A driver mutation is an alteration that gives a cancer cell a fundamental growth advantage for its neoplastic transformation. It differs from passenger mutations in that these do not necessarily determine the development of the cancer. Genomic instability and high mutation rates cause cancer to acquire numerous mutations and chromosomal alterations during its somatic evolution; most are termed passengers because they do not confer cancer phenotypes. Studies suggest that mildly deleterious passengers accumulate and can collectively slow cancer progression. Clinical data also suggest an association between passenger load and response to therapeutics, yet no causal link between the effects of passengers and cancer progression has been established. Although in the biology of cancer, driver mutations have been given more importance, the new evidence shows that passenger mutations are more important because they impact areas such as epigenetics, in mitochondrial DNA, immunogenicity or in the response to chemotherapy. We present an extensive review of the scientific literature on the role of passenger mutations in the evolution of cancer.
passenger, mutation, driver, cancer
Tumorigenesis is the result of the accumulation of genomic alterations and is driven by somatic evolution: alterations that occur because of defects in the regulatory circuits governing normal cell proliferation and homeostasis [1,2]. There are many kinds of cancer and tumor subtypes, in every location in the body and this complexity means that many questions about tumorigenic processes remain to be answered. For instance, many distinct regulatory circuits within each type of target cell must be disrupted for them to become cancerous [3]. However, even though carcinogenesis is very complex, genomic instability (i.e., a high frequency of genetic, epigenetic, and chromosomal alterations, collectively referred to as ‘mutations’) is a hallmark of this process [4].
Over the past decade, next generation sequencing (NGS) has allowed the integration of cancer genomics into clinical care. This has facilitated several major mass-sequencing genome projects—such as the International Cancer Genome Consortium (ICGC) and The Cancer Genome Atlas (TCGA)—for almost every key type of cancer and has identified tens of thousands of tumor mutations (including lung, kidney, and breast cancer mutations) [5]. In addition, studies conceptualizing the clonal hierarchy and phylogeny of cancer have demonstrated that intra-tumoral mutational and chromosomal heterogeneity is high [6-8]. Within this context, NGS technology has allowed the scientific community to characterize the molecular classification of cancer to define mutations as ‘drivers’ or ‘passengers’, depending on their proliferative and invasive capacity [9], also providing evidence that genomic instability is the cornerstone of cancer initiation.
Driver mutations are usually defined as mutations that induce cell proliferation and tumour growth, while passenger or ‘hitchhiker’ mutations, which represent approximately 97% of all cancerous mutations do not [10]. However, the role of passenger mutations has recently become more controversial, with some authors describing them as ‘mini drivers’ [11], also referred to as latent drivers of neutral mutations [12]. This contrasts with the prevailing hypothesis that the accumulation of passenger mutations is detrimental to cancer by slowing tumor growth and reducing metastatic progression [13,14]. In this review we discuss recent evidence in support of this latter hypothesis, the main arguments against the importance of passenger mutations, and the clinical consequences of tumor evolution based on their mutational rate. We argue that current approaches should be applied in new targeted cancer therapies.
The ability to detect genomic variations in cancer by genome, exome, and transcriptome sequencing analysis has led to the increased use of these technologies in large-scale molecular characterization projects such as the ICGC [5] and TCGA [15], facilitating the discovery of oncogenic drivers and candidate drug targets. The genomic characterization of cancer, progress in the understanding of cancer biology, and new ways of identifying its etiology of cancer, stratifying patients, managing the disease, and monitoring its responses have been especially beneficial.
Genomic instability is considered the cornerstone of the molecular classification of cancer; the acquisition of higher mutational rates caused by inducing genomic instability leads to the accelerated accumulation of ‘adaptive drivers’. In turn, this causes an increased passenger load that can cancel out the effects of these drivers, thus modulating tumorigenesis and tumor progression [16]. Chromosomal instability, the occurrence of a high rate of chromosome structural alterations in tumor cells, is the most common type of genomic instability. Another form is characterized by an increased nucleotide mutation rate; microsatellite instability is a special case of this type of genomic instability and is characterized by the expansion or contraction of the oligonucleotide repetitions present in microsatellite sequences [17,18].
Current molecular cancer classifications divides detected mutations into driver and passenger mutations. Within this paradigm, driver mutations confer a growth advantage to cancer cells and are positively selected for in the cancer-tissue microenvironment and are therefore causally involved in oncogenesis. Conversely, passenger mutations do not confer the advantage of clonal growth and therefore, do not contribute to the development of cancer. A driver mutation is not required for the maintenance of a cancer, but must have been present at some point during the cancer’s evolution. Passenger mutations are present in cancer genomes because they often occur during somatic cell division and have no functional consequences. Therefore, any cell that acquires a cancerous driver mutation already contains biologically-inert somatic passenger mutations in its genome which, through clonal expansion, will be duplicated in every subsequent daughter cancer cell [19]. Genomics-driven discovery of novel driver mutations and the molecular classification of cancer have accelerated the design of rational strategies for cancer prevention, patient stratification, the development of new drugs, and treatment options in clinical settings, thereby establishing the concept of precision medicine in cancer.
Only a small fraction of the total mutations present in a tumor are thought to be driver mutations. Tumors typically contain 40-100 gene-coding alterations, including 5-15 driver mutations [20-22], some of which may be important for tumor initiation (e.g., APC in colon cancer) [23], while others could play a role in tumor growth (e.g., VEGF) or metastasis ( e.g., TWIST1) [24]. In addition, it is important to note that there is a fundamental difference between a driver gene and a driver gene mutation. A driver gene produces driver mutations but may also produce passenger mutations. For example, APC is a large driver gene, but only mutations that truncate the protein encoded in the 1,600 amino acids in its N-terminal are driver-gene mutations; missense mutations throughout the gene, as well as protein-truncating mutations in the 1,200 amino acids in its C-terminal, are normally passenger mutations [13] (Figure 1).
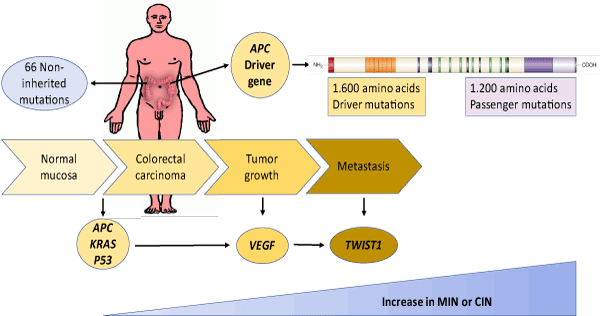
Figure 1. Mutational evolution associated with genomic instability in colorectal cancer
Because driver events are so important for cancer progression, the primary goal of cancer sequencing is usually their discovery throughout the genome [22]. Therefore, most research aims to isolate and analyse driver mutations, although for most types of cancer, these alterations in the early stages of tumorigenesis are poorly understood. Conversely, little attention has been paid to passenger mutations, which constitute the vast majority of the somatic alterations present in cancer. Evolutionary and genomic simulation studies in cancer suggest that passenger mutations accumulate and, collectively, can decrease cancer progression [13,14,16]. Clinical data also imply an association between therapeutic responses and passenger mutation loads. Furthermore, the antitumor effects of chemotherapy may be because this treatment induces genomic instability and increases the passenger mutation load.
The number of somatic passenger mutations accumulated in a tumor can provide valuable information about its evolutionary history because they can be used as a molecular clock to calculate the approximate age of the tumor lineage. In other words, the number of cell divisions that have occurred in the lineage of the dominant clone from the patient’s birth to the time of biopsy. Passengers can also become immunotherapy targets or cause treatment resistance. Likewise, their accumulation can explain several clinical phenomena such as slow progression, long latency periods, the prevalence of small subclinical cancers, spontaneous regression, and the observed range in cancer growth rates. However, these events are not easily explained without considering deleterious passengers.
In 1976, genetic instability started to be considered a mechanism of tumorigenesis, even in the presence of very few chromosomal changes (such as in acute diploid leukemia and chronic granulocytic leukemia) resulting in a ‘clonal evolution model’. In this paradigm, carcinogen-induced changes in normal progenitor cells produced diploid tumor cells with a growth advantage that allowed their initial clonal expansion [25]. Over the past four decades, this classic model of cancer evolution has focused exclusively on driver mutations to describe how these sequentially-acquired alterations provide an advantage to the growth of tumor cells. However, the integration of mathematical frameworks into the analysis of cancer genome sequencing data has resulted in new models: (a) in the ‘Big Bang model’, clonal and subclonal mutations arise early and the tumor grows as a single intermixed population; (b) in the ‘neutral model’ there is no significant difference between clonal populations; and (c) in the ‘punctuated evolution model’ clones rapidly arise between periods of relative mutational equilibrium [26] (Figure 2).
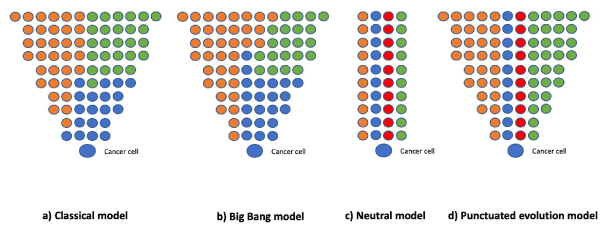
Figure 2. a) In the classical model, sequentially acquired driver mutations offer growth advantage; (b) in the Big bang model, clonal and subclonal mutations arise early, and tumor grows as a single intermixed population; (c) in neutral model, there is no major difference in fitness between the clonal populations, and (d) in the punctuated evolution model, clones arise rapidly between periods of relative mutational equilibrium
There is also a commonly used ‘two-hit model’ in which the first driver mutation produces no fitness benefit but the combination of two driver mutations can have a strong cumulative effect. Furthermore, the recently proposed evolutionary ‘stochastic model’ can explain the dynamics of cancer progression and describes how individual cells can divide and potentially acquire driver or passenger alterations, which may be involved in cell death. The size of tumors changes with the production and death of individual cells, which usually depends on the effect of accumulated drivers and passengers and the cellular environment. Thus, assuming that all driver and passenger mutations have equal fitness advantages and disadvantages, these rates are changed by the number of drivers and passengers and the total hyperplasia, or cell population size [13].
The stochastic evolutionary model of cancer progression indicates that deleterious passengers can accumulate in cancer and that these usually have a negative selection effect on tumor cells; genomic analysis shows that the passengers present in sequenced cancers have harmful phenotypes (to the cancer). Importantly, the origin of many properties of cancer may be explained if their phenotype is considered as a balance between drivers and the deleterious effect of passenger mutations—phenomena not considered in the original model. These include (a) slow initial and rapid late growth; (b) a critical cancer size for dormancy or spontaneous regression; and (c) short-term responses to mutagenic therapies.
In addition, cancers that accumulate alterations in passengers, better respond to treatments are observed. This detrimental effect on cancer is currently an untapped therapeutic target which could be leveraged by increasing either the overall mutation rate (to augment the rate of passenger mutation accumulation) or the deleterious effect of these passengers [13,14]. This evolutionary model led to the discovery of useful treatments via molecular oncology and has facilitated the proposal of new therapeutic targets for the field of precision medicine. In the following sections, we discuss how the effects of passenger mutations could be enhanced in current and future therapies.
The number of passenger mutations accumulated in each tumor lineage can provide information about its approximate age which also corresponds to the age of the tumor itself. The mean age of ovarian and lung tumor lineages is 1,113 and 749 cell divisions, respectively [26]. This is because the ovarian epithelium self-renews regularly [27] while the pulmonary epithelium renews slowly but this process is stimulated after injury [28]. Given that the cell division time for lung tumor cells is about 8 days [29], by multiplying this rate by the average lung tumor lineage age, we can say that most cancers of this type are detected 16.4 years after they first started.
Using computational methods and cell division rates for different tumor types (based on mutational signatures from genome sequencing data), approximations of tumor chronological age could offer important clues into their biology. These include information about the intrinsic and extrinsic mutagenic influences present during the period of tumor initiation and the clonal evolution of the tumor. This data may also help our understanding of intratumoral heterogeneity and differences in patient prognosis and responsiveness to treatment. Together with clinical data, this can help elucidate the time required for benign tumors to become invasive and metastatic, and help design methods for their early detection and treatment [26].
The mutator-phenotype hypothesis suggests that the mutation rate of normal cells is too slow to produce the large number of alterations found in human tumors and that the elevated mutation rate of human tumor cells increases their likelihood of acquiring advantageous mutations. The hypothesis predicts that tumors contain cells harboring hundreds of thousands of mutations rather than only a few specific driver mutations [30], and that malignant cells within a tumor therefore constitute a highly heterogeneous population [31,32]. Probabilistic mathematical models can identify mutator genes that cause point mutations or increased chromosomal alteration rates and can estimate their effect during carcinogenesis.
Data for ovarian cancer reveals that alterations in the genes or regions that result in a mutator phenotype tend to occur early. For non-mutator genes and regions, those containing MYC, KRAS, CCNE1, and RB1 tend to be altered early, while CSMD3, USH2A, and the region containing MECOM and WWOX tend to be altered late. In lung cancer, TP53 and PRKDC increase the rate of mutations; EGFR, KRAS, STK11, and TP53 tend to mutate early while LRP1B and PTPRD tends to become mutated late. However, this method of identifying altered driver genes by correlating them to passenger mutations generates many false positives because mutator genes cannot be distinguished from genes that alter later [26].
Genomic instability can be caused by dysfunction of DNA repair and cell-cycle checkpoint control genes. DNA repair genes altered in cancers include BRCA1/2, MSH2/6, MLH1/2, BLM, RAD50, MRE11, NBS1, PRKDC, NBS1, BLM, RECQL4, BAP1, WRN, RAD51L3, RAD52, FANCA, and PALB2 [18,33]. In lung and ovarian cancer, BRCA1/2, PRKDC, and PPP2R2A in the 8p21.2 region are mutator genes, and PPP2R2A plays a role in inducing chromosomal instability in ovarian cancer. Cell-cycle checkpoint pathway genes that are altered in cancers include TP53, ATM, MDM2/4, BUB1, and STK12, of which, TP53 is a mutator gene.
NGS has helped to promote knowledge of epigenetics and its standing as a field. Epigenetics is the study of transmissible chanes that does not involve DNA sequences. The three major types of epigenetic regulators are post-translational modification of histone tails, DNA methylation by covalent modification of cytosine-5’, and the regulation of microRNA gene expression [34]. In particular, DNA methylation has been extensively assessed in breast, colon, esophageal, lung, pancreatic, ovarian, prostate, and other cancers [35]. Because epigenetic changes affect genomic stability and gene expression, they influence every stage of carcinogenesis over a person’s whole life, and sometimes even across generations [36]. The challenge is now to identify methylation changes that are crucial to the processes of tumor initiation, progression, or metastasis and to distinguish these from non-carcinogenic passengers accompanying the transformation process [37].
Global DNA hypomethylation and gene-specific hypermethylation are among the prominent hallmarks of cancer genomes [38]. Some hypermethylated genes in cancer may be tumor suppressor genes, but it is unlikely that all of these numerous methylation changes play a causative role in tumorigenesis; rather, the majority of promoter CpG islands are probably methylated as a consequence of, or in association with, carcinogenesis (passenger methylation). Thus, key genes that are susceptible to methylation-associated gene silencing and that are functionally important in preventing tumorigenesis (driver methylation) must be pinpointed. This is perhaps an analogous situation to that of mutational changes in cancer: genome-wide DNA sequencing of either a large number of coding sequences or of entire cancer genomes have revealed the presence of a staggering number of mutational changes [39].
Driver methylation can be considered a methylation event which promotes tumorigenesis. If the tumor-driving or initiating event is a methylation change, it is more likely to occur during the early stages of tumorigenesis. In mouse models and in early-stage human tumor specimens and premalignant lesions, methylation changes can be observed from preneoplastic tissues up until late malignant disease [40]. Thus, early changes in methylation most probably drive the cancer phenotype, and later changes may simply reflect the transformed phenotype. Driver methylation can both directly and indirectly inactivate suppressor genes or activate oncogenes and this methylation-based gene silencing mechanism can be considered one of the hallmarks of cancer (4).
Mitochondrial DNA (mtDNA) is a small, circular, double-stranded DNA molecule approximately 16.6 Kb long that encodes 2 ribosomal RNAs (12S and 16S), 22 transfer RNAs required for protein synthesis, and 13 protein subunits that are essential for oxidative phosphorylation [41]. mtDNA is more susceptible to mutations than nuclear DNA because it lacks histones and chromatin-protective structures, has very few introns, its mtDNA repair mechanisms are inefficient, and because it is exposed to high levels of deleterious reactive oxygen species generated during ATP synthesis [42]. In 1956, Otto Warburg defined mitochondrial dysfunction as a hallmark of cancer progression; this led to the proposal of the Warburg effect—that cancer cells favor aerobic glycolic metabolism over oxidative phosphorylation. Genetic and pharmacological studies have conclusively shown that this effect is required for tumor growth, although the reason for this remains controversial [43,44]. Nonetheless, targeting damaged mtDNA could represent a promising anticancer therapy target [45].
Another characteristic cancer marker is the ability of tumor cells to reprogram their own metabolism to cope both with the abnormal protein-building requirements of their uncontrolled cell proliferation and to adapt to their everchanging microenvironments (4). Demonstration that mitochondrial metabolism can be triggered by activation of oncogenes such as BRAF and c-Myc [46,47], loss of tumor suppressors such as p53 [48], and activation of the mTORC1 pathway [49] resulted in the redefinition of the Warburg effect. Even so, it has become evident that accelerated mitochondrial function, including ATP production, is required for cell proliferation and tumor progression [50], at least in specific phases such as adaptation to nutrient and oxygen deprivation [51].
To maintain physiological energy levels, cells have developed a sensitive molecular system that integrates multiple upstream inputs and regulates enzyme activity and transcriptional responses. The core enzyme in this system is the AMP-activated protein kinase, AMPK. This enzyme restores energy levels when intracellular ATP drops, for instance, in response to mitochondrial dysfunction or stress. AMPK has been widely implicated in tumor initiation, progression, and metastasis [52]. However, genetic ablation resulting in AMPK loss is not sufficient to induce cell transformation in vitro or in vivo models [53]. To further complicate this scenario, AMPK is differentially expressed and activated in different cancer types and disease stages and is associated with varying outcomes and prognoses [51].
It is commonly believed that mtDNA variants arise due to positive selection of those “driver” variants conferring clonal growth advantage. Accordingly, we observed that likely non-pathogenic mtDNA variants (“passengers”) reverted to the wild-type homoplasmic status dur- ing tumor progression in colorectal cancer patients [54]. On the contrary, the mtDNA variants that are positively selected during the tumor progression might be considered the most tolerable alterations for neoplastic cells. However, a deleterious impact of mtDNA passenger variants on cancer progression may not be completely excluded, as it has previously been evidenced in nuclear DNA passenger alterations [55].
The association between immunotherapy response the passenger mutation load has been widely studied. Furthermore, numerous studies have estimated the mutational load in different tumor types: melanoma, lung squamous carcinoma, lung adenocarcinoma, and bladder cancer are usually associated with a higher mutational load, while pilocytic astrocytoma, acute lymphoblastic leukemia, medulloblastoma, and acute myeloid leukemia are commonly associated with lower mutational loads [56]. In addition to the overall frequency of mutations, there is also a different spectrum of mutations in each tumor type. For instance, C→A mutations related to exposure to polycyclic aromatic hydrocarbons in tobacco smoke are associated with lung cancers [57] melanomas often show C→T mutations caused by the misrepair of ultraviolet-induced DNA breaks [58] gastrointestinal tumors, show high frequencies of CpG dinucleotide transition mutations, which may reflect higher methylation levels in these tumor types [59] and finally, tumors with a mutator phenotype caused by microsatellite instability bear a mutational load that far exceeds even that of melanomas [60,61].
Another hallmark of cancer is genomic instability, which causes many chromosomal disorders and cellular lineage mutations [62] and both driver and passenger mutations. Passengers account for an estimated 97% of tumor cell mutations (11); they have always been presumed to be neutral and have largely been ignored in cancer research. Passengers are potential biomarkers for patient responses to mutagenic therapies, and increasing evidence now suggests that they might be deleterious to cancer cells, making them important both in clinical and cancer progression outcomes. Because of the limitations in whole-genome analyses, the properties of passenger mutations remain unclear although varying arguments suggest that they are ‘mini-drivers’ (12), ‘latent drivers’ [63], neutral (13), or potentially deleterious to cancer (14)(15).
The ‘tug-of-war’ resulting both from the cumulative effect and presence of high numbers of passengers and drivers in tumor cells may explain some paradoxical cancer treatment outcomes. Furthermore, their accumulation can even cause tumor extinction by mutational meltdown [64], although this is not yet fully understood. Some hypotheses that can explain better prognoses resulting from the accumulation of passengers include: (a) increased tumor immunogenicity [65]; (b) the correlation of high genomic instability with improved clinical outcomes [66]; and (c) reduced cell proliferation [67]. More research is still required, but preliminary studies indicate that clinical phenomena such as long periods of dormancy, slow progression, growth-rate heterogeneity, spontaneous regression, and the prevalence of small subclinical cancers, could be the result of high deleterious passenger accumulation altering the dynamics of cancer progression. For example, budding yeast [68], primary mouse cells (63), and human aneuploid cells [69] with high passenger burdens all show evidence of a proliferative tumor cell growth disadvantage.
Another indirect example is Lynch syndrome which results from a germline mutation in the MLH1, MSH2, MSH6, or PMS2 DNA mismatch repair (MMR) genes or in EPCAM. The DNA MMR system maintains genomic integrity by correcting base substitutions and small insertion-deletion mismatches generated by base-pairing errors during DNA replication. Inactivation of both alleles of an MMR gene leads to defective MMR and may result in high mutability and target-gene inactivation. These mutations better survive in Lynch syndrome patients than in patients without MMR defects [70]. Simultaneously, many studies suggest that single-agent adjuvant fluoropyrimidine-based chemotherapy is less beneficial, or is even potentially harmful, to patients with microsatellite instable or MMR tumors [71,72]. However, the prognostic influence of microsatellite instable is less clear in patients with metastatic colorectal cancer , a population in which the prevalence of MSI-H disease is low (approximately 3.5 percent) . One hypothesis that can explain it, is the adverse influence of the higher frequency of BRAF mutations in this population [73].
Tumor cell growth and survival depends on several mechanisms, including angiogenesis and immune-system avoidance; limitless replication potential is a major factor because constant biosynthesis requires continued genetic material. Thus, classical chemotherapy and radiotherapy aims to impede tumor growth by damaging DNA [74] and this may also allow the accumulation of mutations that can become neoantigens—new targets for immune-system detection. In support of this hypothesis, new data suggests that targeted tumor irradiation combined with dual CTLA-4/PD-1 blockade in melanoma is a promising treatment option. Similarly, cisplatin and checkpoint blockade has proven a successful first-line treatment for non-small cell lung cancer (NSCLC). Another strategy is the development of drug combinations such as olaparib with immunotherapy [75]. Colorectal cancer patients with germline loss-of-function mutations in DNA mismatch repair genes have a 4 to 7-fold greater response rate to pembrolizumab than patients without them. Furthermore, DNA damage response (DDR)-deficient tumors harbor 10 to 100 times more somatic mutations than DDR-proficient patient tumors.
Nitrogen mustard, an analogue of the sulfur mustard gas used as a weapon during the First World War, was introduced in 1942 as the first clinically useful alkylating agent [76] its discovery was the first step towards cancer chemotherapy (17). As a general rule, the cell cycle is always affected by drugs (such as chemotherapy agents) that interact with DNA, although final outcomes depend on the extent of the interaction and the speed at which DNA reparation can overcome any negative effects. Indeed, one of the mechanisms of tumor cell resistance to alkylating and platinum agents is attributed to enhanced DNA cross-link repair. Therefore a new interpretation for the effectiveness of traditional genotoxic chemotherapy is that, aside from directly inhibiting tumor cell growth, these agents increase passenger load which may at least temporarily decrease the malignant potential of cancer development [77].
The accumulation of genetic alterations in cancer cells during tumorigenesis results in tumor neoantigens; these are increasingly considered to be immuno-determinants and in the context of immunotherapy treatment, there is abundant proof that they are related to early tumor recognition and destruction by antigen-specific T cells [78,79]. The expression of neoantigens in cancer cells highlights the ‘foreignness’ of cancer within the human body. Specifically, mutational load (a surrogate marker for tumor neoantigen load) correlates with the expected result of experiments to block T-cell checkpoint inhibitors in melanoma and NSCLCs [80].
Two classes of antigens provide cancer-rejection epitopes: the first are created by nonmutated proteins that are not completely tolerated by T-cells (partly because of their restricted tissue expression patterns). The second type—neoantigens—are created by proteins that are missing from normal human genome, usually in tumors without a viral etiology in which tumor-specific DNA alterations cause the formation of new protein sequences. In virus-associated tumors such as cervical cancer and a subgroup of head and neck cancers, antigens derived from viruses also add to the pool of neoantigens [81]. Compared with non-mutated self-antigens, neoantigens may be especially important in tumor control because the quality of the T cell pool available for these antigens is not affected by central T-cell tolerance [82].
Deep-sequencing technologies make it relatively easy to identify mutations present that are potential neoantigens and they can be confidently predicted [83]. Two studies in mouse models provided proof that this approach can identify neoantigens recognizable by T cells [84,85]. To leverage this phenomena, neoantigens would ideally be derived from essential oncogenes common to most cancers, thus reducing the probability of their escape from the immune system. For instance, in MHC class I- and class II-restricted neoantigens [86] in validated oncogenes shared between patient subgroups [87] which are known to produce T-cell responses. Cancers with substantial exogenous mutagenic exposure, such as ultraviolet light in the case of melanoma or exposure to tobacco smoke carcinogens in lung cancers, have very high mutation rates. A single case report of a melanoma tumor found 187 non-synonymous mutations [57] and an average of 201 mutations across 14 other melanoma cases [88], and another study identified 94 non-synonymous mutations in a lung cancer cell line [89] and 4,300 mutations in one primary tumor (78). In addition, tumors with mismatch repair deficiencies also carry large numbers of mutations [90].
Most of these mutations are ‘neutral passengers’, implying that T-cell reactivity towards neoantigens is usually directed against mutated gene products dispensable for tumor growth. Indeed, selective attack by the immune system may cause the loss of mutated genes; in line with this, intriguing work by Schreiber et al. in a murine model demonstrated the loss of expression of a passenger mutation after T-cell exposure (47,75). It is unknown whether T-cell pressure during human cancer development is sufficient to lead to a similar immune selection, and so this important question should be addressed in future research. There are also ‘essential passenger’ mutations which occur in essential (housekeeping) genes in cases where the wild-type copy is lost, then T-cell reactivity against the neoepitope can only lead to immune escape by mutation reversal. Coulie et al. were the first to describe an essential passenger, by identifying a mutant malate dehydrogenase enzyme epitope recognized by autologous T cells [91].
The formation of any one neoantigen by a given mutation is a probabilistic ‘lottery’. This means that although tumor foreignness can likely be guaranteed for tumors with very high mutational loads, the odds of tumor foreignness can only likely be inferred for tumors with an intermediate or low mutational load. Nevertheless, mutational load represents an imperfect biomarker, even specific cases where neoantigen reactivity is the only tumor-specific T cell reactivity relevant to tumor control. Additionally, the success of the immune system attacks on cancer cells depends on several factors, including the formation of tumor-specific antigens. This concept was well described by the myriad of inhibitory and stimulatory factors involved in the cancer-immunity cycle introduced by Chen and Mellman [92].
Traditionally, cancer research has paid less attention to passengers, even though these represent the overwhelming majority of mutations. However, these may now take the spotlight in early diagnosis and in the improvement of future treatments because they appear to slow tumor growth and reduce metastatic progression. Cancers with the highest burden of chromosomal alterations have the best prognoses which is thought to be because passenger mutations interfere with the acquisition of new drivers and thus, reduce genetic diversity. This information will be very useful for devising new anti-cancer therapies, and therefore, we argue that current approaches in targeted cancer therapy should be carefully reconsidered. For instance, the induction of genomic instability and increased passenger-mutation loads is now considered alternative therapeutic possibilities for cancer treatment in immunotherapy. However a greater understanding of the complexity of tumors with mutations, both driver and passenger mutations, can help us to better manage the treatment of patients with cancer, increasing their survival. The introduction of the Next Generation Sequencing is allowing this greater integration of all players present in the tumor.
Aparisi F and Amado-Labrador H are the main authors of the study in the search for information and development of the review. Calabuig S, Jantus-Lewintre E, Blasco A, Iranzo V, Herreros-Pomares A and Camps C have contributed in the process to obtain articles, revision of the manuscript and the supervision of the process.
This work was supported by Fondo de Investigación Sanitaria, Instituto de Salud Carlos III and Fundación Arnal Planelles. Hector Amado is supported by a Fundación Carolina/ BBVA grant.
- Loeb LA, Springgate CF, Battula N (1974) Errors in DNA replication as a basis of malignant changes. Cancer Res 34: 2311-2321. [Crossref]
- Pepper JW, Findlay CS, Kassen R, Spencer SL, Maley CC (2009) Cancer research meets evolutionary biology. Evol Appl 2: 62-70. [Crossref]
- Hanahan D, Weinberg RA (2000) The Hallmarks of cancer review evolve progressively from normalcy via a series of pre. Cell 100: 57-70.
- Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144: 646-674. [Crossref]
- Hudson TJ, Anderson W, Aretz A, Barker AD, Bell C, et al. (2010) International network of cancer genome projects. Nature 464: 993-998. [Crossref]
- Jamal-Hanjani M, Hackshaw A, Ngai Y, Shaw J, Dive C, et al. (2014) Tracking genomic cancer evolution for precision medicine: the lung TRACERx study. PLoS Biol 12: e1001906. [Crossref]
- Gerlinger M, Rowan AJ, Sc B, Horswell S, Math M (2016) Europe PMC funders group intratumor heterogeneity and branched evolution revealed by multiregion sequencing. 366: 883-892.
- Shah Sohrab P, Roth Andrew, Goya Rodrigo, Oloumi Arusha HG, Zhao Y, et al. (2013) NIH Public Access. Nature 486: 1-13.
- Bozic I, Antal T, Ohtsuki H, Carter H, Kim D, et al. (2010) Accumulation of driver and passenger mutations during tumor progression. Proc Natl Acad Sci U S A 107: 18545-18550. [Crossref]
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, et al. (2013) Cancer genome landscapes. Science 339: 1546-1558. [Crossref]
- Castro-Giner F, Ratcliffe P, Tomlinson I (2015) The mini-driver model of polygenic cancer evolution. Nat Rev Cancer 15: 680-685. [Crossref]
- Bozic I, Gerold JM, Nowak MA (2016) Quantifying clonal and subclonal passenger mutations in cancer evolution. PLoS Comput Biol 12: 1-19.
- McFarland CD, Korolev KS, Kryukov GV, Sunyaev SR, Mirny LA (2013) Impact of deleterious passenger mutations on cancer progression. Proc Natl Acad Sci U S A 110: 2910-2915. [Crossref]
- McFarland CD, Mirny LA, Korolev KS (2014) Tug-of-war between driver and passenger mutations in cancer and other adaptive processes. Proc Natl Acad Sci 111: 15138-15143.
- Cancer Genome Atlas Research Network JN, Weinstein JN, Collisson EA, Mills GB, Shaw KRM, et al. (2013) The cancer genome atlas pan-cancer analysis project. Nat Genet 45: 1113-1120.
- McFarland CD, Yaglom JA, Wojtkowiak JW, Scott JG, Morse DL, et al. (2017) The damaging effect of passenger mutations on cancer progression. Cancer Res 77: 4763-4772. [Crossref]
- Yao Y1, Dai W2 (2014) Genomic instability and cancer. J Carcinog Mutagen 5. [Crossref]
- Lengauer C, Kinzler KW, Vogelstein B (1998) Genetic instabilities in human cancers. Nature 396: 643-649. [Crossref]
- Stratton MR, Campbell PJ, Futreal PA (2009) The cancer genome. Nature 458: 719-724. [Crossref]
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, et al. (2014) Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505: 495-501. [Crossref]
- Parsons DW, Jones S, Zhang X, Lin JC-H, Leary RJ, et al. (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321: 1807-1812.
- Futreal PA, Coin L, Marshall M, Down T, Hubbard T, et al. (2004) A census of human cancer genes. Nat Rev Cancer 4: 177-183. [Crossref]
- Li Q, Ishikawa TO, Oshima M, Taketo MM (2005) The threshold level of adenomatous polyposis coli protein for mouse intestinal tumorigenesis. Cancer Res 65: 8622-8627.
- Benjamin LE, Keshet E (1997) Conditional switching of vascular endothelial growth factor (VEGF) expression in tumors: induction of endothelial cell shedding and regression of hemangioblastoma-like vessels by VEGF withdrawal. Proc Natl Acad Sci U S A 94: 8761-8766.
- Nowell PC (1976) The clonal evolution of tumor cell populations. Science 194: 23-28. [Crossref]
- De S, Ganesan S (2017) Looking beyond drivers and passengers in cancer genome sequencing data. Ann Oncol 28: 938-945. [Crossref]
- Tomasetti C, Vogelstein B, Parmigiani G (2013) Half or more of the somatic mutations in cancers of self-renewing tissues originate prior to tumor initiation. Proc Natl Acad Sci U S A 110: 1999-2004. [Crossref]
- Stripp BR, Reynolds SD (2008) Maintenance and repair of the bronchiolar epithelium. Proc Am Thorac Soc 5: 328-333. [Crossref]
- Tinnemans MM, Schutte B, Lenders MH, Ten Velde GP, Ramaekers FC, et al. (1993) Cytokinetic analysis of lung cancer by in vivo bromodeoxyuridine labelling. Br J Cancer 67: 1217-1222. [Crossref]
- Kinzler KW, Vogelstein B (1996) Lessons from hereditary colorectal cancer. Cell 87: 159-170. [Crossref]
- Salk JJ, Fox EJ, Loeb LA (2012) NIH Public Access. 51-75.
- Fox EJ, Salk JJ, Loeb LA (2009) Cancer genome sequencing - An interim analysis. Cancer Res 69: 4948-4950. [Crossref]
- Loeb LA (2001) A mutator phenotype in cancer. Cancer Res 61: 3230-3239. [Crossref]
- Callinan PA, Feinberg AP (2006) The emerging science of epigenomics. Hum Mol Genet 15 Spec No 1: R95-R101. [Crossref]
- Verma M, Srivastava S (2002) Epigenetics in cancer: Implications for early detection and prevention. Lancet Oncol 3: 755-763.
- Brasset E, Chambeyron S (2013) Epigenetics and transgenerational inheritance. Genome Biol 14: 306. [Crossref]
- Takeshima H, Ushijima T (2019) Accumulation of genetic and epigenetic alterations in normal cells and cancer risk. NPJ Precis Oncol 3: 7. [Crossref]
- Ushijima T (2005) Detection and interpretation of altered methylation patterns in cancer cells. Nat Rev Cancer 5: 223-231.
- Pfeifer GP, Besaratinia A (2009) Mutational spectra of human cancer. Hum Genet 125: 493-506. [Crossref]
- Chen SS, Raval A, Johnson AJ, Hertlein E, Liu T-H, et al. (2009) Epigenetic changes during disease progression in a murine model of human chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 106: 13433-13438. [Crossref]
- Taylor RW, Turnbull DM (2005) Mitochondrial DNA mutations in human disease. Nat Rev Genet 6: 389-402. [Crossref]
- Hanes JW, Thal DM, Johnson KA (2006) Incorporation and replication of 8-oxo-deoxyguanosine by the human mitochondrial DNA polymerase. J Biol Chem 281: 36241-36248. [Crossref]
- Fantin VR, St-Pierre J, Leder P (2006) Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 9: 425-434.
- Shim H, Chun YS, Lewis BC, Dang C V (1998) A unique glucose-dependent apoptotic pathway induced by c-Myc. Proc Natl Acad Sci 95: 1511-1516.
- Warburg O (1956) On the origin of cancer cells. Science 123: 309-314. [Crossref]
- Kang HB, Fan J, Lin R, Elf S, Ji Q, et al. (2015) Metabolic rewiring by oncogenic BRAF V600E links ketogenesis pathway to BRAF-MEK1 signaling. Mol Cell 59: 345-358. [Crossref]
- Fan Y, Dickman KG, Zong WX (2010) Akt and c-Myc differentially activate cellular metabolic programs and prime cells to bioenergetic inhibition. J Biol Chem 285: 7324-7333.
- Ma W, Sung HJ, Park JY, Matoba S, Hwang PM (2007) A pivotal role for p53: Balancing aerobic respiration and glycolysis. J Bioenerg Biomembr 39: 243-246.
- Sun Q, Chen X, Ma J, Peng H, Wang F, et al. (2011) Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc Natl Acad Sci 108: 4129-4134.
- Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, et al. (2010) Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci 107: 8788-8793.
- Smolkova K, Plecita-Hlavata L, Bellance N, Benard G, Rossignol R, et al. (2011) Waves of gene regulation suppress and then restore oxidative phosphorylation in cancer cells. Int J Biochem Cell Biol 43: 950-968.
- Hardie DG (2015) Molecular pathways: Is AMPK a friend or a foe in cancer? Clin Cancer Res 21: 3836-3840. [Crossref]
- Laderoute K (2006) 5’-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol Cell Biol 26: 5336-5347.
- Errichiello E, Balsamo A, Cerni M, Venesio T (2015) Mitochondrial variants in MT-CO2 and D-loop instability are involved in MUTYH-associated polyposis. J Mol Med 93: 1271-1281.
- Errichiello E, Venesio T (2017) Mitochondrial DNA variants in colorectal carcinogenesis: Drivers or passengers? J Cancer Res Clin Oncol 143: 1905-1914. [Crossref]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, et al. (2013) Signatures of mutational processes in human cancer. Nature 500: 415-424.
- Pleasance ED, Stephens PJ, O’Meara S, McBride DJ, Meynert A, et al. (2010) A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature 463: 184-190.
- Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, et al. (2010) A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 463: 191-196.
- Cancer T, Atlas G (2012) Comprehensive molecular characterization of human colon and rectal cancer. Nature 487: 330-337.
- Dung T. Le , Jennifer N. Uram , Hao Wang , Bjarne Bartlett , Holly Kemberling (2015) PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med 372: 2509-2520.
- Adjiri A (2017) DNA mutations may not be the cause of cancer. Oncol Ther 5: 85-101. [Crossref]
- Burrell RA, McGranahan N, Bartek J, Swanton C (2013) The causes and consequences of genetic heterogeneity in cancer evolution. Nature 501: 338-345.
- Nussinov R, Tsai CJ2 (2015) 'Latent drivers' expand the cancer mutational landscape. Curr Opin Struct Biol 32: 25-32. [Crossref]
- Neher RA, Shraiman BI (2012) Fluctuations of fitness distributions and the rate of Muller's ratchet. Genetics 191: 1283-1293. [Crossref]
- Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N (2015) Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160: 48-61. [Crossref]
- Birkbak NJ, Eklund AC, Li Q, McClelland SE, Endesfelder D, et al. (2011) Paradoxical relationship between chromosomal instability and survival outcome in cancer. Cancer Res 71: 3447-3452.
- Williams BR, Prabhu VR, Hunter KE, Glazier CM, Whittaker CA, et al. (2008) Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science 322: 703-709.
- Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, et al. (2007) Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 317: 916-924. [Crossref]
- Segal DJ, McCoy EE (1974) Studies on down's syndrome in tissue culture. I. Growth rates and protein contents of fibroblast cultures. J Cell Physiol 83: 85-90. [Crossref]
- Phipps AI, Limburg PJ, Baron JA, Burnett-Hartman AN, Weisenberger DJ, et al. (2015) Association between molecular subtypes of colorectal cancer and patient survival. Gastroenterology 148: 77-87. [Crossref]
- Elsaleh H, Joseph D, Grieu F, Zeps N, Spry N, et al. (2000) Association of tumour site and sex with survival benefit from adjuvant chemotherapy in colorectal cancer. Lancet 355: 1745-1750.
- Hutchins G, Southward K, Handley K, Magill L, Beaumont C, et al. (2011) Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J Clin Oncol 29: 1261-1270.
- Seymour MT, Richman SD, Cheadle JP, Ylstra B, Nagtegaal ID, et al. (2014) Mismatch repair status and braf mutation status in metastatic colorectal cancer patients: a pooled analysis of the CAIRO, CAIRO2, COIN, and FOCUS studies. Clin Cancer Res 20: 5322-5330.
- Kim D, Li R, Dudek SM, Wallace JR, Ritchie MD (2015) Binning somatic mutations based on biological knowledge for predicting survival: an application in renal cell carcinoma. Pacific Symp Biocomput 0: 96-107.
- Gilman A, Philips FS (1946) The biological actions and therapeutic applications of beta-chloroethyl amines and sulfides. Science 103: 0-415.
- Lowenstein EB (2011) A history of sulfur mustard. Skinmed 9: 310-311. [Crossref]
- [No authors listed] (2016) Cancer Mutational Load: The more the merrier for immune checkpoint blockade therapies. EBioMedicine 13: 1-2. [Crossref]
- Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, et al. (2014) Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344: 641-645. [Crossref]
- Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, et al. (2014) Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 371: 2189-2199. [Crossref]
- Blank CU, Haanen JB, Ribas A, Schumacher TN (2016) CANCER IMMUNOLOGY. The "cancer immunogram". Science 352: 658-660. [Crossref]
- Schumacher TN, Schreiber RD (2015) Neoantigens in cancer immunotherapy. Science 348: 69-74. [Crossref]
- Gilboa E (1999) The makings of a tumor rejection antigen. Immunity 11: 263-270. [Crossref]
- Segal NH, Parsons DW, Peggs KS, Velculescu V, Kinzler KW, et al. (2008) Epitope landscape in breast and colorectal cancer. Cancer Res 68: 889-892.
- Castle JC, Kreiter S, Diekmann J, Löwer M, van de Roemer N, et al. (2012) Exploiting the mutanome for tumor vaccination. Cancer Res 72: 1081-1091. [Crossref]
- Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, et al. (2013) Cancer exome analysis reveals a t cell dependent mechanism of cancer immunoediting. Nature 482: 400-404. [Crossref]
- Wölfel T, Hauer M, Schneider J, Serrano M, Wölfel C, et al. (1995) A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science 269: 1281-1284.
- Schumacher T, Bunse L, Pusch S, Sahm F, Wiestler B, et al. (2014) A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 512: 324-327.
- Lee W, Jiang Z, Liu J, Haverty PM, Guan Y, et al. (2010) The mutation spectrum revealed by paired genome sequences from a lung cancer patient. Nature 465: 473-477.
- Xiaomu Wei, Vijay Walia, Jimmy C Lin, Jamie K Teer, Todd D Prickett, et al. (2011) Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nat Genet 43: 442-446.
- Greenman C, Stephens PJ, Smith R, Dalgliesh GL, Hunter C, et al. (2007) UKPMC funders group patterns of somatic mutation in human cancer genomes. Nature 446: 153-158.
- Karanikas V, Colau D, Baurain JF, Chiari R, Thonnard J, et al. (2001) High frequency of cytolytic T lymphocytes directed against a tumor-specific mutated antigen detectable with HLA tetramers in the blood of a lung carcinoma patient with long survival. Cancer Res 61: 3718-3724.
- Chen DS, Mellman I (2013) Oncology meets immunology: the cancer-immunity cycle. Immunity 39: 1-10. [Crossref]