Abstract
The present paper presents electronic structure and optical properties of the GaxAsx clusters for x= 10, 13, 15, 18, 20, 25 and 30 using density functional theory (DFT) method. The Ga-Ga bond length lies in range of 2.50-2.69 A0, the As-As bond length lies in range 2.58-2.62 A0 and Ga-As bonds length is in range 2.46-2.52 A0. The variation of polarizability, which reflects optical activity, with increase cluster size is presented and the study suggests that Ga25As25 cluster has more optical activity in comparison with other studied clusters.
Key words
density functional theory, micro-cluster, GaAs clusters
Introduction
Low-dimensional semi-conducting material is of great interest to researchers in Nano science [1-4]. Micro-clusters present a new phase of a solid with novel properties and are formed by aggregation of atoms or molecules, and also exhibit properties significantly different from that of the bulk solid state of the constituent molecules or atoms. The dependence of electronic structure of the micro-cluster on its physical size gives rise to new physical, optical [5] electronic [6] chemical as well as magnetic properties [7]. Since the properties of a micro-cluster depend on its composition so it enables to fine tune different properties of micro-clusters, particularly in case of semiconductor clusters, resulting in novel optical and electronic capabilities. Investigation of clusters also enables to study the emergence of bulk crystalline properties from atomic or molecular scale. In order to obtain some desired property from a cluster, a detailed study about the properties of the cluster and its flexibility to control its size. The present day computational resources and techniques have enabled to model and investigate these clusters with ease and accuracy.
Fullerenes that have a special structure which consists only of pentagonal and hexagonal rings with hollow cage like structure and belong to the class of micro-clusters. With all possible arrangement of collections of atom between these pentagon and hexagon many isomers can be constructed by varying the number of atoms [7]. After the discovery of Carbon fullerene the interest has now shifted towards the search of new fullerene like structures formed by other elements/combination of different elements because fullerene type structure offers several unique properties that can be used in different Nano-technology applications like synthesis of Nano-structured semiconducting magnets [6]. With the rapid increase in the density of the electrons with the reduced size of components in the electronic devices such as quantum dots and quantum clusters. Currently available technology has already made it possible to fabricate semiconductor clusters by using laser vaporization technique and mass spectroscopy. A great interest in the field of clusters has arisen because of the advancement in computational and theoretical techniques along with development of advanced spectroscopic techniques.
III – V group semiconductor materials have been the focus of numerous experiments [8-9]. Due to high electron mobility of III – V compounds such as GaAs, these have the capability of replacing Si in the electronics industry. GaAs is a direct band gap semiconductor with band gap energy of ~1.43 eV. In the recent years several experimental and theoretical investigations of GaAs clusters have been done. These studies have enabled to know more about the structural stability, reactivity and nature of bonding in these clusters. It is expect that gallium arsenide cluster may exhibit different bonding characteristics than their bulk because of their strong anion-anion bonds [10]. Investigations based on density functional techniques (DFT) have proved to be very useful for studying these type of micro-clusters. The DFT scheme is accurate enough to explain the physical properties of the cluster which depend on several parameters like the size, charge or the stoichiometric composition of the cluster [11-13].
The present work is focused on the structures, as well as electronic properties of fullerene type GaxAsx micro-clusters for x=10, 13, 15, 18, 20, 25 and 30 atoms.
Computational method
The fullerene like GaxAsx structures were generated using Gauss View 5.0The optimization and study of the structures and characteristics of these clusters has been carried out by using the DFT with unrestricted B3LYP exchange-correlation potential as implemented in Guassion 09 program [14-15]. For describing the good geometrical and bonding feature for heavy atom, the basis set with effective core potential, LanL2DZ [16] is used for Gallium (Ga ) and Arsenic(As) atoms. The basis set LanL2DZ describes the outermost electrons of 3s2 3p6 4s2 3d10 4p1for Ga and As atoms. The interaction between core and valence electron was handled by Torullier-Martins norm conserving pseudo potential in their fully separable form and the pseudo potential also includes d orbitals in the valance configuration [17,18].
Optimized geometries were obtained without any symmetry constraints and continued until the force components on each atom are less than 0.01 eV/Ȧ using a conjugated gradient algorithm. These optimized geometries were subsequently verified by frequency calculations. The presence of real imaginary frequencies confirms the stability of obtained geometries. The gap between energy of the highest occupied molecular orbital and lowest unoccupied molecular orbital levels and the binding energy is also used to determine the stability of the micro-clusters. The method and the basis set used are well established for the similar type study metal clusters [19].
Results and discussion
The optimized structures for gallium arsenide fullerenes (GaXAsX) are presented in Figure 1 for X= 10, 13, 15, 18, 20, 25 and 30, the dark red colours represent the Ga atom and violet colours represent the As atoms. In each structure has 12 pentagons and six edges are shared by adjacent pentagons. Common edges of pentagons consist of Ga-Ga and As-As homo nuclear bonds. The Ga-Ga bond length lies in range of 2.50-2.69 A0, the As-As bond length lies in range 2.58-2.62 A0 and Ga-As bonds length is in range 2.46-2.52 A0. The variation of bond lengths for Ga-Ga, As-As and Ga-As is shown Figure 2. The figure 2 clearly indicates that the dependency bond lengths in clusters are insignificant of cluster size. The homo-nuclear Ga-Ga and As-As bonds are almost constant, except for Ga-Ga bond in case of Ga10As10, which confirms their bonding. The Ga-As-Ga bond angle is 1000-1090 but the As-Ga-As bond angles tends to have larger values 1130-1300. The As-Ga-Ga bond angle at the shared pentagon edge is ≈1110 and Ga-As-As bond angles at shared pentagon edge is ≈ 980 but lies between the above bond angle ranges.
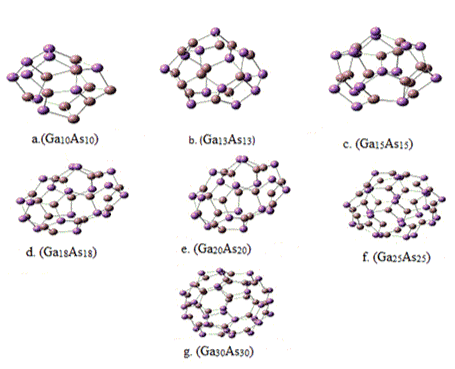
Figure 1. The optimized structures GaxAsx micro-clusters for x= 10, 13, 15, 18, 20, 25 and 30.
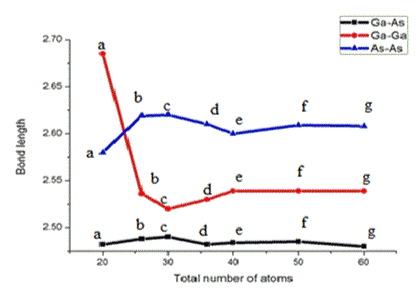
Figure 2. Variation of bond lengths in Å the GaxAsx clusters for x = 10, 13, 15, 18, 20, 25 and 30.
The variation of formation energy in eV for GaxAsx clusters for X=10, 13, 15, 18, 20, 25 and 30 is shown in Figure 3. The formation energy of the clusters lies between 0.1078-0.114976 eV. The binding energy in eV for GaxAsx clusters for X=10, 13, 15, 18, 20, 25 and 30 is shown in Figure 4. The binding energy increases monotonically with increase in cluster size.
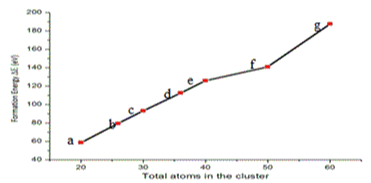
Figure 3. Variation of formation energy in eV for GaxAsx clusters for x = 10, 13, 15, 18, 20, 25 and 30.
The energetic of possible reaction from reactants Ga and As to the products i.e. cluster depending on the stoichiometric the clusters are obtained by the reaction
x(GaAs) ® GaxAsx
The binding energy change ∆E shown in figure 4 is a function of total number of atoms X+X in GaxAsx. It is obvious from figure 4 that the binding energy increases as the cluster size increases.
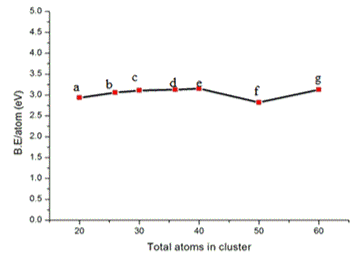
Figure 4. The Variation of binding energy in eV for GaxAsx clusters for x = 10, 13, 15, 18, 20, 25 and 30.
Electronic structure of the gallium arsenide fullerenes
The energy gap Eg is considered as the difference between energy highest occupied orbital (HOMO) and energy of lowest unoccupied orbital (LUMO), this also represents the chemical stability of clusters. The Eg of GaxAsx clusters are lie between 2.20-2.50 eV. The variation of Eg of GaxAsx cluster for x = 10, 13, 15, 18, 20, 25 and 30 is shown in Figure 5. The optical activity of a cluster is related to its corresponding polarizabillity. The variation of polarizability in au for GaxAsx clusters for x= 10, 13, 15, 18, 20, 25 and 30 is presented in Figure 6. The polarizabilty increase gradually till x=25 in GaxAsx cluster and it falls for Ga30As30. From figure 6 is clear that Ga25As25 is higher than other studied clusters.
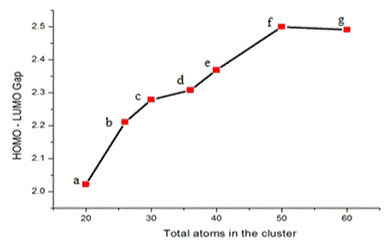
Figure 5. The variation of homo-lumo gap (Eg) in eV for GaxAsx clusters for x = 10, 13, 15, 18, 20, 25 and 30.
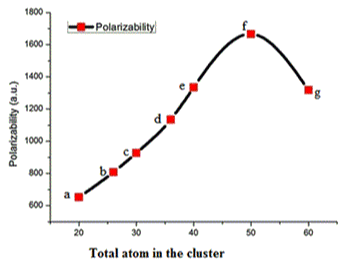
Figure 6. The variation of polarizability in au for GaxAsx clusters for x= 10, 13, 15, 18, 20, 25 and 30.
Conclusions
The electronic structure and optical properties of the GaxAsx clusters for x=10, 13, 15, 18, 20, 25 and 30 were studied using DFT method. The binding energy and HOMO-LUMO gap (Eg) confirms the stability of the clusters. The variation of polarizability, which reflects optical activity, with increase cluster size is presented and the study suggests that Ga25As25 cluster has more optical activity in comparison with other studied clusters.
References
- Dobwolski W, Kossut J, Story T (2003) II–VI and IV–VI Diluted Magnetic Semiconductors – New Bulk Materials and Low-Dimensional Quantum Structures. Handb Magn Mater 15: 289-377.
- Kamat PV (1993) Photochemistry on nonreactive and reactive (semiconductor) surfaces. Chem Rev 93: 267-300.
- Wang Y, Herron N (1988) Photoluminescence and relaxation dynamics of cadmium sulfide superclusters in zeolites. J Phys Chem 92: 4988-4994.
- Cox SD, Gier TE, Stucky GD, Bierlein J (1988) Inclusion tuning of nonlinear optical materials: switching the SHG of p-nitroaniline and 2-methyl-p-nitroaniline with molecular sieve hosts. J Am Chem Soc 110: 2986-2987.
- Javan MB (2013) Optical properties of GaAs nanocrystals: influence of an electric field. J Mol Model 19: 2273-2283. [Crossref]
- Li-Ren L, Heng-Jiang Z, Zhi-Feng L, Peng W (2012) Structures, stabilities, and electronic properties of GaAs tubelike clusters and single-walled GaAs nanotubes. Chin Phy B 21: 123601.
- Galica M, Buczko R (2013) Physica status solidi B-basic solid state. Physics pp: 739.
- Sabirov DS, ÅŒsawa E (2015) Information Entropy of Fullerenes. J Chem Inf Model 55: 1576-1584. [Crossref]
- Obrien SC, Liu Y, Zhang Q (1986) Supersonic cluster beams of III–V semiconductors: GaxAsy. J Chem Phys 84: 4074.
- Schäfer R, Becker JA (1996) Photoabsorption spectroscopy on isolated GaNAsM clusters. Phys Rev B Condens Matter 54: 10296-10299. [Crossref]
- Wang L, Chibante LPF, Tittel FK (1990) Ammonia chemisorption on gallium arsenide clusters. Chem Phy Lett 172: 335-340.
- Sarkar P, Springborg M (2003) Density-functional study of size-dependent properties of CdmSen clusters. Phys Rev B 68: 235409.
- Ghosh C, Pal S, Goswami B (2005) Theoretical studies on size-dependent properties of GanAsn clusters. Chem Phys Lett 407: 498-503.
- Brena B, Ojamae L (2008) Surface Effects and Quantum Confinement in Nanosized GaN Clusters: Theoretical Predictions. J Phys Chem C 112: 13516-13523.
- Becke AD (1988) Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A Gen Phys 38: 3098-3100. [Crossref]
- Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B Condens Matter 37: 785-789. [Crossref]
- Wadt RW, Hay JP (1985) Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J Chem Phys 82: 284.
- Fowler PW, Rogers KM, Seifert G (1999) Pentagonal rings and nitrogen excess in fullerene-based BN cages and nanotube caps. Chem Phys lett 299: 359-367.
- de Visser SP, Kumar D, Danovich M, Nevo N, Danovich D, et al. (2006) Ferromagnetic bonding: high spin copper clusters (n+1)Cu(n); n = 2-14) devoid of electron pairs but possessing strong bonding. J Phys Chem A 110: 8510-8518. [Crossref]