Abstract
Cardiomyopathies define a heterogeneous group of heart muscle disorders. They are an important cause of heart failure and a leading indication for heart transplantation in pediatric patients. Research on cardiomyopathy has however focused on the adult populations with fewer dedicated clinical trials investigating pediatric cardiomyopathy. Diagnosis and clinical management of pediatric cardiomyopathy are also challenging because of the lack of specific methods for diagnosis and treatment. Assessment of ventricular structure using absolute values in adults are not useful in children because of their constantly changing body surface area. Therapies developed for adults applied to pediatric patients also have had varied clinical outcomes. This paper therefore reviews existing research evidence on pediatric cardiomyopathy, with the objective of broadening its clinical understanding and management.
Key Words
Cardiomyopathy, pediatric cardiomyopathy, pediatric patients, heart failure
Despite advancements in medical therapy and increased availability of cardiac transplantation, cardiomyopathies remains a leading cause of pediatric mortality. The true incidence of pediatric cardiomyopathies remains unknown mostly attributed to the lack of clinical recognition and under-reporting. In a majority of cases, the cause of pediatric cardiomyopathy is unknown and prognosis is usually ominous. A reduction in the morbidity and mortality associated with pediatric cardiomyopathies requires a better understanding of the causes and pathogenesis to support the development of etiologic specific treatment. Thus, this paper presents a comprehensive review of the status of scholarly and practitioner knowledge on pediatric cardiomyopathies and the major phenotypes involved. The review is organized into seven sections: clinical description, epidemiology, clinical manifestation, prognosis, etiopathogenesis, differential diagnosis and clinical management. Also included are two meta-analyses of diagnostic methods and of clinical management approaches.
Dedicated research on cardiomyopathies in pediatric population has been much more recent compared to adult and geriatric populations. Prior to the mid-1990s, there was a paucity of research evidence for epidemiologic features and natural history of pediatric cardiomyopathies. However, the establishment of Pediatric Cardiomyopathy Registry (PCMR) in 1995, which is an ongoing population-based multi-center observational study of primary and idiopathic cardiomyopathies in children and adolescents, inspired current research interest in pediatric cardiomyopathies [1]. The National Heart, Lung and Blood Institute (NHLBI) funded the initial research between September 1995 and August 1999. The objective of the PCMR was to provide a precise estimate of incidence rates and a description of the natural course of selected phenotypes of cardiomyopathies in pediatric populations, and facilitate the development of etiology-specific approaches to clinical management [2].
The development of PCMR data involved 61 private and institutional health care facilities handling pediatric cardiomyopathies in the U.S and Canada [3]. As of 1999, the PCMR consisted of a prospective, population-based cohort of 337 pediatric patients diagnosed between 1996 and 1999 from New England and Central Southwestern U.S., and a retrospective cohort of 990 pediatric patients diagnosed between 1990 and 1995 identified through a review of medical records [2]. Since its establishment, the PCMR has been the basis of several subsequent studies on pediatric cardiomyopathies including the National Australian Childhood Cardiomyopathy Study (NACCS). The PCMR has made significant contributions to the current understanding of incidence, natural history, clinical symptoms, diagnosis and clinical management of pediatric cardiomyopathies.
Pediatric cardiomyopathies is not a distinct clinical entity but an umbrella term for uncommon and heterogeneous group of myocardial disorders with multifactorial etiologies and many different clinical outcomes in individuals aged 18 years or younger [4]. The World Health Organization (WHO) classifies cardiomyopathies into four distinct types based on morphological and functional abnormalities: (i) dilated cardiomyopathy (DCM); (ii) hypertrophic cardiomyopathy (HCM); (iii) restrictive cardiomyopathy (RCM); and (iv) arrhythmogenic right ventricular cardiomyopathy (ARVD) [5]. Pediatric cardiomyopathies mainly includes three of these four major phenotypes of cardiomyopathy: DCM, HCM and RCM. Of the three types, DCM and HCM have been reported as the most frequently encountered in clinical practices as well as extensively studied [3,6]. In addition to pediatric DCM, HCM and RCM phenotypes, LV non-compaction (LVNC) has been recently classified as another common type of pediatric congenital (also spongy) cardiomyopathy primarily involving the LV apex. The WHO [5] and the National Organization of Rare Diseases (NORD) [7] also classify cardiomyopathies into etiologic (primary or secondary) and pathologic (ischemic or non-ischemic) cardiomyopathies. Idiopathic and non-ischemic cardiomyopathies are the most common types in pediatric populations [2]. Based on the available classification systems, pediatric cardiomyopathies can be classified based on their (a) morphology (DCM, HCM, RCM, ARVD or LVNC); (b) etiology (primary or secondary); and (c) pathology (ischemic or non-ischemic) (Table 1).
Table 1: Classification and Distinguishing Features of Types of Pediatric Cardiomyopathies
Classification |
Major Types |
Major Distinguishing Feature |
Morphological/ Structural |
Dilated |
Left ventricular (LV) or bi-ventricular dilatation and global systolic dysfunction in the absence of abnormal loading conditions (such as hypertension or valvular disorders) or coronary artery disease (CAD) [8] |
Hypertrophic |
LV thickening with non-dilated chambers in the absence of flow-limiting CAD or abnormal loading conditions [9] |
Restrictive |
Stiffened ventricles impairing ventricular filling with normal or decreased diastolic volume affecting one or both ventricles [10]. |
Arrhythmogenic Right Ventricular Dysplasia |
Fibrofatty replacement of the right ventricular (RV) myocardium mainly in the RV inflow, outflow and apex [11]. |
LV Non-Compaction |
Deep inter-trabecular recesses and thickened myocardium separated into two distinct compacted and non-compacted layers [1]. |
Etiologic |
Primary |
Develops by itself or by idiopathic (unknown) etiologies [5]. |
Secondary |
Develops in the setting of known cardiac abnormalities such as viral or bacterial myocardial inflammation or from chronic/prolonged exposure to environmental toxins such as heavy metals [5]. |
Pathologic |
Ischemic |
Caused by hardening of coronary arteries leading to insufficient blood flow and oxygen in the myocardium [7]. |
Non-ischemic |
Caused by structural or electrical dysfunction of the myocardium [7]. |
Prior to the establishment of PCMR in 1995, epidemiologic features of pediatric cardiomyopathies remained vaguely defined. Although a large number of early studies investigated the natural history of cardiomyopathies in infants and children, they only offered fragmentary evidence on the incidence of pediatric cardiomyopathies [8-19]. The main limitation of these early studies was small or ethnically homogenous samples limiting statistical support of findings to other excluded pediatric populations [2]. A population-based study in Olmsted County in Minnesota suggested an increased incidence in the duration of the study (1975-1984) but lacked sufficient sample to support the findings to all pediatric age groups – neonates (birth - 1 month), infants (1 month – 2 years), developing children (2 to 12 years) and adolescents (12 to 16 years) [18]. The earliest and largest population-based study in Finland conducted between 1980 and 1991 also indicated increased incidence of pediatric cardiomyopathy, reporting an incidence rate of 0.65 in 100,000 persons aged 20 years or younger [19]. Another large population-based study conducted in Australia 1987-1996 reported a higher incidence of 1.24 in 100,000 persons aged 10 years or younger [20].
The establishment of the PCMR provided precise estimates of pediatric cardiomyopathies in the U.S. The PCMR data, gathered from 18 and 20 pediatric centers in New England and Central Southwest U.S. respectively, reported an annual incidence of pediatric cardiomyopathies of 1.13 cases per 100,000 children [1]. The incidence was significantly higher in infants < 1 year estimated at 8.34 incidence in 100,000, in boys (1.32) compared to girls (0.92), and in blacks (1.47) compared to whites (1.06), which were determined using DCM and HCM functional phenotypes [21,22]. However, the incidence might have been underestimated because the definition of pediatric cardiomyopathies excluded children who suffered from sudden cardiac death, findings from pathologists and medical examiners in the original protocol, and children and adolescent asymptomatic of left ventricular dysfunction who did not seek medical attention [22].
Table 2 provides a summary of major epidemiological studies on incidence rates and percentages of the two major types of pediatric cardiomyopathies. The summary reveals wide variations in the age-ranges of pediatric patients included in the studies ranging from younger than ten years to younger than 20 years, which affects the ability to compare incidence rates or to calculate global incidence rates of pediatric cardiomyopathies. Six studies [18-21,23,24] with a combined pediatric population of 2,510 indicate the overall incidence rate of pediatric cardiomyopathies ranged between 0.57 and 1.24 per 10,000 children, with DCM being the most prevalent phenotype followed by hypertrophic (Table 2).
Table 2: Incidence of Pediatric Cardiomyopathies from Major Population-based Studies
First Author [Ref#] |
Study Period |
Study Region |
Sample |
Age-range (Yrs.) |
Overall Incidence per 100,000 |
Percentage (%) |
DCM |
HCM |
Others |
Codd et al. [18] |
1975-1984 |
Minnesota |
64 |
< 15 |
0.9 |
67 |
30 |
3 |
Arola et al. [19] |
1980-1991 |
Finland |
118 |
< 20 |
0.65 |
52 |
37 |
11 |
Nugent et al. [20] |
1987-1996 |
Australia |
314 |
< 10 |
1.24 |
59 |
26 |
15 |
Malcic et al. [23] |
1988-1998 |
Croatia |
121 |
< 18 |
38.1* |
43 |
36 |
5 |
Lipshultz et al. [21] |
1996-1999 |
U.S. |
467 |
< 18 |
1.13 |
51 |
42 |
7 |
Towbin et al. [24] |
1996-2003 |
U.S. |
1426 |
< 18 |
0.57** |
NA |
NA |
NA |
* per 10,000 patients examined in the participating healthcare facilities; ** Studied only dilated cardiomyopathy
The hallmark of pediatric cardiomyopathies is ventricular impairment or dysfunction. Despite having multiple etiologies, the development of cardiomyopathies in children and adolescent is progressive occurring in three main stages (Figure 1).
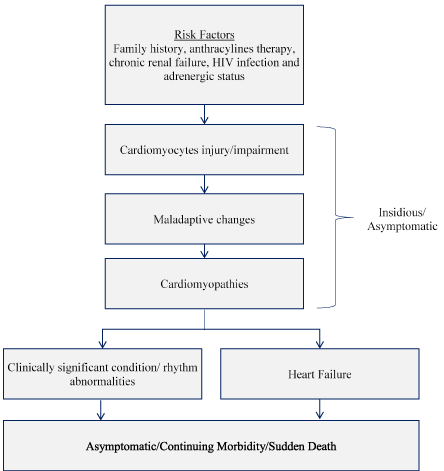
Figure 1: Clinical Stages in Pediatric Ventricular Dysfunction/Impairment [25]. Pediatric ventricular dysfunction begins with exposure to risk factors, leading to cardiomyocytes injury/impairment, maladaptive changes and finally cardiomyopathy. Cardiomyopathy presents as clinically significant rhythm abnormalities or heart failure leading to morbidity or sudden cardiac death.
Pediatric ventricular dysfunction begins with chronic exposure to risk factors such as family history of sudden death, anthracylines therapy, chronic renal failure, HIV infection and adrenergic status. The exposure leads to impairment of cardiac myocytes inducing maladaptive changes in cardiac functions, which results into ventricular dysfunctions (cardiomyopathy). These initial stages are largely asymptomatic or insidious. Cardiomyopathy may lead to rhythm abnormalities, clinically significant condition or heart failure, which may be asymptomatic or characterized with continuing morbidity. The ultimate stage is usually sudden cardiac death [25].
Clinical signs and symptoms of pediatric cardiomyopathies may present at birth or develop at any age before the eighteenth year of life [17]. These often always vary depending on the type of cardiomyopathy present but some children and adolescent may remain asymptomatic for many years or even throughout life. Despite the variation, clinical symptoms common to all forms of pediatric cardiomyopathies are fatigue, exertional dyspnea and chest pain [11]. Other symptoms common to a majority of individuals with pediatric cardiomyopathies include arrhythmias (tachycardia or bradycardia), endocarditis (inflammation of the endocardium), congestive heart failure (CHF) or sudden cardiac death (SCD). In DCM, sudden cardiac death may be the first and only clinical manifestation in about 5% of children [26]. Other symptoms may differ depending on the underlying type of cardiomyopathy HCM, RCM, ARVD and LVNC) (Table 3).
Table 3: Major Clinical Symptoms of Different Types of Pediatric Cardiomyopathies
Cardiomyopathy |
Major Clinical Signs and Symptoms |
Dilated Cardiomyopathy |
Fatigue, exertional dyspnea, peripheral edema, chest pain, irritability, persistent cough due to pulmonary congestion, syncope, abdominal pain and arrhythmias. |
Hypertrophic Cardiomyopathy |
Exertional dyspnea, fatigue, excessive sweating, poor appetite, chest pain, arrhythmias, syncope during exertion, pulmonary congestion, congestive heart failure or sudden cardiac death. |
Restrictive Cardiomyopathy |
Dyspnea, chest pain, poor appetite, ascites, peripheral edema, pulmonary congestion, hepatomegaly, arrhythmias, thrombus, heart block, congestive heart failure or sudden cardiac death. |
Arrhythmogenic Right Ventricular Dysplasia |
Arrhythmias, dyspnea, swollen neck veins, abdominal discomfort, syncope, cardiac arrest or sudden cardiac death. |
LV Non-Compaction |
Dyspnea, New York Heart Association (NYHA) Class III/IV heart failure, chest pain, chronic atrial fibrillation and heart failure in the setting of sustained/paroxysmal arrhythmias and thromboembolic events. |
Increased understanding of etiopathogenesis of pediatric cardiomyopathies has contributed to the current shift towards etiology-specific treatment, and together with increased availability of heart transplantation, have had a significant improvement in clinical management. However, pediatric cardiomyopathies remain an important cause of death in children and adolescents [27]. In a majority of cases (two-thirds), the cause remains idiopathic, and in some cases, diagnosis has been usually incidental during assessment of unrelated illness or during routine check-up, which has contributed to the current poor prognosis [1]. Predictor of poor prognosis also varies depending on the type of cardiomyopathy. DCM: age at diagnosis > 6 years, severely reduced FS Z-score and LVEF; HCM: age of diagnosis at < 12 months of age, malformation syndromes, certain inborn errors of metabolism, low LV FS and exercise-related abnormal blood pressure; RCM: family history, ischemia, chest pain and syncope; and LVNC: family history, delayed diagnosis, progressive LV dysfunction, arrhythmias or embolic events (Table 4).
Table 4: Predictors of Poor Prognosis in Pediatric Cardiomyopathies
Type |
Predictors of Poor Prognosis (Heart Transplantation/Cardiac Death) |
Dilated |
Age at diagnosis > 6 years, severely reduced FS Z-score and LVEF, Familial (idiopathic) DCM, absence of lymphocytic myocarditis, spherical LV, and CHF at diagnosis [24,26,28,29] |
Hypertrophic |
Diagnosis at < 12 months of age, malformation syndromes, certain inborn errors of metabolism, low LV FS [6]; exercise-related abnormal blood pressure [30], mixed functional phenotypes, higher LVED posterior wall thickness or end-diastolic ventricular septal thickness, or two or more risk factors [31]. |
Restrictive |
Has the poorest prognosis: 66-100% undergo heart transplantation or death a few years after diagnosis. Predictors of poor prognosis are family history, ischemia, chest pain and syncope [31]. |
LV Non-Compaction |
Family history, delayed diagnosis, progressive LV dysfunction, arrhythmias or embolic events [31] |
FS: Fractional Shortening; HR: Hazard Ratio; CHF: Congestive Heart Failure; LV: Left Ventricular; LVED: Left Ventricular End-Diastolic
The list excludes paediatric cardiomyopathies due to neuromuscular disease and inborn errors of metabolism because heart transplantation is usually not indicated as a treatment for this cohort
Causes of pediatric cardiomyopathies are classified into two broad categories: genetic (also referred to as primary or idiopathic), and non-genetic (also referred to as secondary), caused by a known underlying cardiac or non-cardiac condition [27].
In pediatric cardiomyopathies, genetic etiologies may fall into four main categories [21,27-31]:
- Inborn errors of metabolism;
- Malformation syndromes;
- Neuromuscular diseases; and
- Familial Isolated disorders.
Inborn Errors of Metabolism (IEM) are hereditary conditions with an incidence of 1 in 4,000 newborns. In the Online Mendelian Inheritance of Man database (https://www.omim.org/), IEMs account for more than 1,000 distinct disorders. About 5% of IEM have been linked with pediatric cardiomyopathies [32]. The main pattern of inheritance is autosomal recessive with a few having X-linked recessive pattern. The PCMR data reveals IEM accounts for 26.8% of HCM, 6.8% of DCM, 26.9% of mixed or other phenotypes of pediatric cardiomyopathies and the remaining are idiopathic causes [1]. Understanding IEMs has an important clinical value because many IEMs have a disease-specific treatment [32]. IEM presents the largest group of conditions associated with pediatric cardiomyopathies, which consists infiltrative storage disorders, energy metabolism disorders, and disorders producing cardiotoxic intermediary metabolites [33-43]. The exact processes that IEMs may induce the onset of pediatric cardiomyopathies remains tenuous. Three hypothesis have been advanced to explain the pathophysiological mechanisms of IEM disorders leading to the development of pediatric cardiomyopathies. These are storage and infiltration non-metabolized substrates, impaired energy production and the production of toxic metabolites [32] (Figure 2).
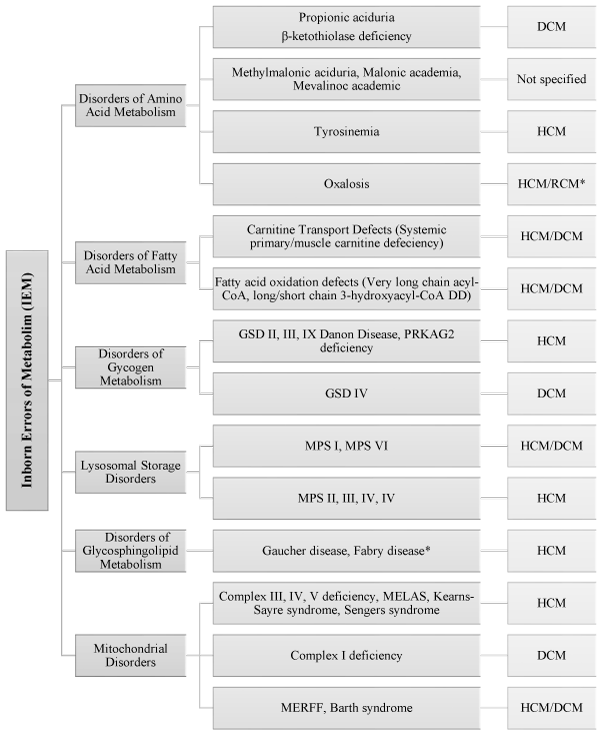
Figure 2: Types of IEMs Associated with Pediatric Cardiomyopathies [32]. Summarizes the main sub-types of IEMs associated with pediatric cardiomyopathies.
Storage and infiltration of substrate: Bulk storage and infiltration of non-metabolized substrate are postulated to convey a debilitating mechanical effect on the efficient functioning of cardiomyocytes (cardiac muscle cells). Bulk substrate deposition on cardiomyocytes may disrupt the normal alignment of myofibrils, which form the basis of cardiac contraction, impairing their contractile ability. This pathophysiologic mechanism usually involves large macromolecules mostly triglycerides (fatty-acid oxidation and carnitine transport defects), glycogen and lysosomal substrates. Ultimately, triglycerides and glycogen accumulate on the cytoplasm while lysosomes enlarge and multiply in numbers [32].
Impaired energy production: Impaired energy production leading to inadequate adenosine triphosphate (ATP) to support energy requirements at the cellular level is the second hypothesis proposed to explain the development of pediatric cardiomyopathies. The principal sources of cardiac energy are fatty acids and glycogen, which are broken down to release hydrogen molecules. Mitochondrial respiratory processes extract energy from the hydrogen molecules and oxidative phosphorylation process converts the energy into ATP. Impairment in oxidative phosphorylation process reduces the ability to convert any energy source into ATP. Since cardiomyocytes require high amounts of energy for myofibril contraction obtained from hydrolysis of ATP, inadequate energy means inefficient contractility, whose adaptive process is hypertrophy, and thus the reason behind the association of many IEMs to HCM [32].
Production of toxic metabolites: The third hypothesized pathophysiologic mechanism for IEM-related pediatric cardiomyopathies is the production of toxic metabolites by defects in oxidative phosphorylation, organic acidemias and aminoaciduria. Deposition and accumulation of these toxic metabolites lowers cellular pH of acids, inhibits intermediary metabolism or oxidizes components of the mitochondria such as DNA, lipids and proteins structures [32].
Malformation syndromes represent another category of genetic etiologies postulated to lead to the development of pediatric cardiomyopathies. Clinical presentation include minor or major physical abnormalities, which are characterized by distinctive facial features. Malformation syndromes occur due to inherited genetic mutation with autosomal dominant, autosomal recessive or X-linked patterns of transmission. The syndrome could also result from a defect (deletion or duplication) in specific chromosomes [21]. The main types of hereditary malformation syndromes associated with pediatric cardiomyopathies are autosomal dominant, autosomal recessive, X-linked recessive, chromosomal defect and unknown gene syndrome (Figure 3).
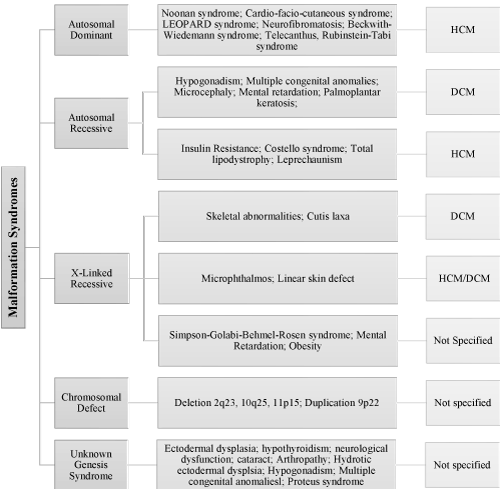
Figure 3: Malformation Syndrome Associated with Pediatric Cardiomyopathies [27]. The main types of hereditary malformation syndromes associated with pediatric cardiomyopathies are autosomal dominant, autosomal recessive, X-linked recessive, chromosomal defect and unknown gene syndrome and their associated genetic disorders.
Neuromuscular diseases affecting nervous or skeletal muscles is another category of genetic etiologies associated with the development of pediatric cardiomyopathies. The main types of neuromuscular disease are muscular dystrophies especially Duchenne’s and Becker type, congenital myopathies and ataxias. Typical symptoms include reduced muscle relaxation, bulk and tone, general weakness and loss of motor control [21] (Figure 4).

Figure 4: Neuromuscular Disorders Associated with Pediatric Cardiomyopathies [27]. Neuromuscular disorders associated with pediatric cardiomyopathy include muscular dystrophies especially Duchenne’s and Becker type, congenital myopathies and ataxias while their typical symptoms include reduced muscle relaxation, bulk and tone, general weakness and loss of motor control.
Familial isolated disorders are considered genetic etiologies of pediatric cardiomyopathies when there is a determined family history of cardiomyopathy but the child does not present with any signs and symptoms of metabolic or muscular disorders [21,27]. The major patterns of inheritance are autosomal dominant, autosomal recessive, X-linked or maternal (mitochondrial) transmission. Although the same genetic mutation may affect family members, disease expression widely varies from asymptomatic to mild symptoms and to severe complications [27] (Figure 5).

Figure 5: Familial Isolated Disorders Associated with Pediatric Cardiomyopathies [27]. The main familial disorders (HCM, DCM, RCM and LVNC) causing pediatric cardiomyopathy are transmitted through autosomal dominant, x-linked, autosomal recessive and maternal or mitochondrial patterns.
Pediatric cardiomyopathies may also result from non-genetic etiologies consisting of both cardiac and non-cardiac conditions. Although many of these cardiac and non-cardiac conditions may have a genetic basis in their natural development, cardiomyopathy in pediatrics usually occurs as a secondary effect. However, the exact pathogenesis of non-genetic etiologies remains vaguely understood requiring additional clinical trials. The main cardiac conditions include atherosclerotic CAD, Kawasaki disease, hypertension, dysrhythmia, congenital heart defect, and chronic modification in circulatory volume. Others cardiac causes are related to therapy such as heart transplantation, major cardiac or invasive cardiothoracic surgery. Non-cardiac conditions include pulmonary parenchymal disease of vascular origin, pregnancy/3-month postpartum, infection, toxin/drug, radiation, immunologic disease, connective tissue disease, endocrine disease, nutritional deficiency, granulomatous disease or malignancy (Figure 6).

Figure 6: Non-Genetic Etiologies of Pediatric Cardiomyopathies [27]. Non-genetic causes are classified into cardiac conditions or effect of cardiac therapies such as heart transplantation or major cardiac or invasive cardiothoracic surgery, and non-cardiac conditions including pulmonary diseases, pregnancy, infection, toxin, immunologic disease, exposure to radiation and nutritional deficiency.
The hallmark of differential diagnosis of pediatric cardiomyopathies is the assessment of myocardial abnormalities in systolic function (contractile ability), diastolic function (relaxation and compliance), and myocardial growth (hypertrophy and atrophy) [43-47]. Still, diagnostic evaluation of pediatric cardiomyopathies remains an important clinical challenge because of a large number of idiopathic genetic etiologies, a broad range of clinical manifestations and large numbers of specialized clinical and biochemical tests complicating confirmatory diagnosis [27]. Unlike in adults, where the assessment of ventricular dysfunction is the absolute LV wall thickness or hypertrophy, in children, absolute values are not useful because of their constantly changing body surface area. Instead, diagnosis depends on the association of LV dysfunction indexed to body surface area, defined as fractional shortening Z-score [30].
Electrocardiography (ECG) is an important test aimed to provide initial information prompting for further or specific diagnostic assessment for pediatric cardiomyopathies. ECG tests assess the presence of left or right, or both, ventricular contractile dysfunction (arrhythmic disturbance) and repolarizing parameters relative to normal values and their standardized interpretation for children [48,49]. ECG measures myocardial abnormalities based on abnormal Q-wave, QT interval, isolated ST segment changes, left/right bundle branch block, premature contractions and supraventricular or ventricular ectopic activities [12]. Together with clinical signs and symptoms, ECG abnormalities provide valuable information to guide further tests for confirmatory diagnosis of the specific phenotype of pediatric cardiomyopathies [27].
Echocardiographic-defined ventricular dysfunction is considered the gold standard for the assessment and diagnostic evaluation of different forms of cardiomyopathies in children and adolescents. Echocardiographic measurement and semi-quantitative patterns provide important information about abnormalities in ventricular function and structure for differential diagnosis [2]. Schwartz et al. [27] corroborates the value of echocardiography-defined ventricular dysfunction in a multi-disciplinary diagnostic assessment of pediatric cardiomyopathies due to genetic etiologies. Echocardiographic measures and patterns were used in the selection of children and adolescent during the development of the PCMR, which have been accepted and re-used by several subsequent studies as standardized criteria for diagnosis and understanding the clinical course of pediatric cardiomyopathies [27]. The main echocardiography measurements are LV fractional shortening, LV posterior wall thickness, and LV end-diastolic dimension/volume (Table 5).
Table 5: Echocardiographic Findings for Diagnosis of Pediatric Cardiomyopathies
Echocardiographic Measurement |
Abnormal LV Outcomes |
LV Fractional Shortening |
> 2 SD less than normal mean value of age |
LV posterior wall thickness at end-diastole |
> 2 SD less than the normal mean value for BSA |
LV end-diastolic dimension/volume |
> 2 SD more than the normal mean value for BSA |
SD: Standard Deviation; BSA: Body Surface Area
The presence of LV functional or structural abnormalities should prompt further investigation of the cause and diagnostic assessment for pediatric cardiomyopathies. While any one of the criteria for diagnosis is consistent with myocardial abnormalities in pediatric cardiomyopathies, the presence of more than one criteria strengthens diagnosis [27]. Additional echocardiographic semi-quantitative patterns alongside their clinical description, which strongly suggest the need for further diagnostic evaluation for specific types of pediatric cardiomyopathies include localized ventricular hypertrophy, RCM, contracted endocardial firbroelastosis, ventricular dysplasia, concentric hypertrophy or LVNC [2] (Table 6).
Table 6: Echocardiographic Patterns Suggesting the Need for Further Evaluation
Echocardiographic Patterns |
Description |
Localized ventricular hypertrophy |
Septal thickness > 1.5 times LV posterior wall thickness with or without outflow obstruction. |
Restrictive cardiomyopathy |
Enlarged atrium; small/normal sized ventricles; impaired diastolic filling |
Contracted endocardial firbroelastosis |
Same to RCM but with echo-dense endocardium |
Ventricular dysplasia |
Thinned RV, dilated atrium better assessed by MRI |
Concentric hypertrophy |
In the absence of hemodynamic causes |
LV non-compaction |
Spongy LV myocardium with trabeculae and multiples interstices. |
Endomyocardial biopsy can be safely performed in children to provide valuable information for diagnosing cases of pediatric cardiomyopathies especially of idiopathic (genetic) and some non-genetic etiologies. Microscopic and histologic features of endomyocardial biopsy specimens provide valuable information for confirmatory diagnosis of pediatric cardiomyopathies as well as diseases presenting with similar characteristics [50-53]. Some of these histologic features may be specific for a certain group of disorders such as infiltrative storage disorders or a certain disease such as Fabry disease. Biopsy may also assess tissues other than endomyocardium to detect associated disorders such as storage and mitochondrial disorders as well as disorders due to accumulation of neutral fat. Only cardiac phosphorylase kinase deficiency can be diagnosed from cardiac tissue specimen using biopsy [27].
Pediatric cardiomyopathies remain a leading cause of death in pediatric population. In adequate research recognition and under-reporting has undermined precise guidelines for its diagnosis. This meta-analysis review published peer-reviewed articles on the diagnosis of pediatric cardiomyopathy intended to improve its diagnosis and inform the implementation of etiologic-specific clinical management.
Search for studies for inclusion in this meta-analysis was conducted on online databases: PubMed, EMBASE, Ovid MEDLINE, and Google Scholar. The search employed broad search terms, “pediatric” OR “children” AND “cardiomyopathies” OR “heart diseases” AND “diagnosis” OR “prognosis”. Then use of broad search terms was to ensure the search outcomes includes as much as possible relevant studies. The search then used a hierarchical approach beginning with title, then abstract and finally perusal of the entire article to screen all the retrieved studies for inclusion.
The inclusion criteria was the study was (i) prospective or retrospective; (ii) studied at least one phenotype of pediatric cardiomyopathy; and (iii) included at least one diagnostic method for pediatric cardiomyopathies. Studies were excluded if only abstract was available, they were conference papers (because they are not final) and data was presented in a format that was not easy to extract.
Individual studies were each screened for the inclusion criteria and all relevant data extracted from the included studies. Extracted data was recorded on Microsoft Excel Worksheet. The extracted data included first author, study period, number of patients, diagnosis method, diagnosed phenotype of cardiomyopathy, mortality rate, and conclusion (Table 7).
Table 7: Meta-Analysis of Diagnosis and Prognosis of Pediatric Cardiomyopathies
1st Author [Ref. #] |
Study Period |
No. of Patients |
Diagnosis Method |
Phenotype |
Predictors Poor Prognosis |
Mortality (%) |
Conclusion |
Yeomen et al. [12] |
1958-1997 |
99 |
Echocardiography
|
HCM |
Increased QT interval; tachycardia; myocardial bridging. |
18 |
ECG and angiography results help identify children at risk of death. |
Towbin et al. [24] |
1990-1995 |
1,426 |
Echocardiography/Biopsy |
DCM |
Older age; CHF; Lower LVFS Z-score; and Cause |
31 |
Outcome depends on cause, age and heart failure status. |
Daubeney et al. [26] |
1987-1996 |
184 |
Echocardiography/Histology |
DCM |
Baseline age > 5 yrs.; familial DCM; lo LVFS Z-score; no increase in Z-score |
5 |
High early mortality but good long-term prognosis. |
Decker et al. [30] |
1985-2006 |
96 |
Echocardiography |
HCM |
Low LVFS Z-score; abnormal exertional blood pressure |
7 |
Medication and exercise restriction in isolated HCM have favorable prognosis. |
Colan et al. [54] |
1996-2000 |
855 |
Echocardiography |
HCM |
IEMs; Malformation Syndromes; idiopathic HCM diagnosed > 1 year |
1 |
Outcomes depend on etiology and age. |
Lipshultz et al. [31] |
1990-2006 |
1,085 |
Echocardiography
|
HCM |
IEMs; mixed functional phenotypes; Diagnosis > 1 year of age. |
16 |
Outcome depends on age, cause, heart failure status and extent of LV dysfunction. |
Connuck et al. [55] |
1990-1995 |
455 |
Echocardiography |
DCM |
Presence of DMD/BMD; Lower LVFS/NO changes in LVFS-Z-score |
10 |
DMD-related CM has higher mortality rate/heart transplantation |
Alvarez et al. [56] |
1990-2007 |
1,192
|
Echocardiography |
DCM |
Diagnosis > 6years; CHF; lower LVFS Z-score; Lower height for age Z-score; cause |
10 |
Poor prognostic predictors were age at diagnosis, etiology, extent of LV dysfunction. |
Yang et al. [57] |
2005 |
50 |
Genetic analysis; Biopsy |
HCM |
Mutation in lysosomal associated protein-2 |
NA |
Danon disease is under recognized entity in familial HCM |
Yetman et al. [58] |
1956-1997 |
99 |
Angiography |
HCM |
Myocardial bridging in 28% of patients with more chest pains, cardiac arrest and LV tachycardia |
NA |
Myocardial bridging is a predictors of poor prognosis and a risk factors for SCD in HCM |
Pignatelli et al. [59] |
1997-2002 |
36 |
Echocardiography
|
LVNC |
Age of diagnosis; Extent of LV dysfunction. |
14 |
LVNC lacks a fatal course if diagnosed within neonatal period |
Rivenes et al. [60] |
1967-1988 |
18 |
Echocardiography |
RCM |
Girls; chest pain; syncope without CHF |
22 |
RCM in children presents a risk of ischemia-related complications and death. |
Everitt et al. [61] |
1990-2012 |
868 |
Echocardiography |
DCM |
Older age; higher LVEDD Z-score |
NA |
Children with idiopathic DCM can recover LV function despite marked LV dilation at diagnosis |
Webber et al. [62] |
1990-2008 |
152 |
Echocardiography |
RCM |
Heart failure; low LCFS Z-score no decrease in Z-score; high posterior wall thickness. |
NA |
Transplant-free survival is poor in RCM children |
Biagini et al. [63] |
1964-2002 |
42 |
Echocardiography |
HCM |
Younger age; family history; greater wall thickness |
NA |
Dilated hypokinetic HCM indicated poor prognosis. |
Cetta et al. [64] |
2021 Copyright OAT. All rights reserv
1975-1993 |
8 |
Echocardiography |
RCM |
Short mitral deceleration time; greater pulmonary vein atrial reversal velocity and duration. |
63 |
Prognosis in idiopathic RCM in children is generally ominous |
Total/ Average |
|
6,665 |
|
|
|
12 |
|
LVNC: Left Ventricular Non-Compaction; CHF: Congestive Heart Failure; LVFS: Left Ventricular Fractional Shortening; DMD: Duchenne Muscular Dystrophy; BMD: Becker Muscular Dystrophy
Sixteen (16) prospective or retrospective studies meeting the inclusion criteria were included in this meta-analysis [12,24,26,30,31,54-64]. Altogether, the studies investigated a total sample of 6,665 patients aged younger than 18 years with pediatric cardiomyopathies having mean high mortality rate of 12% within five years of diagnosis. The study period ranged from 1958 [12] to 2012 [61]. Echocardiographic defined left ventricular dysfunction (also used in the recruitment of pediatric patients constituting the PCMR data), was the dominant method of diagnosis adopted by 14 studies (87.5%) [12,24,26,30,31,54-56,59-64]. The remaining two studies adopted genetic testing and biopsy [57] and angiography [58]. A majority of the studies (44%), investigated pediatric HCM phenotype, 31% investigated pediatric DCM, 19% pediatric RCM and the remaining 6% investigated left ventricular non-compaction.
The common diagnostic method for the three frequently encountered phenotypes of pediatric cardiomyopathies in clinical practice – HCM, DCM and RCM – is the echocardiographic-defined left ventricular dysfunction used by 87.5% of the studies. Endomyocardial biopsy and histology [24,26,57], angiography [58], and genetic testing [57] may provide additional valuable information to support the echocardiograph method in the confirmatory diagnosis of pediatric cardiomyopathy.
Findings from the 16 studies demonstrate echocardiographic defined prognostic predictors of poor clinical outcomes: low left ventricular fractional shortening Z-score, no increase in the Z-score during follow-up (44%) [24,26,30,55,56,61,62] and other clinical predictors such as age of diagnosis at one year or older (38%) [26,31,54,59,61], type of etiology (idiopathic, IEMS or malformation syndrome), mixed phenotypes (25%) [24,54,56,57] and status of the heart such as presence of congestive heart failure (25%) [24,56,60,62]. Other poor predictors of prognosis include myocardial bridging [12,58], family history, greater wall thickness [62,64], chest pain, syncope without congestive heart failure [60] and presence of muscular dystrophinopathies such as Duchenne and Becker muscular dystrophy [55].
Diagnosis of cardiomyopathies generally focus on evaluating intrinsic myocardial abnormalities of systolic function (contractility), diastolic function (relaxation and compliance) and growth (hypertrophy and atrophy) [27]. Standardized objective diagnosis criteria have since been implemented for the three most frequently encountered cardiomyopathy phenotypes: DCM, HCM and RCM but these guidelines have not specifically addressed pediatric patients. Such standardized and objective diagnostic guidelines would have improved the accuracy of diagnosis as well as a greater understanding of the clinical course of pediatric cardiomyopathies [2,27]. Thus, the present meta-analysis aimed to review the current methods used in the diagnosis of cardiomyopathies in pediatric populations to determine the most appropriate method.
In the present analysis, echocardiography is the most used and researched imaging modality for the diagnosis of cardiomyopathies in pediatric populations. A greater majority of the studies (87.5%) used echocardiography to assess and characterize LV dysfunction for the diagnosis of pediatric cardiomyopathy. The current guidelines based on small-scale clinical trials and a review of literature support the use of echocardiography in the diagnosis of pediatric cardiomyopathies [27]. According to Grenier and associates [2], echocardiography provides important quantitative and semi-quantitative measurement of the LV structure and function to support diagnosis or to suggest the need for additional confirmatory tests. In the present findings, echocardiographic defined LV fractional shortening, wall thickness at end-diastole and LV end-diastolic volume provide important diagnostic clues. However, LV fractional shortening emerged as the most useful measurement for cardiac systolic function in pediatrics because it is influenced by preload and afterload. Moreover, LV contractile function provide more accurate measurement reflecting the intrinsic health of the myocardium and are thus preferred for assessing LV dysfunction.
These measurements are consistent with those defined in echocardiography-based diagnostic criteria proposed by Schwartz et al. [27]. Whereas these quantitative measures of LV dysfunction may support diagnosis, the criteria emphasizes on other semi-quantitative measures such as septal thickness, enlarged atrium, thinned right ventricular, contracted endocardial firbroelastosis or spongy LV myocardium [27]. While echocardiography is the preferred imaging modality, other tests such as endomyocardial diagnosis and ECG abnormalities are important to provide additional diagnostic clues for the identification of an underlying cardiac condition [50,51]. Endomyocardial biopsies is significant in identifying the underlying cause of pediatric cardiomyopathy such as infiltrative storage disorders as well as assess surrounding tissues to detect mitochondrial disorders [27].
In summary, the standard diagnosis approach of pediatric cardiomyopathy is echocardiography-imaging modality, which is able to define LV systolic dysfunction (alterations in contractility) defined as LV fractional shortening, wall thickness at end-diastole and LV end-diastolic volume. Other semi-quantitative measures such as septal thickness, enlarged atrium, thinned right ventricular, contracted endocardial firbroelastosis or spongy LV myocardium provide supporting diagnostic clues. Other diagnostic methods such as endomyocardial biopsies are useful in analyzing surrounding tissues to detect underlying cardiac conditions.
Despite advances in the management of pediatric heart failure, limited evidence-based options for clinical management for children with cardiomyopathies exist. The ability to restore normal cardiac functions remains limited in pediatric populations with many adopting protocols modelled for adults with very few children-specific therapies [21]. However, nutritional, medical and heart transplantation have been widely used as clinical management therapies to prevent or even treat cardiomyopathies in pediatric populations.
Under-nutrition, defined as BMI < 10% for age and sex, and over-nutrition defined as BMI > 95%, are independent indications of poor prognosis in pediatric cardiomyopathies. This could be attributed to metabolic myopathies, which is one of the key underlying genetic etiologies of pediatric cardiomyopathies or exacerbates an already existing condition. Metabolic myopathies underlies the proposal that nutritional therapies could be an important ancillary treatment for improving the overall health and quality of life of children with cardiomyopathies [27]. In addition, chronic pediatric cardiomyopathy often always causes energy imbalance, where added metabolic demands increase faster than the ability of the child to consume adequate calories. The aim of nutrition is to provide sufficient dietary energy and other nutrients for proper cardiac functioning. Gastrointestinal malabsorption, suboptimal dietary intake of macro/micro nutrients, antioxidants, amino acids and vitamins, and psychosocial problems could also confer an important synergistic effect causing growth problems. Importantly, increasing the efficacy and clinical outcomes of nutritional or dietary therapy requires early detection and prompt initiation [65].
Medical treatment of pediatric cardiomyopathy is the same to that of heart failure, whose clinical objective is to manage inborn chronic and maladaptive physiological responses. Data from clinical trials of medical treatment of adult patients support the use of diuretics, beta-blockers, angiotensin-converting-enzyme (ACE) inhibitors and antiarrhythmic drugs for chronic treatment of heart failure [66]. Since pathophysiology of heart failure replicates that of adults, medical therapy used in adults has been applied to children with cardiomyopathies but with varying degrees of success. However, differences in etiologies, pharmacodynamics, duration of therapy and therapeutic goals between children and adults with cardiomyopathies requires additional studies to clarify the efficacy and value of medical therapy in pediatric cardiomyopathies [6]. While medical treatment may be not necessary for children with non-obstructive HCM, it has beneficial preventive effect against sudden cardiac death for asymptomatic children with obstructive HCM [67].
Clinical trials in adults with cardiomyopathies demonstrate that automatic cardioverter-de?brillation conveys a preventive secondary effect against sudden cardiac death. However, there is little evidence or consensus in studies of the value of automatic cardioverter-de?brillation in pediatric cardiomyopathies [68,69]. Although unexplained syncope linked with an elevated risk of sudden cardiac death is an indication for ICD as a primary or secondary prophylaxis, it lacks clear guidelines for pediatric populations [70,71].
Heart transplantation is the only definite treatment for children with end-stage heart failure with favorable prognosis indicated by one- and five-year survival rates of 92% and 80% respectively [72,73]. However, heart transplantation has important limitations, which include life-long immune suppression, limited graft survival, potential need for a repeat surgery and availability only in developed countries having advanced healthcare systems with transplantation capabilities. However, RCM phenotype has no universally accepted evidence-based criteria for indication for heart transplantation, which usually depends on local listing of pediatric heart centers [6].
Although current outcomes of clinical trials of human gene therapy have yielded disappointing findings, murine studies suggest that direct gene manipulation could be a promising future treatment strategy for children with cardiomyopathies. Evaluating the safety and effectiveness of potential vectors in humans is warranted to enable gene transfer to terminally differentiated myocardial cells [74]. Another viable treatment option to improve cardiac function is incorporating healthy cardiomyocytes into a dysfunctional myocardium [75]. Already, fetal cardiomyocytes and skeletal myoblasts engrafted into murine hearts have shown promising results of differentiating into adult myocardium myocytes, which may be beneficial in clinical practice to assist myocardial remodeling and repair [76,77].
Pediatric cardiomyopathies lack specific clinical management guidelines and many of the current approaches re-use therapies developed and validated for efficacy on adult populations. Since the pathophysiology of cardiomyopathies in adults and children may have vary, the lack of specific therapies may undermine effective clinical management of pediatric cardiomyopathies, which is believed to contribute to the increased cases of pediatric mortality and morbidity caused by cardiomyopathy. This meta-analysis reviews studies on clinical management of pediatric cardiomyopathies to help identify the most appropriate approaches to prevent and management pediatric cardiomyopathies.
Studies investigating clinical management of pediatric cardiomyopathies were searched from online databases (PubMed, EMBASE, Ovid MEDLINE) and Google Scholar. The broad search terms were “medical therapy” OR “clinical management” AND “pediatric” OR “children” AND “cardiomyopathies”.The search process was hierarchical, beginning with title search, then abstract and finally perusal of the entire document before consideration for inclusion.
Studies were included if they (a) studied cardiomyopathies in children (patients < 18 years); (b) investigated at least one clinical management method; (c) its outcomes included at least one clinical outcome (survival rate and/or mortality rate). Publication language was not considered as an inclusion criteria since all studies on pediatric cardiomyopathies were included irrespective of the publication language used. Studies that included both adults and children and did not provide separate analysis for children were also excluded. Conference papers, case series and studies with only abstract available (insufficient data for extraction) were also excluded. The extracted data included first author, study period, number if patients included, mean age of the patients, type of cardiomyopathy, clinical management method, follow-up period, five-year survival rate and mortality rate (Table 8).
Table 8: Meta-Analysis of Clinical Management of Pediatric Cardiomyopathies
1st Author [Ref. #] |
Study Period |
No. of Patients |
Mean Age (Yrs.) |
Type |
Clinical Management Method |
Follow-up (Months) |
5-yr Survival Rate (%) |
Mortality (%) |
Tsirka, et al. [63] |
1990-1999 |
91 |
5.1 |
DCM |
Diuretics; ACE-Inhibitors; HTx |
12 |
58 |
12 |
Maron et al. [78] |
1987-2011 |
224 |
14.5 |
HCM |
ICD |
51 |
NA |
4 |
Kantor et al. [79] |
1976-2005 |
189 |
1.0 |
DCM |
ACE-Inhibitors/BB |
42 |
54 |
29 |
Ostman-Smith et al [80] |
1969-1999 |
26 |
6.6 |
HCM |
High dose BB |
144 |
65 |
0 |
Fenton et al. [81] |
1988-2004 |
18 |
6.1 |
RCM |
HTx |
12 |
NA |
11 |
Russo et al. [82] |
1970-2002 |
21 |
3.8 |
RCM |
Without HTx |
-- |
39 |
38 |
Bograd et al. [83] |
1986-2006 |
23 |
8.8 |
RCM |
HTx |
6 |
86 |
13 |
Levi et al. [84] |
1985-2001 |
28 |
8.0 |
Not-Specified |
LVAD/ECMO bridge to HTx |
22 |
89 |
11 |
Murtuza et al. [85] |
2000-2011 |
159 |
8.6 |
DCM/ RCM |
HTx |
54 |
82 |
NA |
Hoskote et al. [86] |
2000-2006 |
102 |
9.5 |
DCM/ RCM |
HTx |
44 |
86 |
11 |
Kimberling et al. [87] |
2002* |
8 |
6.3 |
RCM |
HTx |
12 |
NA |
0 |
Fiser et al. [88] |
2003* |
32 |
1.2 |
Not Specified |
ECMO bridge to HTx |
-- |
59 |
12 |
Lewis et al. [89] |
2003* |
27 |
|
DCM |
ACE-Inhibitors |
NA |
Improves Survival |
NA |
Shaddy et al. [90] |
1999* |
15 |
8.6 |
DCM |
ACE-Inhibitors |
22 |
Improves LVFS |
NA |
Rusconi et al. [91] |
2004* |
24 |
7.2 |
DCM |
BB – Carvedilol + Standard therapy |
23 |
Improves LVEF |
4 |
Oflaz et al. [92] |
2013* |
34 |
7.4 |
DCM |
BB – Carvedilol + Standard therapy |
9.5 |
Manages heart rate/ rhythm |
NA |
Total/ Average |
|
1021 |
7.73 |
|
|
6.8 |
69 |
12 |
*Study period not specified; ACE: Angiotensin-Converting Enzyme; BB: Beta-Blockers; ICD: Implantable Cardioverter-De?brillators; IRCM: Idiopathic RCM; LVAD: Left Ventricular Assist Devices; ECMO: Extracorporeal Membrane Oxygenation; HTx: Heart Transplantation
Sixteen (16) studies conducted between 1969 and 2013 meeting the inclusion criteria were included in this meta-analysis [63,78-92]. The total pediatric patients in all the 16 studies was 1,021 with mean age 7.73 years and mean follow-up period of 6.8 months. Combined five-year survival rate was 69% and mortality rate of 12%. The most commonly studied phenotype of pediatric cardiomyopathy was DCM by 50% of the studies [63,79,85,86,89-92] followed by RCM (37.5% of the studies) [81-83,85-87] and finally HCM (12.5% of the studies) [78,80]. Two studies did not specify the type of pediatric cardiomyopathy [84,88]. The dominant clinical management methods were heart transplantation assessed by 44% of the studies [63,81-83,85-87] followed by pharmacotherapy, which was similarly assessed by 44% of the studies [63,79,80,89-92], and finally mechanical support (left ventricular assist devices [LAVD], extracorporeal membrane oxygenation [ECMO], implantable cardioverter-de?brillators [ICD]) assessed by 18% of the studies [78,84,88]. The main clinical outcomes assessed in the studies was five-year survival rate and/or reported mortality rate but the period differed in individual studies.
Conventional pharmacotherapy using digoxin, diuretics and ACE inhibitors is the initial therapy prescribed mainly to DCM phenotype in children [63,79,89-92]. The addition of beta-blocker (carvedilol) has a beneficial effect of improving heart rate and rhythm (left ventricular ejection fraction or fractional shortening), and survival rates among DCM pediatric patients [91,92]. In addition , adjunct therapy using high dose beta-blocker on the HCM phenotype in children reveals superior clinical outcomes and survival rates compared to using conventional pharmacotherapy alone [80]. Mechanical support using LVAD and ECMO conveys effective short-term circulatory support but the benefits wane for long-term support. However, they are an effective short-term bridge for pediatric patients waiting for heart transplantation [84,88]. Heart transplantation remains the most effective preventive therapy for pediatric patients with cardiomyopathy-related end-stage heart failure. Reported long-term mortality rates range between 0% [87] and 13% [83] compared with 38% without heart transplantation [82]. The most frequent indication for heart transplantation is pediatric patients diagnosed with the RCM phenotype [81-83,85-87] since pharmacotherapy has not demonstrated preventive effect against cardiac death in the long-term [81].
Currently, research on clinical management of cardiomyopathies have focused extensively on adult populations with only a brief mention on pediatrics. This had led to management of pediatric cardiomyopathies adopting therapeutic protocols developed for adults despite pediatrics having differences in etiologies, pharmacodynamics, duration of therapy and therapeutic goals compared to adults [6,21]. In pediatrics, the present meta-analysis reveals DCM is the dominant phenotype affecting half of diagnosis, followed by RCM (37.5%) and HCM (12.5%). Despite the phenotypic heterogeneity of pediatric cardiomyopathies, the most researched and used clinical management methods are pharmacotherapy and heart transplantation each studied by 44% of the included articles.
Same to adults, the main pharmacotherapy drugs are that prescribed for the conventional management of heart failure - diuretics, beta-blockers, angiotensin-converting-enzyme (ACE) inhibitors and antiarrhythmic drugs. For the DCM phenotype, pharmacotherapy is the standard therapy for improving heart rate and rhythm. Consistent with the present findings, Gersh, et al. [67] reports medical therapy is mostly indicated for DCM, RCM and non-obstructive HCM phenotypes. Moreover, according to Pahl et al. [66], the aim of medical therapy in pediatric patients is to manage inborn and maladaptive physiological responses and treatment for heart failure symptoms, and the prevention of sudden cardia death in non-obstructive HCM.
Heart transplantation on the other hand is the most common and effective therapy especially for pediatric patients with end stage heart failure with improved long-term survival. Its most common indication if the ECM phenotype [81,83,85]. In addition to pharmacotherapy and heart transplantation, other heart failure therapies such as mechanical circulatory support and LC assist devices (LVAD) are common clinical management therapies assessed by 18% of the studies. The main use of device therapies in pediatric patients is providing a preventive secondary effect against sudden cardiac death. The clinical value of device therapy seems to reduce on the long-term. However, device therapy lacks clear guidelines in the management of cardiomyopathies in pediatric patients compared to adult patients [70,71].
Despite clinical value of pharmacotherapy, heart transplantation and device therapy in the management of pediatric cardiomyopathies, prognosis remains ominous. Five-year survival rate for the 1,021 pediatric patients was 69% and mortality rate of 12%. The rate was higher in patients receiving medical therapy followed by device therapy, suggesting reduced efficacy in the long-term. Only heart transplantation therapy conveyed a protective long-term effect. In sum, clinical management of pediatric cardiomyopathy adopts protocols developed for adults mainly for the management of heart rate, heart rhythm and heart failure. Despite treatment, prognosis remains ominous for chronic use of medical and/or device therapies.
Pediatric cardiomyopathies represent a heterogeneous group of serious myocardial conditions affecting children aged < 18 years. They are an important cause of heart failure and a leading indication for heart transplantation in children and adolescents. The approximate incidence in the general population is 1.13 per 100,000 children with higher incidence reported in infants. Its clinical course begins with chronic exposure to risk factors (genetic, anthracyclines therapy, renal failure or HIV infection), which impairs myocytes and incudes maladaptive changes in cardiac function leading to ventricular dysfunction, rhythm abnormalities and eventually cardiac death. Although clinical signs and symptoms vary based on the underlying phenotype, the main ones are fatigue, exertional dyspnea and chest pain. Its etiopathogenesis has a predominant genetic basis associated with inborn errors of metabolism, malformation syndromes, neuromuscular diseases and familial isolated disorders. Confirmatory diagnosis of the disease is a challenge because etiology may overlap or the disease may change from one phenotype to another in its clinical course and the applicability of many diagnostic techniques for adults have varying results on children due to constantly changing body surface area. The main diagnostic criteria relies on echocardiography measurement and semi-quantitative patterns of ventricular dysfunction. Clinical management is complicated by fewer dedicated studies on pediatric populations while the applicability of therapies developed for adult population has variable outcomes due to differences in etiology, pharmacodynamics, duration of therapy and therapeutic goals. Pharmacotherapy and mechanical support is effective for short-term treatment of heart failure symptoms. Heart transplantation remains the only effective therapy with acceptable positive long-term effect on survival rates especially for idiopathic RCM phenotype that has shown poor long-term prognosis with pharmacotherapy.
References
- Wilkinson JD, Landy DC, Colan SD, Towbin JA, Sleeper LA, et al. (2010) The pediatric cardiomyopathy registry and heart failure: Key results from the first 15 years. Heart Fail Clin 6: 401-413. [Crossref]
- Grenier MA, Osganian SK, Cox GF, Towbin JA, Colan SD, et al. (2000) Design and implementation of the North American pediatric cardiomyopathy registry. Am Heart J 139: s86-s95. [Crossref]
- Lipshultz SE, Towbin JA, Grenier MA, Osganian V, Colan SD, et al. (1998) Etiology and family history of pediatric cardiomyopathy: the early pediatric cardiomyopathy registry (PCMR) experience. J Am Col Cardiol 31: 30.
- Bharucha T, Lee KJ, Daubeney PE, Nugent AW, Turner C, et al. (2015) Sudden death in childhood cardiomyopathy: results from a long-term national population-based study. J Am Coll Cardiol 65: 2302-2310. [Crossref]
- Richardson P, McKenna W, Bristow M (1996) Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the definition and classification of cardiomyopathies. Circulation 93: 841-842. [Crossref]
- Lipshultz SE, Cochran TR, Briston DA, Brown SR, Sambatakos PJ, et al. (2013) Pediatric cardiomyopathies: Causes, epidemiology, clinical course, preventive strategies and therapies. Future Cardiology 9: 817-848. [Crossref]
- National Organization of Rare Diseases (2017) Pediatric Cardiomyopathy. Retrieved from https://rarediseases.org/rare-diseases/pediatric-cardiomyopathy/ on 15 October 2017.
- Pinto YM, Elliott PM, Arbustini E, Adler Y, Anastasakis A, et al. (2016) Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Euro Heart Jour 37: 1850-1858. [Crossref]
- Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, et al. (2011) 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy. Circulation CIR-0b013e318223e2bd.
- Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, et al. (1996) Report of the 1995 World Health organization/International Society and Federation of cardiology Task Force on the Definition and Classification of Cardiomyopathies. Circulation 93: 841-842. [Crossref]
- Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, et al. (2007) Classification of the cardiomyopathies: A position statement from the European Society pf Cardiology Working Group on Myocardial and Pericardial Diseases. Euro Heart Jour 29: 270-276. [Crossref]
- Yetman AT, Hamilton RM, Benson LN, McCrindle BW (1998) Long-term outcome and prognostic determinants in children with hypertrophic cardiomyopathy. Journal of the American College of Cardiology, 32, 1943-1950. [Crossref]
- Greenwood RD, Nadas AS, Fyler DC (1976) The clinical course of primary myocardial disease in infants and children. Am Heart J 92: 549-560. [Crossref]
- Taliercio CP, Seward JB, Driscoll DJ, Fisher LD, Gersh BJ, et al. (1985) Idiopathic dilated cardiomyopathy in the young: clinical profile and natural history. J Am Coll Cardiol 6: 1126-1131. [Crossref]
- Chen SC, Nouri S, Balfour I, Jureidini S, Appleton RS (1990) Clinical profile of congestive cardiomyopathy in children. J Am Col Cardiol 15: 189-193. [Crossref]
- Lewis AB, Chabot M (1991) Outcome of infants and children with dilated cardiomyopathy. Am J Cardiol 68: 365-369. [Crossref]
- Akagi T, Benson LN, Lightfoot NE, Chin K, Wilson G, et al. (1991) Natural history of dilated cardiomyopathy in children. Am Heart J 121: 1502-1506. [Crossref]
- Codd MB, Sugrue DD, Gersh BJ, Melton LJ 3rd (1989) Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. A population-based study in Olmsted County, Minnesota, 1975-1984. Circulation 80: 564-572. [Crossref]
- Arola A, Jokinen E, Ruuskanen O, Saraste M, Pesonen E, et al. (1997) Epidemiology of idiopathic cardiomyopathies in children and adolescents: A nationwide study in Finland. Am J Epidemiol 146: 385-393. [Crossref]
- Nugent AW, Daubeney PE, Chondros P, Carlin JB, Cheung M, et al. (2003) The epidemiology of childhood cardiomyopathy in Australia. N Engl J Med 348: 1639-1646. [Crossref]
- Lipshultz SE, Sleeper LA, Towbin JA, Lowe AM, Orav EJ, et al. (2003) The incidence of pediatric cardiomyopathy in two regions of the United States. New Eng J Med 348: 1647-1655. [Crossref]
- Wilkinson JD, Sleeper LA, Alvarez JA, Bublik N, Lipshultz SE; Pediatric Cardiomyopathy Study Group (2008) The pediatric cardiomyopathy registry: 1995–2007. Prog Pediatr Cardiol 25: 31-36. [Crossref]
- MalciÄ I, JelusiÄ M, Kniewald H, BarisiÄ N, JelasiÄ D, et al. (2002) Epidemiology of cardiomyopathies in children and adolescents: a retrospective study over the last 10 years. Cardiol Young 12: 253-259. [Crossref]
- Towbin JA, Lowe AM, Colan SD, Sleeper LA, Orav EJ (2006) Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA 296: 1867-1876. [Crossref]
- Lipshultz SE, Wilkinson JD (2012) Beta-adrenergic adaptation in idiopathic dilated cardiomyopathy: differences between children and adults. Europ Heart Journ 35: 10-12. [Crossref]
- Daubeney PE, Nugent AW, Chondros P, Carlin JB, Colan SD, et al. (2006) Clinical features and outcomes of childhood dilated cardiomyopathy. Circulation 114: 2671-2678.
- Schwartz ML, Cox GF, Lin AE, Korson MS, Perez-Atayde A, et al. (1996) Clinical approach to genetic cardiomyopathy in children. Circulation 94: 2021-2038.
- Lewis AB, Chabot M (1991) Outcome of infants and children with dilated cardiomyopathy. Am J Cardiol 68: 365-369. [Crossref]
- Alvarez JA, Wilkinson JD, Lipshultz SE (2007) Outcome predictors for pediatric dilated cardiomyopathy: A systematic review. Prog Pediatr Cardiol 23: 25-32. [Crossref]
- Decker JA, Rossano JW, Smith EO, Cannon B, Clunie SK, et al. (2009) Risk factors and mode of death in isolated hypertrophic cardiomyopathy in children. J Am Coll Cardiol 54: 250-254. [Crossref]
- Lipshultz SE, Orav EJ, Wilkinson JD, Towbin JA, Messere JE, et al. (2013) Risk stratification at diagnosis for children with hypertrophic cardiomyopathy: an analysis of data from the Pediatric Cardiomyopathy Registry. Lancet 382: 1889-1897. [Crossref]
- Cox GF (2007) Diagnostic approaches to pediatric cardiomyopathy of metabolic genetic etiologies and their relation to therapy. Prog Pediatr Cardiol 24: 15-25. [Crossref]
- Coleman RA, Winter HS, Wolf B, Gilchrist JM, Chen YT (1992) Glycogen storage disease type III (glycogen debranching enzyme deficiency): correlation of biochemical defects with myopathy and cardiomyopathy. Ann Intern Med 116: 896-900. [Crossref]
- Miller CG, Alleyne GA, Brooks SE (1972) Gross cardiac involvement in glycogen storage disease type 3. Br Heart J 34: 862. [Crossref]
- Gross DM, Williams JC, Caprioli C, Dominguez B, Howell RR (1988) Echocardiographic abnormalities in the mucopolysaccharide storage diseases. Am J Cardiol 61: 170-176. [Crossref]
- Colucci WS, Lorell BH, Schoen FJ, Warhol MJ, Grossman W (1982) Hypertrophic obstructive cardiomyopathy due to Fabry's disease. N Engl J Med 307: 926-928. [Crossref]
- Figarella-Branger D, Pellissier JF, Scheiner C, Wernert F, Desnuelle C (1992) Defects of the mitochondrial respiratory chain complexes in three pediatric cases with hypotonia and cardiac involvement. J Neurol Sci 108: 105-113. [Crossref]
- Cruysberg JR, Sengers RC, Pinckers A, Kubat K, van Haelst UJ (1986) Features of a syndrome with congenital cataract and hypertrophic cardiomyopathy. Am J Ophthalmol 102: 740-749. [Crossref]
- Colin AA, Jaffe M, Shapira Y, Ne'eman Z, Gutman A, et al. (1987) Muscle carnitine deficiency presenting as familial fatal cardiomyopathy. Arch Dis Child 62: 1170-1172. [Crossref]
- Treem WR, Stanley CA, Hale DE, Leopold HB, Hyams JS (1991) Hypoglycemia, hypotonia, and cardiomyopathy: the evolving clinical picture of long-chain acyl-CoA dehydrogenase deficiency. Pediatrics 87: 328-333. [Crossref]
- Edwards MA, Green A, Colli A, Rylance G (1987) Tyrosinaemia type I and hypertrophic obstructive cardiomyopathy. Lancet 1: 1437-1438. [Crossref]
- Rocchiccioli F, Wanders RJ, Aubourg P, Vianey-Liaud C, Ijlst L, et al. (1990) Deficiency of long-chain 3-hydroxyacyl-CoA dehydrogenase: a cause of lethal myopathy and cardiomyopathy in early childhood. Pediatric Research 28: 657-662. [Crossref]
- Bautista J, Rafel E, Martinez A, Sainz I, Herrera J, Segura L, et al. (1990) Familial hypertrophic cardiomyopathy and muscle carnitine deficiency. Muscle & Nerve 13: 192-194. [Crossref]
- Olsen JF, Oakley CM, Pisa Z (1981) Report of the WHO/ISFC Task Force on the definition and classification of cardiomyopathies. Circulation 64: 437A-438A.
- Boffa GM, Thiene G, Nava A, Dalla Volta S (1991) Cardiomyopathy: a necessary revision of the WHO classification. Int J Cardiol 30: 1-7. [Crossref]
- Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, et al. (1996) Report of the 1995 World Health Organization/International Society and Federation of Cardiology Taskforce on the Definition and Classification of Cardiomyopathies. Circulation 93: 841-842. [Crossref]
- Perloff JK (1985) Cardiomyopathies in infants and children. In: Moss AJ (Ed) Pediatrics Update: Reviews for Physicians. New York, NY: Elsevier Science Publishing Co; 1985:187-204.
- Davignon A, Rautaharju P, Boisselle E, Soumis F, Megelas M, Choquette A (1979) Percentile charts: ECG standards for children. Pediatric Cardiomyopathy 1: 133-152.
- Garson A (1983) The electrocardiogram in infants and children: A systemic approach. 1st Ed. Philadelphia: Lea & Febiger.
- Schmidt-Achert M, Fischer P, Pongratz D (1992) Myocardial evidence of dystrophin mosaic in a Duchenne muscular dystrophy carrier. Lancet 340: 1235-1236. [Crossref]
- Luri PR (1992) Endomyocardial biopsies in cardiomyopathies of children. Prog Pediatr Cardiol 1: 71-81.
- Shaddy RE, Bullock EA (1993) Efficacy of 100 consecutive right ventricular endomyocardial biopsies in pediatric patients using the right internal jugular venous approach. Pediatr Cardiol 14: 5-8. [Crossref]
- Leatherbury L, Chandra RS, Shapiro SR, Perry LW (1988) Value of endomyocardial biopsy in infants, children, and adolescents with dilated or hypertrophic cardiomyopathy and myocarditis. J Am Coll Cardiol 12: 1547-1554. [Crossref]
- Colan SD, Lipshultz SE, Lowe AM, Sleeper LA, Messere J, et al. (2007) Epidemiology and cause-specific outcome of hypertrophic cardiomyopathy in children. Circulation 115: 773-781. [Crossref]
- Connuck DM, Sleeper LA, Colan SD, Cox GF, Towbin JA, et al. (2008) Characteristics and outcomes of cardiomyopathy in children with Duchenne or Becker muscular dystrophy: a comparative study from the Pediatric Cardiomyopathy Registry. Am Heart J 155: 998-1005. [Crossref]
- Alvarez JA, Orav EJ, Wilkinson JD, Fleming LE, Lee DJ, et al. (2011) Competing risks for death and cardiac transplantation in children with dilated cardiomyopathy: results from the pediatric cardiomyopathy registry. Circulation CIRCULATIONAHA-110. [Crossref]
- Yang Z, McMahon CJ, Smith LR, Bersola J, Adesina AM, et al. (2005) Danon disease as an underrecognized cause of hypertrophic cardiomyopathy in children. Circulation 112: 1612-1617. [Crossref]
- Yetman AT, McCrindle BW, MacDonald C, Freedom RM, Gow R (1998) Myocardial bridging in children with hypertrophic cardiomyopathy: A risk factor for sudden death. New Engl Journ Med 339: 1201-1209. [Crossref]
- Pignatelli RH, McMahon CJ, Dreyer WJ, Denfield SW, Price J, et al. (2003) Clinical characterization of left ventricular noncompaction in children: a relatively common form of cardiomyopathy. Circulation 108: 2672-2678. [Crossref]
- Rivenes SM, Kearney DL, Smith EO, Towbin JA, Denfield SW (2000) Sudden death and cardiovascular collapse in children with restrictive cardiomyopathy. Circulation 102: 876-882. [Crossref]
- Everitt MD, Sleeper LA, Lu M, Canter CE, Pahl E, et al. (2014) Recovery of echocardiographic function in children with idiopathic dilated cardiomyopathy: Results from the pediatric cardiomyopathy registry. J Am Coll Cardiol 63: 1405-1413. [Crossref]
- Webber SA, Lipshultz SE, Sleeper LA, Lu M, Wilkinson JD, et al. (2012) Outcomes of restrictive cardiomyopathy in childhood and the influence of phenotype: a report from the Pediatric Cardiomyopathy Registry. Circulation 126: 1237-44. [Crossref]
- Biagini E, Coccolo F, Ferlito M, Perugini E, Rocchi G, et al. (2005) Dilated-hypokinetic evolution of hypertrophic cardiomyopathy: prevalence, incidence, risk factors, and prognostic implications in pediatric and adult patients. J Am Coll Cardiol 46: 1543-1550. [Crossref]
- Cetta F, O'Leary PW, Seward JB, Driscoll DJ (1995) Idiopathic restrictive cardiomyopathy in childhood: diagnostic features and clinical course. Mayo Clin Proc 70: 634-40 [Crossref]
- Davos CH, Doehner W, Rauchhaus M, Cicoira M, Francis DP (2003) Body mass and survival in patients with chronic heart failure without cachexia: the importance of obesity. J Card Fail 9: 29-35. [Crossref]
- Pahl E, Sleeper LA, Canter CE, Hsu DT, Lu M, et al. (2012) Incidence of and risk factors for sudden cardiac death in children with dilated cardiomyopathy: a report from the Pediatric Cardiomyopathy Registry. J Am Coll Cardiol 59: 607-615. [Crossref]
- Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, et al. (2011) 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: Executive summary: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Thorac Cardiovasc Surg 142: 1303-1338. [Crossref]
- Moss AJ, Hall WJ, Cannom DS, Daubert JP, Higgins SL, et al (1996) Improved survival with an implanted defibrillator in patients with coronary disease at high risk for ventricular arrhythmia. N Engl J Med 335: 1933-1940. [Crossref]
- Lenarczyk R, Kowalski O, Kukulski T, Kowalczyk J, Kalarus Z (2007) Resynchronization or dyssynchronization – successful treatment with biventricular stimulation of a child with obstructive hypertrophic cardiomyopathy without dyssynchrony. J Cardiovasc Electrophysiol 18: 542–544. [Crossref]
- Spirito P, Autore C, Rapezzi C, Bernabò P, Badagliacca R, et al. (2009) Syncope and risk of sudden death in hypertrophic cardiomyopathy. Circulation 119: 1703-1710. [Crossref]
- Ostman-Smith I (2010) Hypertrophic cardiomyopathy in childhood and adolescence: Strategies to prevent sudden death Fundam Clin Pharmacol 24: 637–652. [Crossref]
- Larsen RL, Canter CE, Naftel DC, Tressler M, Rosenthal DN, et al. (2011) The impact of heart failure severity at time of listing for cardiac transplantation on survival in pediatric cardiomyopathy. J Heart Lung Transplant 30: 755–760. [Crossref]
- Tsirka AE, Trinkaus K, Chen SC, Lipshultz SE, Towbin JA, et al. (2004) Improved outcomes of pediatric dilated cardiomyopathy with utilization of heart transplantation. J Am Coll Cardiol 44: 391-397. [Crossref]
- Marian AJ, Mares A Jr, Kelly DP, Yu QT, Abchee AB, et al. (1995) Sudden cardiac death in hypertrophic cardiomyopathy: Variability in phenotypic expression of ß-myosin heavy chain mutations. Eur Heart J 16: 368-376. [Crossref]
- Nabel EG1 (1995) Gene therapy for cardiovascular disease. Circulation 91: 541-548. [Crossref]
- Soonpaa MH, Koh GY, Klug MG, Field LJ (1994) Formation of nascent intercalated disks between grafted fetal cardiomyocytes and host myocardium. Science 264: 98-101. [Crossref]
- Koh GY, Klug MG, Soonpaa MH, Field LJ (1993) Differentiation and long-term survival of C2C12 myoblast grafts in heart. J Clin Invest 92: 1548-1554. [Crossref]
- Maron BJ, Spirito P, Ackerman MJ, Casey SA, Semsarian C, et al. (2013) Prevention of sudden cardiac death with implantable cardioverter-defibrillators in children and adolescents with hypertrophic cardiomyopathy. J Am Coll Cardiol 61: 1527-1535. [Crossref]
- Kantor PF, Abraham JR, Dipchand AI, Benson LN, Redington AN (2010) The impact of changing medical therapy on transplantation-free survival in pediatric dilated cardiomyopathy. J Am Coll Cardiol 55: 1377-1384. [Crossref]
- Ostman-Smith I, Wettrell G, Riesenfeld T (1999) A cohort study of childhood hypertrophic cardiomyopathy: Improved survival following high-dose beta-adrenoceptor antagonist treatment. J Am Coll Cardiol 34: 1813-1822. [Crossref]
- Fenton MJ, Chubb H, McMahon AM, Rees P, Elliott MJ, et al. (2006) Heart and heart–lung transplantation for idiopathic restrictive cardiomyopathy in children. Heart 92: 85-89. [Crossref]
- Russo LM, Webber SA (2005) Idiopathic restrictive cardiomyopathy in children. Heart 91: 1199-1202. [Crossref]
- Bograd AJ, Mital S, Schwarzenberger JC, Mosca RS, Quaegebeur JM (2008) Twenty-Year Experience With Heart Transplantation for Infants and Children With Restrictive Cardiomyopathy: 1986–2006. Am J Transplant 8: 201-207. [Crossref]
- Levi D, Marelli D, Plunkett M, Alejos J, Bresson J (2002) Use of assist devices and ECMO to bridge pediatric patients with cardiomyopathy to transplantation. J Heart Lung Transplant 21: 760-770. [Crossref]
- Murtuza B, Fenton M, Burch M, Gupta A, Muthialu N, et al. (2013) Pediatric heart transplantation for congenital and restrictive cardiomyopathy. Ann Thorac Surg 95: 1675-1684. [Crossref]
- Hoskote A, Carter C, Rees P, Elliott M, Burch M, et al. (2010) Acute right ventricular failure after pediatric cardiac transplant: predictors and long-term outcome in current era of transplantation medicine. J Thorac Cardiovasc Surg 139: 146-153. [Crossref]
- Kimberling MT, Balzer DT, Hirsch R, Mendeloff E, Huddleston CB, et al. (2002) Cardiac transplantation for pediatric restrictive cardiomyopathy: presentation, evaluation, and short-term outcome. J Heart Lung Transplant 21: 455-459. [Crossref]
- Fiser WP, Yetman AT, Gunselman RJ, Fasules JW, Baker LL, et al. (2003) Pediatric arteriovenous extracorporeal membrane oxygenation (ECMO) as a bridge to cardiac transplantation. J Heart Lung Transplant 22: 770-777. [Crossref]
- Lewis AB, Chabot M (1993) The effect of treatment with angiotensin-converting enzyme inhibitors on survival of pediatric patients with dilated cardiomyopathy. Pediatr Cardiol 14: 9-12. [Crossref]
- Shaddy RE, Tani LY, Gidding SS, Pahl E, Orsmond GS, et al. (1999) Beta-blocker treatment of dilated cardiomyopathy with congestive heart failure in children: a multi-institutional experience. J Heart Lung Transplant 18: 269-274. [Crossref]
- Rusconi P, Gómez-Marín O, Rossique-González M, Redha E, Marín JR, et al. (2004) Carvedilol in children with cardiomyopathy: 3-year experience at a single institution. J Heart Lung Transplant 23: 832-838. [Crossref]
- Oflaz MB, Balli S, Kibar AE, Ece I, Akdeniz C, et al. (2013) Effects of carvedilol therapy on cardiac autonomic control, QT dispersion, and ventricular arrhythmias in children with dilated cardiomyopathy. Med Sci Monit 19: 366-72. [Crossref]