Introduction: Progressive cavitational leukoencephalopathy (PCL) is a recently described pathology, with few cases reported in the world literature. It is characterized by progressive neurological deterioration and characteristic neuroimaging findings, which distinguish this disorder as a unique entity given the massive cystic degeneration.
Patients and methods: A case of a female infant with regression of motor skills and speech is described at 27 months, followed by progressive neurological deterioration during the following year.
Results: In cranial MR findings, white matter compromise and cystic degeneration with contrast enhancement were observed, along with a brain MRS with double negative lactate spikes. Enzymatic study of leukodystrophies, genetic panel for leukodystrophies, and negative mitochondrial exome.
Conclusions: Due to the progressive form of the disease and paraclinical criteria, it is concluded that the patient meets the diagnostic criteria for PCL. This entity does not have treatment; only supportive care is available. The prognosis is fatal, with an average life of up to 14 years in the case studies described.
leukoencephalopathy, mitochondrial disorders, hypomyelination, leukodystrophy, lactate
Early childhood leukoencephalopathies are a group of diseases of heterogeneous etiology that affect the white matter of the central nervous system (CNS). A distinctive group of these leukodystrophies presents degenerative cystic changes, and a particular semiology [1].
In 2005 Naidu et al. characterized an infantile neurodegenerative syndrome with pictures of acute clinical deterioration associated with asymmetric patchy leukoencephalopathy in neuroimaging studies along with cavities and vascular permeability, found initially on corpus callosum and centrum semiovale, and developing cystic degeneration; This entity was called progressive cavitational leukoencephalopathy (PCL) [2]. Contrast enhancement and involvement of the corpus callosum makes it different from other pathologies, such as Vanishing White Matter Disease (VWMD) [3].
Until now, an autosomal recessive inheritance related to a history of first-degree consanguinity has been suggested; even though its pathophysiology is not yet clear, mitochondrial DNA mutations may be involved [1].
Clinical course ranges from rapidly progressive deterioration to long periods of clinical stability; in newborns it causes severe neurological and systemic alterations, including seizures, hypotonia, growth retardation, lactic acidosis and respiratory failure [4].
Given the non-specific neurological symptoms and neuroimaging features, a differential diagnosis with infections or immunological conditions that involve the CNS is essential, an important finding being the increase of serum lactate in CSF, or in affected brain regions identified through MRS [5].
The appropriate diagnosis allows us to evaluate the risk of possible complications, determine prognosis and focus the interdisciplinary care required for each comorbidity [6].
The objective of this article is to describe the clinical case of a patient with clinical evolution and imaging findings related to progressive cystic leukoencephalopathy.
Female infant was evaluated in a fourth level pediatric hospital located in the city of Bogotá, Colombia. Following the protocols established by the hospital, those responsible for the patient were asked to authorize the publication of clinical data, which they accepted by signing a written informed consent.
Literature search was carried out in multiple databases and indexed journals, such as PubMed, Elsevier, JAMA, Neurology, among others.
Female patient, 2 years and 3 months old, hispanic, first pregnancy of young consanguineous parents (first-degree cousins), with no significant pre or perinatal history.
Psychomotor development was normal up to 24 months, at which time she presented sudden regression in neurodevelopment, left eye strabismus, and ipsilateral hemiparesis; deterioration in the following 4 months showing gait and speech abnormalities, and loss of sphincter control, as well as mixed consistency dysphagia. The patient was evaluated by the pediatric emergency department, finding age-appropriate body measures, no systemic alterations, minor non-syndromic features, and discarding infectious, traumatic, or ictal triggers.
During the neurological examination she presents irritability, sporadic responsiveness to commands or instructions, normal fundoscopy, left third cranial nerve palsy, and facial symmetry with no compromise of lower pairs; axial hypotonia and quadriparesis of left predominance are also observed, as well as musculotendinous hyporeflexia without presence of pathological reflexes, normal head control, sitting position with support, no standing posittion, partial grasp reflex, and no meningeal or cerebellar involvement.
Based on the clinical findings, further paraclinic studies are requested (Table 1), and systemic pathologies are discarded. During neuroimaging studies with contrast MRI, significant white matter compromise is evidenced, along with asymmetric deterioration on posterior limbs of the internal capsule, and cystic degeneration with contrast enhancement that compromises the corpus callosum but spares basal ganglia and cortical gray matter (Figure 1) brain MRS shows double negative spikes of lactate, increased levels of choline, and creatine-choline ratio of 3:1 (Figure 2).
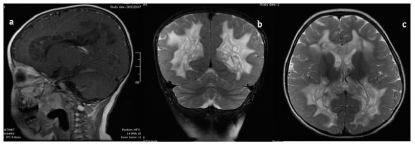
Figure 1. Contrast enhanced brain MRI (1a) Sagittal plane with cystic degeneration that compromises the callous body (1b) Coronal plane with lesions in deep white matter (1c) Axial plane with asymmetric deterioration on posterior limbs of the internal capsule and sparing the basal ganglia and cortical grey matter

Figure 2. Brain MRS. Double negative spikes of lactate, with increased levels of choline and creatine-choline ratio of 3:1
Table 1. Initial paraclinics
General paraclínic studies |
Blood oxygen |
Lumbar puncture |
LDH (U/L) |
330 |
330 |
209 |
Total Bilirubin (mg/dL) |
0.33 |
pH |
7.4 |
Appearance |
transparent |
BUN, mg/dL |
23 |
14.99 |
15 |
Alkaline Phosphatase (Ul/L) |
104 |
PCO2 |
30 |
Red blood cells |
1/mm3 |
Creatinine (mg/dl) |
0.52 |
0.3 |
0.45 |
GOT (u/i) |
41.1 |
HCO3 |
16 |
Fresh |
100% |
Sodium (mEq/L) |
138.6 |
137 |
138 |
GPT (u/i) |
16 |
B5 |
-5.1 |
Crenated |
0% |
Potassium (mEq/L) |
4.5 |
4.8 |
4.3 |
TSH (mlU/L) |
0.61 |
|
|
Leukocytes |
0/mm3 |
Chlorine (mEq/L) |
107 |
96.8 |
96 |
Free T4 (mU/L) |
0.73 |
|
|
Glucose |
58 mg/dl |
Magnesium (mEq/L) |
2.5 |
2.23 |
2.4 |
CKMB |
13.8 |
|
|
Proteins |
41 md/dl |
Glycemia (mg/dl) |
61 |
67 |
69 |
CPK (mg/dl) |
24 |
|
|
|
|
Ammonium (m/dl) |
54.8 |
|
|
|
|
|
|
|
|
Neoplastic infectious processes are initially ruled out; neuroimaging abnormalities, initial diagnosis, and absence of pleocytosis or High concentrations of CSF proteins discard a demyenlinating disease. During admission she receives maintenance therapy and shows clinical improvement, so outpatient services are performed.
Four months later the patient shows a loss of head control, torso control, and visual tracking; bilateral optic nerve atrophy, spastic quadriparesis, and generalized musculotendinous dysreflexia are also observed. Given the rapid progression of the disease brain imaging is carried out. The results show cystic lesions and contrast enhancement of significant size (Figure 3); plasma and CSF studies are performed again, showing a non-significant increase of lactic acid (Table 2), and no compromise of liver, kidney, hydroelectrolytic or respiratory functions.
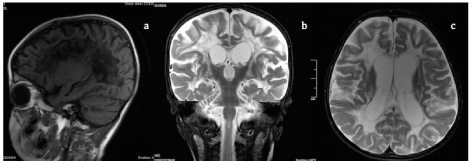
Figure 3. Contrast enhanced brain MRI (1a) Sagittal plane with significant increase in cortical atrophy (1b) Coronal plane with significant increase in cystic lesions in the posterior arms of the internal capsule (1c) Axial plane with increase in corpus callosum and deep white matter lesions, and their contrast enhancement therein
Table 2. Lactic acid and pyruvate values
Study |
Value |
Reference value |
Lactic acid (p) |
8.03 |
0.5-2.0 mmol/L |
Lactic acid (p) |
6.41 |
0.5-2.0 mmol/L |
Lactic acid (csf) |
2.75 |
1.3-1.9 mmol/L |
Pyruvate (p) |
0.11 |
0.04-0.14 mmol/L |
csf/p ratio |
24 |
|
Lactate (csf) |
4.0 |
|
Pyruvate (csf) |
0.12 |
|
csf/p ratio |
33 |
|
Based on the results, paraclinic studies of leukodystrophies with short-, medium-, and long-chain fatty acids, Arylsulfatase A (ARSA), Galactocerebrosidase in Leukocytes and Beta galactosidase are performed and show normal results; Genetic panel for leukodystrophy and mitochondrial exoma is requested and shows no alterations (Table 3).
Table 3. Metabolic studies for leukodystrophies
Study |
Value |
Reference value |
Short- and medium-chain organic acids |
Slight excretion of 3-hydroxybutyric and acetoacetic acid |
NA |
ARSA |
36.5 |
Control at 29.2 |
Galactocerebrosidase in leukocytes |
6.61 |
|
Beta-galactosidase |
28.63 nmol/h/mg |
Control at 75.73 nmol/h/mg |
C22 |
99. 1 |
Under 96.3 |
C24 |
80. 9 |
Under 91.4 |
C26 |
0. 92 |
Up to 1.30 |
C24/C22 ratio |
0. 82 |
Under 1.39 |
C26/C22 ratio |
0. 009 |
0.023 |
Pristanic acid |
1. 02 |
Under 2.98 |
Phytanic acid |
7. 96 |
Under 9.88 |
Pristatic/Phytanic Acid |
0. 07 |
0.39 |
Second plasma and CSF studies, showing non-significant increase of lactic acid. Source: Developed by authors.
A medical board of metabolic diseases is held and based on symptomatology, acute deterioration and evident changes in neuroimaging, the patient meets the criteria for PCL diagnosis. Pharmacological treatment with coenzyme Q10 and multivitamins is requested.
Actually, our patient has progressive deterioration. She is currently on interdisciplinary follow-up and support for palliative care.
Leukodystrophies are part of a heterogeneous group of genetic diseases that cause white matter lesions; thanks to scientific, neuroimaging, and clinical advances, a new classification of this group of diseases was carried out in 2017, based on pathological changes and pathophysiologic mechanisms; in this manner Knaap and Burgiani propose these new categories (Table 4) [7].
Table 4. Classification of Leukodystrophies
Type |
Pathophysiologic features |
Diseases linked |
Myelin Disorders |
Sever loss of oligodendrocytes and myelin.
Hypomyelinating disorders
Demyelinating disorder
Myelin vacuolization disseases |
Pelizaeus-Merzbacher Disease (Hypomyelinating)
Metachromatic leukodystrophy (Demyelinating)
Progressive cavitating leukoencephalopathy (Myelin Vacuolization Disorder) |
Astrocytopathies |
Mutations in astrocyte-specific gene products |
Alexander disease
Megalencephalic leukoencephalopathy with subcortical cysts |
Leuko-axonopathies |
Defects in neuron- or axon-specific gene products
White substance degeneration by abnormal axo-glia interaction. |
Hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC)
GM1 gangliosidosis |
Microgliopathies |
Genetic alterations of microglia-specific gene products |
Hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS) |
Leuko-vasculopathies |
Alteration of white matter in which the main disease mechanisms involve the brain small blood vessels |
Cathepsin A-related arteriopathy with strokes and leukoencephalopathy (CARASAL) |
LCP is a clinical and neuroimaging syndrome, proposed in 2005 by Naidu [2], characterized by cavitating lesions and acute neurological dysfunction, and recently added to the myelin disorder group according to the new classification of leukodystrophies [7].
The genetic basis of LCP is not fully clarified;however, current data suggest an autosomal recessive inheritance pattern [3]. Multiple genes related to mitochondrial function have been suggested. In 2017 Ishiyama et al. reported two pediatric patients with LCP related to mutations of the Iron-Sulfur Cluster Assembly Factor gene IBA57 [8]. The first patient was a male who at 6 months showed alterations in visual development, as well as axial hypotonia, optic atrophy and low vision at 17 months, with an MRI showing diffuse leukoencephalopathy with large cavitated areas and alteration of the callous body; by age 7, the patient developed decreased visual acuity, spastic quadriplegia, and refractory epilepsy. The second patient was a female born to consanguineous parents, who showed rapid regression in motor and visual abilities later than 6 months, as well as evolutionary spastic quadriparesia, decreased visual acuity and pupillary reflex; her MRI showed extensive bilateral leukocyencephalopathy with corpus callosum and middle cerebellar peduncles involved, and large cavitaties in the deep white matter. Both patients presented heterozygous mutations of IBA57 gene in their molecular tests [8,9], thus showing that the clinical picture and neuroimaging of our patient are similar to that described in the literature.
Mutations in the LYRM7 gene are mentioned as well, which associates a drastic change in a highly conserved amino acid residue that leads to serious defects in mitochondrial respiratory chain complex III, relating it to multifocal abnormalities of the periventricular and deep cerebral white matter, progressively coalescing to a cystic lesion [10]. PCL has also been related to the NFU1 gene, which contributes to the assembly of lipoate synthase, and the formation of four enzymes involved in the mitochondrial respiratory chain complex II (RCC): pyruvate dehydrogenase complex (PDHc), α-ketoglutarate dehydrogenase (α-KGDH), branched chain ketoacid dehydrogenase (BCKDH), and glycine cleavage system H protein [11,12].
Even though leukodystrophies affect people of all ages, their onset is usually during childhood or adolescence in previously healthy patients, with a progressive deterioration that leads the majority of patients to a premature death [13].
The semiology of the entity is not very specific, generally developing motor and cognitive deterioration without systemic compromise; Therefore, some specific clinical and paraclinical factors that help focusing the differential diagnosis, such as the adrenal involvement in X-linked adrenoleukodystrophy, macrocephaly in Alexander-Canavan disease and Van Der Knaap syndrome, increased serum lactate and CSF lactate in mitochondrial diseases, peripheral neuropathy in PCL, among others, should be taken into account [9].
The clinical course of PCL has been classified as follows:
- Stabilization/improvement pattern: Children with an acute episode who remain stable and even show improvement.
- Progressive deterioration pattern: Patient with rapid deterioration and a fatal outcome, like the patient described in our case report.
- Paroxysmal deterioration pattern: Paroxysmal episodes related to external factors (e.g. infectious diseases); it is associated with a progressive deterioration.
MRI has become a valuable tool for the differential diagnosis of leukoencephalopathies; the specific distribution of lesions in the white matter (affecting mostly the U-shaped fibers, periventricular deep white matter) and moving to other structures (cortical gray matter, basal ganglia), and the identification of cystic lesions has allowed the identification of specific genetic forms [14].
The cystic changes in PCL neuroimaging mainly involve the corpus callosum, cerebral and cerebellar white matter, and the spinal cord [15]. In advanced stages, lesions in centrum semiovale have been observed, progressing to a cystic degeneration of the aforementioned areas, until it covers almost the entire SB, sparing U-fibers and grey matter [12]. Zhang et al. categorized PCL injuries as follows [1]:
- Diffuse white matter pattern.
- Deep white matter pattern, as evidenced in our patient.
- Frontal predominant pattern.
- Parieto-occipital predominant pattern.
- Multiple region pattern.
Contrast enhancement has been described as a characteristic of PCL, making it different from other pathologies, such as VWMD [16]. Whenever a patient shows neurological deterioration with leukodystrophic cystic changes in neuroimaging, the following differential diagnoses must be taken into account (Table 5) [2].
Table 5. Differential diagnoses of PCL and its main features
Disease |
Features |
Megalencephalic leukoencephalopathy with subcortical cysts |
- Anterior temporal cysts
- Increased head circumference
|
Alexander disease (fibrinoid leukodystrophy) |
- T2: Visible lesions in bifrontal white matter, usually symmetrical. Caudate nucleus> globus pallidus> thalamus> brainstem
Periventricular border
- T1: Enhanced contrast around the same areas (In pediatric patients)
Obstructive hydrocephalus produced by periaqueductal gray lesions, and inflammation of the basal ganglia |
Acute disseminated encephalomyelitis (ADEM) |
- CSF pleocytosis
- Increased myelin proteins in CSF
- Anti-MOG antibodies present
- Neuroimaging:
- Calloseptal damage is unusual
- Asymmetric bilateral lesions
- Gray matter deterioration
- Absence of diffuse bilateral lesion pattern
- Hypointense lesions
- Two periventricular lesions
|
Autosomal dominant acute necrotizing encephalopathy (ADANE) |
- Bilateral thalamic lesions
- Hypointense lesions on T1 and hyperintense on T2
- Restricted diffusion in affected regions
- Hemorrhage, cavitation and post-contrast enhancement
|
Hemophagocytic lymphohistiocytosis (HLH) |
- Nonspecific periventricular white matter abnormalities
- Cerebral atrophy
- Increased extra-axial CSF spaces
- Orbital myopathy
|
Progressive vacuolating leukoencephalopathy |
- Symmetric cystic leukoencephalopathy mainly affecting the occipital and temporal lobes.
- CSF glycine/Plasma ratio> 0.09
|
Progressive cavitational leukoencephalopathy: |
- Asymmetric patchy areas evolving into cystic degeneration.
- Cystic changes affect corpus callosum, and spinal cord.
|
Leigh syndrome |
- T2: Typical enhancement in brainstem, periaqueductal gray matter, medulla, putamen, corpus striatum, subthalami nuclei; deterioration of cerebral or cerebellar white matter is unusual.
- T1: Abnormal T2 areas are not so clearly observed.
- MRS: Elevated choline, occasionally elevated lactate, reduced NAA.
|
Although MRS does not present a specific pattern, it has been associated with PCL given the spike in lactate, which is correlated with its parallel increase in CSF; in the present case study a double negative spike of lactate was observed, translating into a positive spike and its elevation. This last finding has generated the hypothesis of a possible mitochondrial origin, which has yet to be confirmed [15].
Regarding diagnostic aids, in addition to elevated levels of lactate in the brain, plasma and CSF, alterations have been evidenced in the values of organic acids in urine and changes in the muscle respiratory chain enzymes, which were normal in our patient. Pathologically, there is a severe loss of U-shaped fibers and myelin, axonal disruption, and cavitary lesions without inflammation [17,18].
A clinical focus is essential since the diagnosis, treatment, and prognosis of leukodystrophies vary significantly. An example is in adrenoleukodystrophy, in which the Loes severity score indicates whether patients are eligible for treatments, such as bone marrow transplant [19].
At the moment, there is no cure for PCL, but symptomatic and palliative care is provided for the comorbidities that occur during the clinical deterioration of the patient. In several case reports, megavitamins, systemic corticosteroids, coenzyme Q10, among others, have been prescribed, without significant studies that detail a specific management [20].
Currently, studies of possible associated mutations continue based on the hypothesis that nuclear gene alterations compromise mitochondrial function or axonal myelin interaction, given the autosomal recessive inheritance in these patients [2].
Based on the clinical progression and the paraclinical findings (with emphasis on the diagnostic images and the double negative spike of lactate), it was concluded that the patient reported in this article meets the diagnostic criteria for PCL. In the initial stages, differentiation from acute infectious, postinfectious or immunological diseases is complex, making neuroimaging a key diagnostic point, and thus vital for the clinical approach. Having clinical knowledge about PCL will allow it to be considered in the differential diagnosis when finding a patient with progressive neurological deterioration associated with cystic white matter lesions that enhance with contrast. This allows a better interdisciplinary and rehabilitation approach, since this entity does not have therapeutic treatment and only supportive care is available. The prognosis is fatal, with an average life of up to 14 years in the case studies described [1].
- Zhang J, Liu M, Zhang Z, Zhou L, Kong W, et al. (2019) Genotypic spectrum and natural history of cavitating leukoencephalopathies in childhood. Pediatr Neurol 94: 38-47. [Crossref]
- Naidu S, Bibat G, Lin D, Burger P, Barker P, et al. (2005) Progressive cavitating leukoencephalopathy: A novel childhood disease. Ann Neurol 58: 929-938. [Crossref]
- Sharma S, Singh P, Fernandez-Vizarra E, Zeviani M, Van der Knaap MS, et al. (2018) Cavitating leukoencephalopathy with posterior predominance caused by a deletion in the APOPT1 gene in an Indian boy. J Child Neurol 33: 428-431. [Crossref]
- Neilson DE, Feiler HS, Wilhelmsen KC, Lynn A, Eiben RM, et al. (2004) Autosomal dominant acute necrotizing encephalopathy maps to 2q12.1-2q13. Ann Neurol 55: 291-294. [Crossref]
- Abdelsalam EM, Ashamallah GA, Lateef MA, Fathy K (2015) Proton MR Spectroscopy in leukodystrophies. Egypt J Radiol Nucl Med 46: 1091-1097.
- Seyda A, Newbold RF, Hudson TJ, Verner A, Mackay N, et al. (2001) A novel syndrome affecting multiple mitochondrial functions, located by microcell-mediated transfer to chromosome 2p14-2p13. Am J Hum Genet 68: 386-396. [Crossref]
- van der Knaap MS, Bugiani M (2017) Leukodystrophies: A proposed classification system based on pathological changes and pathogenetic mechanisms. Acta Neuropathol 134: 351-382. [Crossref]
- Ishiyama A, Sakai C, Matsushima Y, Noguchi S, Mitsuhashi S, et al. (2017) IBA57 mutations abrogate iron-sulfur cluster assembly leading to cavitating leukoencephalopathy. Neurol Genet 3: 1-9. [Crossref]
- Stehling O, Wilbrecht C, Lill R (2014) Mitochondrial iron-sulfur protein biogenesis and human disease. Biochimie 100: 61-77.
- Dallabona C, Abbink TEM, Carrozzo R, Torraco A, Legati A, et al. (2016) LYRM7 mutations cause a multifocal cavitating leukoencephalopathy with distinct MRI appearance. Brain 139: 782-794. [Crossref]
- Nizon M, Boutron A, Boddaert N, Slama A, Delpech H, et al. (2014) Leukoencephalopathy with cysts and hyperglycinemia may result from NFU1 deficiency. Mitochondrion 15: 59-64. [Crossref]
- Pippucci T, Maresca A, Magini P, Cenacchi G, Donadio V, et al. (2015) Homozygous NOTCH 3 null mutation and impaired NOTCH 3 signaling in recessive early onset arteriopathy and cavitating leukoencephalopathy. EMBO Mol Med 7: 848-858. [Crossref]
- Biancheri R, Rossi D, Cassandrini D, Rossi A, Bruno C, et al. (2010) Cavitating leukoencephalopathy in a child carrying the mitochondrial A8344G mutation. Am J Neuroradiol 31: 78-79. [Crossref]
- Hong A, Assaad P, Karkare S (2019) Teaching NeuroImages: MRI findings in an infant with cavitating leukoencephalopathy. Neurology 92: e884-e885. [Crossref]
- Kekatpure MV, Karthik GA, Bhat V (2014) Progressive cavitating leukoencephalopathy: Case report of a rare childhood onset neurodegenerative disease. Indian J Child Health 1: 61-64.
- Mayr JA, Zimmermann FA, Fauth C, Bergheim C, Meierhofer D, et al. (2011) Lipoic acid synthetase deficiency causes neonatal-onset epilepsy, defective mitochondrial energy metabolism, and glycine elevation. Am J Hum Genet 89: 792-797. [Crossref]
- Del Toro M, Arranz JA, Macaya A, Riudor E, Raspall M, et al. (2006) Progressive vacuolating glycine leukoencephalopathy with pulmonary hypertension. Ann Neurol 60: 148-152. [Crossref]
- Read IJ (2001) Progressive cavitating leukoencephalopathy: Case report of a rare childhood onset neurodegenerative disease. Can Fam Physician 47: 788-789.
- Shinagawa A, Hugdal S, Babu J, Rangaswamy R (2020) Progressive cavitating leukoencephalopathy associated with a homozygous POLG mutation of 264C>G (p.F88L). Radiol Case Rep 15: 908-913. [Crossref]
- Verity CM, Winstone AM, Stellitano L, Krishnakumar D, Will R, et al. (2010) The clinical presentation of mitochondrial diseases in children with progressive intellectual and neurological deterioration: A national, prospective, population-based study. Dev Med Child Neurol 52: 434-440. [Crossref]
Editorial Information
Editor-in-Chief
Andy Goren
University of Rome, Italy
Article Type
Case Report
Publication history
Received date: January 26, 2021
Accepted date: February 02, 2021
Published date: February 09, 2021
Copyright
©2021 Divahia G. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation
©2021 Divahia G, Natalia M, Sara L, Eugenia E (2021) Progressive cavitational leukoencephalopathy: An enigma. Case Rep Rev, 7: DOI: 10.15761/CCRR.1000497.