment strategy for AD patients, and anti-amyloid therapies remain a rational approach for preventing or delaying AD [7]. Amyloid-β is produced by the proteolytic cleavage of amyloid-β precursor protein (AβPP) by β-and γ-secretases and many novel pharmaceuticals are being developed to prevent the initial cleavage of APP [8,9]. The blood–brain barrier (BBB) has limited or thwarted the success of many of these agents either by preventing the drugs from initially crossing into the brain, or by ensuring the rapid removal of those drugs that can cross the BBB [10]. Immunotherapy targeting amyloid have not been shown to be beneficial in clinical trials to date [11].
Our group has undertaken a series of experiments to test the hypothesis that ionizing radiation therapy (RT) may be used to treat AD. This novel approach is based on previous work that has shown RT to reduce amyloid-like deposits in extra-cranial disease sites and has been used clinically to treat systemic amyloidosis for decades [12-14]. RT has not been considered previously for AD patients because of the potential to exacerbate cognitive impairment. The potential for side effects from brain RT are dependent on the total radiation dose given. Total doses of 30 to 60 Gray (Gy) given in 2 Gy fractions can produce macroscopic tissue destruction [15] and impair cognition [16-20]. However, only minimal cognitive effects are produced (minor compared with the normal cognitive decline associated with AD if the total dose remains low and critical brain structures are spared using image guided RT. The severity of cognitive impairment depends upon the dose delivered to the medial temporal lobes, the site of the hippocampus [21].
Prophylactic cranial irradiation (PCI) [22], to a total dose of 25 Gy in 2.5 Gy fractions given daily, has become the standard-of-care for selected groups of adult patients with small cell lung cancer and in paediatric leukaemia patients to decrease CNS relapse [23], with no mild [24-27] or moderate [28] cognitive impairment. Moreover, whole brain RT can lead to the recruitment of protective glial cells that would be beneficial in an AD patient [18], and the use of recognized protectors or mitigators of RT damage would provide additional clinical safeguards [18,28-33]. RT is a potential novel treatment option for AD that could be rapidly and inexpensively implemented, especially when compared to the time needed and costs associated with developing new pharmaceuticals that are often only partially effective.
Our group sought to test the hypothesis that RT reduces amyloid-b (Ab) plaques and improves influences cognitive function in a transgenic mouse model of AD (B6.Cg-Tg (APPswePSEN1dE9]85Dbo/J). Initial studies used hemi-cranial radiation using the shielded side as a control. Immunohistochemical studies demonstrated a reduction in the number and size of β amyloid plaques. A fractionated radiation dose escalation study identified 2 × 5 Gy as yielding the optimal reduction in β amyloid (Figure 1) [34].
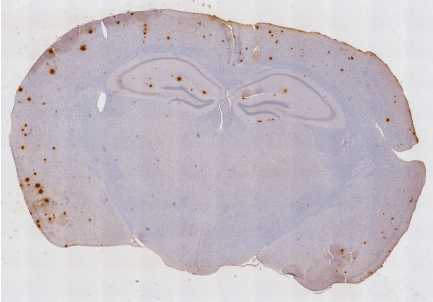
Figure 1. Previously unpublished, unretouched photomicrograph of a 5 × 2 Gy hemi-irradiated B6. Cg-Tg (APPswePSEN1dE9)85Dbo/J transgenic mouse brain at the level of the hippocampus, processed Immunohistochemically for β amyloid (materials and methods). Note marked reduction in β amyloid staining (brown) on hemi-irradiated side.
These hemi-radiation results also demonstrated the following changes in radiated versus non-radiated sides: increase in BACE 2 and decrease in presenilin 1 expression by PCR array analysis; Aβ (A4] precursor like protein 1 (APLP1], APLP2, and apolipoprotein A-1 decrease expression by PCR; increased IL-10 and IL-1β immunostaining; and differential expression of 84 AD specific genes (Mouse Alzheimer’s Disease RT2 ProfilerTM).
Whole brain radiated mice were then used to study episodic memory using a Morris water-maze (MMW) task. No side effects of RT were observed in the experimental group. The radiated mice showed a significant improvement in MMW latencies compared to unirradiated controls This improvement could not be explained by difference in swimming velocity or baseline ambulatory velocity. Following testing, animals were sacrificed, and the radiated brains showed similar reductions in β amyloid plaque size and number as seen in the initial hemi-radiation studies. The results of the hemi- and whole brain radiation studies were published by Marples et al. in 2015 [34].
The exact mechanism by which RT confers beneficial effects in the AD mouse model will be difficult to elucidate because the exact pathobiology of AD is not known. Our experiments point to several molecular and cellular changes which occur in AD which, in combination, could be favourably impacted by RT. Each of these potential mechanisms require further investigation.
Reduction of β amyloid
The reduction of β amyloid plaque is an obvious possible mechanism by which RT exerts its beneficial effect on episodic memory. RT almost certainly does not reduce amyloid by direct action on the 40-42 amyloid β (Aβ) monomer which ultimately aggregates to form plaques. The effects of RT on peptides and proteins are well known. The amount of radiation needed to denature these molecules would cause widespread, immediate cell death.
It is more likely that RT reduces β amyloid plaque by reducing production, increasing clearance or both at the molecular level. Radiation is known to cause cell death by inducing irreparable double strand DNA damage, induction of reactive oxygen species and /or induction of apoptosis. It is now becoming clear that short of inducing cell death, RT may up regulate and or down regulate specific genes giving rise to the field of radiation genomics [66]. Change in the amyloid precursor protein (APP) gene expression or that of any of the genes of molecules involved in processing and deposition of amyloid plaques could result in plaque reduction. Potential targets include genes for β site APP cleavage enzyme 1 (BACE1] and γ secretase. Work in our lab has demonstrated change in expression of 84 genes associated with AD after RT [34]. Increase in BACE 2 expression using microarray analysis was detected. Upregulation of BACE 2 may exert anti amyloidogenic activity [36]. Our work also detected a decrease in presenilin 1 activity. Presenilin 1 is a subunit of the γ secretase complex. Inhibition of this complex has been the target of a number of amyloid-reducing drug studies [6].
Similarly, alteration in gene expression of the ApoE gene due to RT could result in Aβ reduction. The ApoE gene codes for a glycoprotein produced by predominantly by astrocytes and microglia in the brain. It is thought to play a role in clearance of Aβ in the normal brain. The ApoE gene has three common alleles: ε2,ε3 and ε4. The ε4 allele has been demonstrated as a genetic risk factor for AD, while ε2 confers a protective effect [37]. PCR analysis done in our lab suggests a differential expression of Apo ε3 between radiated and unradiated AD mice suggesting another possible mechanism by which RT could alter Aβ levels.
Alteration of Tau
Another hallmark of the AD brain is the accumulation of neurofibrillary tangles (NFT), now known to consist of aggregated Tau protein [38]. Tau interacts with a wide variety of intercellular proteins and is known to be neurotoxic. Evidence suggests that the accumulation of Aβ precedes and contributes to the deposition of Tau. Our lab has demonstrated a 38% reduction in overall Tau staining in RT treated versus non treated sides in the hemi radiation model [64]. The treated side seems to exhibit a mature NFT staining pattern than the untreated which appears more like the pre NFT stage. Further study of the effects of RT on Tau would seem warranted.
Inflammation
The role of inflammation in AD is the subject of intense and voluminous study [39]. Aβ has been shown to activate microglia and induce astrocyte activation. Microglia activation is mediated by cytokines IL-4, IL-10, IL-13 and TGFβ. Exposure of astrocytes in vitro to Aβ results in increased production of IL1β, TNF-α, iNOS and NO. The complement system also appears to be activated by Aβ. Alterations in antigen receptors such as the toll like receptors have been noted in AD models.
Cyclooxygenase-2 (COX-2) conversion of arachidonic acid to prostaglandin H2 occurs during inflammation and results in the production of a number of proinflamatory molecules. The use of non-steroidal anti-inflammatory drugs blocks COX-2. A meta-analysis of epidemiologic studies suggest AD risk may be reduced by half with long term NSAID use [40]. Prospective studies have failed to demonstrate the benefit of NSAIDs in AD.
The immune response in AD is complex time course dependent. Early response to events in AD may prove beneficial while harmful later in the course of the disease. The effects of RT also elicit multiple immune responses which vary with time [67]. Thus far our lab has identified several changes in cytokine expression and microglia activation in the hemi radiated AD model, however much more work remains to elucidate the effect of RT on inflammation over time in our model.
Epigenetic effects
RT has been shown to cause epigenetic alteration of gene expression through DNA methylation and alteration in histone deacetylase activity [41-43]. A recent study suggests blocking histone deacetylase 2 leads to improved memory in a murine model of AD [44]. Studies to detect alterations of either DNA methylation or histone deacetylase as a result of RT in our model are planned.
Heat shock proteins
One of the mechanisms cells may rid themselves of misfolded proteins is through the ubiquitin proteasome system suggesting this as therapeutic target in AD. Molecules to be catabolized are conjugated with ubiquitin then degraded by a proteasome. Heat shock proteins (hsp) are a family of chaperone molecules which aid in this process. HSP70 ubiquitinization is assisted by the interaction of the carboxy terminus of HSP70-interacting protein (CHIP) and the target protein. Increase of HSP70 in tau expressing neuronal culture leads to a reduction in insoluble Tau [45]. HSP90 inhibition has been shown to reduce phosphor-Tau species [46]. RT is having been shown to alter HSP90 expression [47]. Studies using our model to investigate the effect of RT on hsp expression are planned.
Vascular effects
Aβ is found in the brains of AD victims not only in the form of amyloid plaques, but also in the walls of cerebral blood vessels, known as cerebral amyloid angiopathy (CAA). CAA is found in about 80% of AD brains at autopsy [48]. CAA is demonstrated in capillaries, arterioles and small cortical arteries. CAA is associated with micro and gross intracerebral haemorrhages. CAA has been prosed as either contributing to cognitive decline in AD patients or even as a fundamental cause of ADD [49]. Alterations in the AD capillary wall have been shown to contribute blood brain barrier (BBB) dysfunction with reduction of glucose transporter GLUT1 and RAGE which binds Aβ. Blocking RAGE has been suggested as a potential therapeutic target in AD.
The effects of RT on cerebral vasculature have been studied, for the most part using doses in the 40-100 Gy range. At these doses an opening of the BBB has been detected to persist for weeks [50]. It is also known that radiation increases the risk for long term cerebral vasculitis, also at higher doses. The effect of lower doses of RT in the short term is largely unknown. It is possible that there are some molecular changes induced by RT in AD cerebral blood vessels are beneficial; e.g. reduction of Aβ mural deposition or decreased RAGE expression. Studies to investigate such effects are in progress.
“Neuroplasticity can be viewed as a general umbrella term that refers to the brain’s ability to modify, change, and adapt both structure and function throughout life and in response to experience.” [66] The preponderance of neuroplasticity occurs during growth and development of the brain. Neuroplasticity is also involved in learning and memory [51] and in response to neuropathology. When the neuroplasticity is beneficial is referred to as “adaptive”, when harmful, “maladaptive”. Maladaptive neuroplasticity has been implicated in stroke [52], phantom limb syndrome and post cochlear nerve implant syndrome.
Niell published a hypothesis of AD based on maladaptive neuroplasticity in 1995 [53]. He proposed that the loss of memory in AD was due to maladaptive neuroplasticity in the hippocampus and parahippocampal cortex due to the loss of neocortical afferents bought on by normal aging. The hypothesis is consistent with the fact that β amyloid plaques in the neocortex are generally seen to precede plaque formation in the hippocampus and parahippocampal cortex [54]. Based on his hypothesis Neill Concluded: “Treatment strategies might involve the use of drugs which impede the maladaptation of the synaptoplastic response in vulnerable neurons.”
Another form of neuroplasticity may be important in normal hippocampal function: adult neurogenesis [55]. Although neurogenesis in adult mammals has been demonstrated, its role in normal adult hippocampal function, i.e. learning and memory, is uncertain [56,57]. Using a novel technique, Spalding and colleagues were able to determine that a median of 700 new neurons are added to the adult, human hippocampus daily. This corresponds to a turnover rate of 0.004% during adulthood with only a modest decline in the rate with aging. They estimate that one- third of the hippocampal neurons are subject to turnover during human adulthood (versus 10% in the mouse). Since immature neurons demonstrate greater neuritic sprouting and synaptogenesis than mature neurons [58] this suggests neural plasticity plays an important role in normal hippocampal function. Electrophysiologic studies implicated altered synaptic plasticity in the hippocampus of experimental models of AD (see 59 for review). Work suggests an increase of neuronal excitation leads to increased hippocampal neural plasticity in AD [60] and evidence suggests these hyperexcitable adult born neurons exert modulatory effects on more mature neurons which might be maladaptive in AD [65]. A recent paper notes while neural stem and progenitor cells persist in the adult brain and can faithfully integrate into adult brain circuitry constituting structural plasticity, contradictions and controversy in this rapidly evolving field exist and more work is called for [70].
The likely important role of neuroplasticity in normal hippocampal function and maladaptive neuroplasticity in AD suggests another mechanism by which RT may be beneficial. We theorize that in the normal state hippocampal function is maintained through a normal amount of neurogenesis and adaptive plasticity In AD the mature neurons, which constitute the majority of hippocampal neurons, are more likely to be affected by the deleterious effects of β amyloid. This leads to maladaptive neuroplasticity of the immature neurons, with increased sprouting, inappropriate synapse formation, and a decline in memory. RT preferentially targets reproducing neurons in the hippocampus, decreasing maladaptive neuroplasticity and thus restoring episodic memory.
Our theory is consistent with work demonstrating selective loss of immature neurons in the normal hippocampus when exposed to radiation [61]. A number of groups, however, have demonstrated decreased neurogenesis in the APPswe/PS1dE9 murine model of AD [62]. While we have used a different model (B6.Cg-Tg (APPswePSEN1dE9]85Dbo/J), it is sufficiently close so as to call the role of maladaptive neuroplasticity in the pathogenesis of AD into question. Demars et al. [63] found an early, severe reduction in hippocampal progenitor cells and suggest this is the basis of episodic memory loss in AD. The loss of the progenitor cells in the AD model is demonstrated in comparison to normal mice or AD mice treated with environment enrichment. Synaptogenesis of the remaining immature neurons was not examined and may increase in a maladaptive fashion.
This theory suggests several testable hypotheses. If RT restores the balance between immature and mature synapses in our AD model one should be able to measure a difference in markers for neurogenesis and synaptogenesis between radiated and unradiated groups. Immunohistochemistry for Ki 67, DCx and synaptophysin and other techniques could be used to demonstrate these differences [61]. Preliminary studies in our lab suggest synaptophysin is reduced in radiated versus un-radiated hippocampi [69].
We are currently investigating the sex differences in onset of amyloid deposition and response to RT in our model based on clinical observations of AD onset, severity and response to therapy in men and women. Our work has been has prompted similar studies of RT effect and mechanisms in an AD rat model currently underway in Switzerland. Preliminary report, yet unpublished, suggests a similar reduction in β amyloid in treated versus untreated rats and variable response in male & female rats.
Based on our preclinic work our group designed a Phase 1 trial of RT in AD patients. This study has received FDA and local IRB approval. See trials.gov for details of the protocol (https://clinicaltrials.gov/ct2/show/NCT02359864?term=Radiation+therapy&cond=Alzheimer%27s+Disease&rank=2] l. We expect begin subject enrolment in 2019.
Our group has shown hemi-brain RT significantly reduces plaque burden in a murine model of AD. Further we have demonstrated that RT is well tolerated in aged AD mice and results in improved episodic memory function as demonstrated by MWM testing. This beneficial effect of RT may be due to a variety of mechanisms suggested by additional results we have published. This work is being extended to the rat model of AD by another lab. We have undertaken a Phase 1 clinical trial of RT for AD which has received FDA and local IRB approval. We expect to begin patient accrual in 2019.
Support
The authors gratefully acknowledge the support of the following instutions:
Department of Radiation Oncology, Beaumont Health
The Beaumont Foundation
The 21st Century Oncology Group
The Michigan Head & Spine Institute.
- Alzheimer’s Association (2019) 2019 Alzheimer’s Disease Facts and Figures. Alzheimers Dement 15:321.
- Brookmeyer R, Corrada MM, Curriero FC, Kawas C (2002) Survival following a diagnosis of Alzheimer disease. Arch Neurol 59:1764-1767. [Crossref]
- Fan LY, Chiu MJ (2014) Combotherapy and current concepts as well as future strategies for the treatment of alzheimer's disease. Neuropsychiatr Dis Treat 10:439-451. [Crossref]
- 2014-2015 Alzheimer’s Disease Progress Report, Advancing Research toward a Cure. NIA/NIH Dec. Available from: www.nia.nih.gov/alzheimers.
- Masters CL, Selkoe DJ (2012) Biochemistry of Amyloid β Protein and Amyloid Deposits in Alzheimer Disease. Cold Spring Harbor Persect Med 2: a006262. [Crossref]
- Schenk D, Basi GS, Pangalos MN (2012) Treatment strategies targeting amyloid β-protein. Cold Spring Harbor Persect Med 2: a006387. [Crossref]
- Neugroschl J, Sano M (2009) An update on treatment and prevention strategies for alzheimer's disease. Curr Neurol Neurosci Rep 9: 368-376. [Crossref]
- Beher D, Graham SL (2005) Protease inhibitors as potential disease-modifying therapeutics for alzheimer's disease. Expert Opin Investig Drugs 14: 1385-1409. [Crossref]
- Hull M, Berger M, Heneka M (2006) Disease-modifying therapies in alzheimer's disease: How far have we come? Drugs 66: 2075-2093. [Crossref]
- Hossain S, Akaike T, Chowdhury EH (2010) Current approaches for drug delivery to central nervous system. Curr Drug Deliv 7: 389-397. [Crossref]
- Mehta D, Jackson R, Paul G, Shi J, Sabbagh M (2017) Why do trial for Alzheimer’s Disease drugs keep failing? Adiscontinued drug perspective for 2010-2015. Expert Opin Investig Drugs 26: 735–739. [Crossref]
- Kurrus JA, Hayes JK, Hoidal JR, Menendez MM, Elstad MR (1998) Radiation therapy for tracheobronchial amyloidosis. Chest 114: 1489-1492. [Crossref]
- Poovaneswaran S, Razak AR, Lockman H, Bone M, Pollard K, et al. (2008) Tracheobronchial amyloidosis: Utilization of radiotherapy as a treatment modality. Medscape J Med 10: 42. [Crossref]
- Monroe AT, Walia R, Zlotecki RA, Jantz MA (2004) Tracheobronchial amyloidosis: A case report of successful treatment with external beam radiation therapy. Chest 125:784-789. [Crossref]
- Sheline GE, Wara WM, Smith V (1980) Therapeutic irradiation and brain injury. Int J Radiat Oncol Biol Phys 6: 1215-1228.
- Huang Z, Jiang J, Belikova NA, Stoyanovsky DA, Kagan VE, et al. (2010) Protection of normal brain cells from gamma-irradiation-induced apoptosis by a mitochondriatargeted triphenyl-phosphonium-nitroxide: A possible utility in glioblastoma therapy. J Neurooncol 100: 1-8. [Crossref]
- Raber J, Rola R, LeFevour A, Morhardt D, Curley J, et al. (2004) Radiation-induced cognitive impairments are associated with changes in indicators of hippocampal neurogenesis. Radiat Res 162: 39-47. [Crossref]
- Ramanan S, Kooshki M, Zhao W, Hsu FC, Riddle DR, et al. (2009) The PPARalpha agonist fenofibrate preserves hippocampal neurogenesis and inhibits microglial activation after whole-brain irradiation. Int J Radiat Oncol Biol Phys 75: 870-877. [Crossref]
- Rola R, Raber J, Rizk A, Otsuka S, VandenBerg SR, et al. (2004) Radiation-induced impairment of hippocampal neurogenesis is associated with cognitive deficits in young mice. Exp Neurol 188: 316-330. [Crossref]
- Rowe CC, Villemagne VL (2011) Brain amyloid imaging. J Nucl Med 52: 1733-1740. [Crossref]
- Abayomi OK (1996) Pathogenesis of irradiation-induced cognitive dysfunction. Acta oncologica (Stockholm, Sweden) 35: 659-663.
- Marsh JC, Gielda BT, Herskovic AM, Abrams RA (2010) Cognitive sparing during the administration of whole brain radiotherapy and prophylactic cranial irradiation: Current concepts and approaches. J Oncol 2010: 198208. [Crossref]
- Pui CH, Cheng C, Leung W, Rai SN, Rivera GK, et al. (2003) Extended follow-up of long-term survivors of childhood acute lymphoblastic leukemia. N Engl J Med 349: 640-649. [Crossref]
- Waber DP, Turek J, Catania L, Stevenson K, Robaey P, et al. (2007) Neuropsychological outcomes from a randomized trial of triple intrathecal chemotherapy compared with 18 gy cranial radiation as cns treatment in acute lymphoblastic leukemia: Findings from dana-farber cancer institute all consortium protocol 95-01. J Clin Oncol 25: 4914-4921. [Crossref]
- Komaki R, Meyers CA, Shin DM, Garden AS, Byrne K, et al. (1995) Evaluation of cognitive function in patients with limited small cell lung cancer prior to and shortly following prophylactic cranial irradiation. Int J Radiat Oncol Biol Phys 33: 179-182. [Crossref]
- Edelstein K, D'Agostino N, Bernstein LJ, Nathan PC, Greenberg ML, et al. (2011) Long-term neurocognitive outcomes in young adult survivors of childhood acute lymphoblastic leukemia. J Pediatr Hematol Oncol 33: 450-458. [Crossref]
- Slotman BJ, Mauer ME, Bottomley A, Faivre-Finn C, Kramer GW, et al. (2009) Prophylactic cranial irradiation in extensive disease small-cell lung cancer: Short-term health-related quality of life and patient reported symptoms: Results of an international phase iii randomized controlled trial by the EORTC radiation oncology and lung cancer groups. J Clin Oncol 27: 78-84. [Crossref]
- Spiegler BJ, Kennedy K, Maze R, Greenberg ML, Weitzman S, et al. (2006) Comparison of long-term neurocognitive outcomes in young children with acute lymphoblastic leukemia treated with cranial radiation or high-dose or very high-dose intravenous methotrexate. J Clin Oncol 24: 3858-3864. [Crossref]
- Conner KR, Forbes ME, Lee WH, Lee YW, Riddle DR (2011) AT1 receptor antagonism does not influence early radiation-induced changes in microglial activation or neurogenesis in the normal rat brain. Radiat Res 176: 71-83. [Crossref]
- Conner KR, Payne VS, Forbes ME, Robbins ME, Riddle DR (2010) Effects of the AT1 receptor antagonist l-158,809 on microglia and neurogenesis after fractionated whole-brain irradiation. Radiat Res 173: 49-61. [Crossref]
- Motomura K, Ogura M, Natsume A, Yokoyama H, Wakabayashi T (2010) A free-radical scavenger protects the neural progenitor cells in the dentate subgranular zone of the hippocampus from cell death after x-irradiation. Neurosci Lett 485: 65-70. [Crossref]
- Gauthier S, Aisen PS, Ferris SH, Saumier D, Duong A, et al. (2009) Effect of tramiprosate in patients with mild-to-moderate alzheimer's disease: Exploratory analyses of the mri sub-group of the alphase study. J Nutr Health Aging 13: 550-557. [Crossref]
- Zhao W, Payne V, Tommasi E, Diz DI, Hsu FC, et al. (2007) Administration of the peroxisomal proliferator-activated receptor gamma agonist pioglitazone during fractionated brain irradiation prevents radiation-induced cognitive impairment. Int J Radiat Oncol Biol Phys 67: 6-9. [Crossref]
- Marples B, McGee M, Callan S, Bowen SE, Thibodeau BJ, et al. (2016) Cranial irradiation significantly reduces beta amyloid plaques in the brain and improves cognition in a murine model of Alzheimer’s Disease. Radiother Oncol 118: 43-51. [Crossref]
- Patrias LM, Klaver AC, Coffey MP, Finke JM, Digambaranath JL, et al. (2011) Effects of external beam radiation on in vitro formation of Abeta1-42 fibrils and preformed fibrils. Radiat Res 175: 375-81. [Crossref]
- Basi G, Frigon N, Barbour R, Doan T, Gordon G, et al. (2003) Antagonistic effects of beta-site amyloid precursor protein-cleaving enzymes 1 and 2 on beta-amyloid peptide production in cells. J Biol Chem 278: 31512-31520. [Crossref]
- Holtzman DM, Herz J, Bu G (2012) Apolipoprotein E and Apolipoprotein E Receptors: Normal Biology and Roles in Alzheimer Disease. Cold Spring Harbor Persect Med 2: a006312. [Crossref]
- Mandlekow EM, Mandlekow E (2012) Biochemistry and Cell Biology of Tau Protein in Neurofibrillary Degeneration. Cold Spring Harbor Persect Med 2: a006247. [Crossref]
- Wyss-Coray T, Rogers J (2012) Inflamation in Alzheimer Disease-A Brief Review of the Basic Science and Clinical Literature. Cold Spring Harbor Persect Med 2: a006346. [Crossref]
- McGeer PL, Schulzer M, McGeer MC (1996) Arthritis and anti-inflamatory agents as possible protective factors in Alzheimer’s Disease: a review of 17 epidemiologic studies. Neurology 47: 423-432. [Crossref]
- Kuhmann C, Weichenhan D, Rehli M, Plass C, Schmezer P, et al. (2011) DNA methylation changes in cells regrowing after fractioned ionizing radiation. Radiother Oncol 101: 116-21. [Crossref]
- Koturbash I, Zemp F, Kolb B, Kovalchuk O (2011) Sex-specific radiation-induced microRNAome responses in the hippocampus, cerebellum and frontal cortex in a mouse model. Mutat Res 722: 114-118. [Crossref]
- Ma S, Liu X, Jiao B, Yang Y, Liu X (2010) Low-dose radiation-induced responses: focusing on epigenetic regulation. Int J Radiat Biol 86: 517-28. [Crossref]
- Gräff J, Rei D, Guan J-S, Wang W-Y, Seo J, et al. (2012) An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 483: 222–226. [Crossref]
- Petrocelli L, Dickson D, Kehoe K, Taylor J, Snyder H, et al. (2004) CHIP and hsp 70 regulate tau ubiquitinization, degradation and aggregation. Hum Mol Genet 13: 703-714. [Crossref]
- Dickey CA, Kamal A, Lungren K, Klosak N, Baily RM, et al. (2007) The high affinity hsp 90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J Clin Invest 117: 648-658. [Crossref]
- Lim YB, Pyun BJ, Lee HJ, Jeon SR, Jin YB, et al. (2011) Proteomic identification of radiation response markers in mouse intestine and brain. Proteomics 11: 1254-63. [Crossref]
- Sarrano-Pozo A, Frosch MP, Masliah E, Hyman BT (2011) Neuropathological Alterations In Alzheimer’s Disease. Cold Spring Harbor Persect Med 1: a006189. [Crossref]
- Sagare AP, Bell RD, Zlokovic BV (2012) Neurovascular dysfunction in Faulty Amyloid β-Peptide Clearance in Alzheimer Disease. Cold Spring Harbor Persect Med 2. pii: a011452. [Crossref]
- Rubin P, Gash DM, Hansen JT, Nelson DF,Williams JP (1994) Disruption of the blood-brain barrier as the primary effect of CNS irradiation. Radiother Oncol 31: 51-60. [Crossref]
- Cotman CW, Nieto-Sampedro M (1984) Cell biology of synaptic plasticity. Science 225: 1287-1294. [Crossref]
- Takeuchi N, Izumi S (2012) Maladaptive Plasticity for Motor Recovery after Stroke: Mechanisms and Approaches. Neural Plast 2012: 359728. [Crossref]
- Neill D (1995) Alzheimer’s Disease: Maladaptive Synaptoplasticity Hypothesis. Neurodegeneration 4: 217-232. [Crossref]
- Thal DR, Rüb U, Orantes M, Braak H (2002) Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58: 1791-1800. [Crossref]
- Spalding KL, Bergmann O, Alkass K, Bernard S, Salehpour, et al. (2013) Dynamics of Hippocampal Neurogenesis in adult humans. Cell 153: 1219-1227. [Crossref]
- Rakic P (1985) Limits of neurogenesis in primates. Science 227:1054-1056. [Crossref]
- Kempermann G (2012) New neurons for ‘survival of the fittest’. Nat Rev Neurosci 13: 727–736. [Crossref]
- Ge S, Yang CH, Hsu KS, Ming GL, Song H (2007) A critical period for enhanced synaptic plasticity in newly generated neurons of the adult brain. Neuron 54: 559–566. [Crossref]
- Palop JJ, Mucke L (2010) Amyloid-beta-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci 13: 812-818. [Crossref]
- Mucke L, Selkoe DJ (2012) Neurotoxicity of Amyloid β Protein: Synaptic and Network Dysfunction. Cold Spring Harbor Persect Med 2: a006338. [Crossref]
- Rola R, Raber J, Rizk A, Otsuka S, VandenBerg SR, et al. (2004) Radiation-induced impairment of hippocampal neurogenesis is associated with cognitive deficits in young mice. Exp Neurol 188: 316-330. [Crossref]
- Hu YS, Xu P, Pigino G, Brady ST, Larson J, et al. (2010) Complex environment experience rescues impairedneurogenesis, enhances synaptic plasticity, and attenuates neuropathology in familial Alzheimer’s disease linked APPswe/PS1DeltaE9 mice. FASEB J 24: 1667-1681. [Crossref]
- Demars M, Hu YS, Gadadhar A, Lazarov O (2010) Impaired Neurogenesis is an early event in the etiology of Familial Alzheimer’s disease in transgenic mice. J Neurosci Res 88: 2103–2117. [Crossref]
- Wilson GD, Marples B (2016) A new use for an old treatment: Radiation therapy and Alzheimer’s Disease. Radiation Res 185: 443–448. [Crossref]
- Luna VM, Anacker C, Burghardt NS, Khandaker H, Andreu V, et al. (2019) Adult-born hippocampal neurons bidirectionally modulate inputs into the dentate gyrus. Science 364: 578-583. [Crossref]
- Xu L, Osei B, Osei E (2019) A review of radiation genomics: Integrating patient radiation response with genomics for personalised and targeted radiation therapy. J Radiother Pract 18: 198-209.
- Lumniczky K, Szatmarti T, Safrani G (2017) Ionizing Radiation-Induced Immune and Inflammatory Reactions in the Brain. Front Immunol 8: 517. [Crossref]
- Voss P, Thomas ME, Cineras-Franco JM, de Villiers-Sidani E (2017) Dynamic Brains and the Changing Rules of Neuroplasticity: Implications for Learning and Recovery. Front Psychol 8: 1657. [Crossref]
- Michael DB, Hanna A, Hunt RJ, Martinez A, Maddens M, et al. (2014) External Radiation is Associated with Reduction of Aβ Plaque Burden and Hippocampal Synaptophysin Staining in a Murine Model of Alzheimer's Disease. Society for Neuroscience, Abstract. Washington DC.
- Gage FH (2019) Adult neurogenesis in mammals. Science 364: 827-828. [Crossref]