Oxidative Stress (OS) is a condition in which production of reactive oxidative species (ROS) exceeds the capacity of the antioxidant system. In presence of an oxidative stress, the uncontained ROS cause tissue damage/dysfunction. Oxidative stress and chronic inflammation are constant feature of chronic kidney diseases (CKD). Together oxidative stress and inflammation contribute to development and progression of CKD and the associated complications. That is why it is important for the nephrologists and clinical physicians to know the mechanisms and consequences of OS in CKD.
oxidative stress, reactive oxidative species, chronic kidney diseases, inflammation, antioxidant system
In presence of oxidative stress, the reactive oxidative species (ROS) cause tissue damage and dysfunction by:
- Directly attacking, denaturing and modifying structural and functional molecules (e.g. lipids, proteins, carbohydrates, DNA, RNA, NO, etc.)
- Modulating activities of redox-sensitive transcription factors (e.g. NFκB, AP-1) and signal transduction pathways (activation of protein kinases e.g. ERK, Ca- AT-Phosphatase release channels), thereby promoting inflammation, ER stress, fibrosis, apoptosis.
Oxidative stress in chronic kidney disease is both a cause and a consequence of inflammation. They both contribute to development and progression of CKD and the associated complications, including atherosclerosis, cardiovascular disease, erythropoietin-resistant anaemia, immune deficiency, cachexia etc. [1-4].
Mechanisms of oxidative stress in CKD are as follows:
A - Increased production of ROS
B - Impaired antioxidant defense system [1,4]
The main factors, contributing to increased ROS production and dissemination of oxidative stress are: activation of tissue angiotensin system, hypertension, inflammation, uremic toxins (endogenous; exogenous), mitochondrial dysfunction, accumulation of oxidation-prone lipoprotein remnants, underlying conditions (e.g. diabetes, autoimmune diseases), increased tissue iron load (Fe shift, blood transfusion, excess intravenous Fe use), iatrogenic causes (blood/dialyzer interaction, dialysate impurities, excessive use of intravenous Fe, rejected transplant kidney, reaction to failed arteriovenous grafts) [1,4-6].
A)Mechanisms of excess ROS production in CKD
The mechanisms of excess ROS production are well described [1,2,5,7,8], namely:
- Up-regulation/activation of ROS-producing enzymes (e.g. nicotine amid dinucleotide phosphate - NAD(P)H oxidase, cyclooxygenase, lipoxygenase, etc)*.
- Uncoupling of NO synthase (via NOS monomerization, depletion of tetrahydro-biopterin [BH4], accumulation of asymmetric dimethylamine -ADMA)
- Impairment of mitochondrial electron transport chain
- Activation of leukocytes and resident cells
- Dissemination of oxidative stress by circulating oxidized low density lipoproteins (LDL) and phospholipids via oxidation chain reaction
*NAD(P)H Oxidase is the major source of ROS production in endothelial cells vascular smooth muscle cells and renal parenchymal cells. NAD(P)H oxidase activation involves assembly of enzyme’s membrane-associated subunits with cytosolic subunits [1,3,9] (Figure 1).
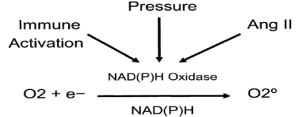
Figure 1. Activation of NAD(P)H Oxidase
B) Impaired antioxidant defense system in CKD.
The factors, contributing to the antioxidant depletion are widely investigated [1,5,6,10] and here they are shown in the next order:
- Reduced Production of endogenous antioxidants (antioxidant enzymes, glutathione sulphate (GSH), ApoA1, Albumin, lecithin-cholesterol acyltransferase (LCAT), Melatonin, etc.)
- Impaired activation of the regulator of genes encoding antioxidant/detoxification molecules
- Depletion of antioxidant molecules by ROS
- Diminished antioxidant activity of high density lipoproteins (HDL)
- Reduced intake of fresh fruits and vegetables (because of potassium restriction, causing vitamins deficit)
- Removal of water-soluble antioxidants by dialysis
- Anemia – decreased red blood cells (RBC) antioxidants: glutathione sulphate (GSH), glutathione peroxidase (GPX), platelet activating factor – acetyl-hydrolase (PAF-AH), Phospholipids)
Free radicals have a very short half-life (in seconds) and their evaluation in clinical practice is difficult. But there are more stable marker molecules with longer half-lives, ranging from hours to weeks, which can be used to assess oxidative stress associated with different diseases, including uremia. A lot of them are molecules- byproducts of ROS interaction with bio-molecules [11-14]. The major biomarkers of the oxidative stress are presented in the next groups:
- Elevated plasma and tissue Malone dialdehyde (MDA)
- Elevated plasma, urine and tissue F2 isoprostane
- Elevated plasma and tissue nitrotyrosine (NO oxidation)
- Increased Protein carbonyls and oxidized thiols
- Increased plasma and urine oxidized nucleic acids
- Elevated plasma and tissue advanced glycoxidation end products (AGE)
Under normal condition, disruption of redox equilibrium by environmental or internal pro-oxidants triggers an adaptive response which results in up-regulation of antioxidant and cyto-protective enzymes and proteins. In mammals, nuclear factor-erythroid 2 p45-related factors 1 and 2 (Nrf2) regulates constitutive expression and orchestrates transcriptional up-regulation of genes encoding these cyto-protective molecules. Nuclear factor-erythroid-2-related factor 2 (Nrf2) is held in the cytoplasm as an inactive complex bound to Keap1 (Kelch-like ECH-associated protein 1), a repressor molecule that facilitates Nrf2 ubiquitination. Keap1 contains several reactive cysteine residues that serve as sensors of the intracellular redox state [7,8,10,14,15]. Oxidative or covalent modification of thiols in some of these cysteine residues leads to conformational changes in Keap1 that result in disruption of one of the two Keap1 interactions with Nrf2 (“hinge and latch” model). By limiting proteasomal degradation of Nrf2 this process results in accumulation of the de novo synthesized Nrf2 and its translocation to the nucleus. Within the nucleus, Nrf2 binds to regulatory sequences (antioxidant response elements) or electrophile response elements, in the promoter regions of genes encoding antioxidant and phase 2 detoxifying molecules. This process entails hetero-dimerization of Nrf2 with other transcription factors within the nucleus. In addition to modification of Keap1, nuclear translocation of Nrf2 may occur via phosphorylation of its threonine or serine residues by upstream kinases, such as protein kinase C, mitogen-activated protein kinases, phosphatidylinositol-3-kinase/Akt, casein kinase-2, and the protein kinase RNA-like endoplasmic reticulum (PERK) [9,10,12,15]. Regulation of cellular antioxidant and anti-inflammatory machinery by Nrf2 plays a central role in defense against oxidative stress. There is strong evidence, supporting the protective role of Nrf2-mediated pathway against oxidative stress and inflammation. For example, the response to activation of nuclear factor B (NF-B) and consequent induction of COX-2, acting on nitric oxide synthase, interleukin-6 (IL-6), and tumor necrosis factor alfa (TNF-alfa) is more intense in Nrf2 knockout mice compared with the wild-type mice [9,10,12]. Hyperglycemia results in more severe renal injury in Nrf2-deficient than in the wild-type mice with induced diabetes [8,12,15]. On the other hand, certain anti-inflammatory and antioxidant activators of Nrf2 signaling, can enhance cellular defense against oxidative and electrophilic insults [16,17]. GSH is the most abundant cellular endogenous antioxidant and is a major factor in the regulation of the cellular redox state. GSH maintains cellular redox systems and affects redox signaling, cell proliferation, and cell death [1,14]. GSH directly is scavenging ROS and serving as the substrate in a number of antioxidant enzymes such as glutathione peroxidase. Selective inhibition of the enzymes of the glutathione redox cycle heightens the susceptibility to ROS-mediated cell injury [15,17,18] and many pathological conditions including atherosclerosis, diabetes, liver disease, myocardial infarction, and stroke are associated with elevated glutamate-cysteine ligase (GCLC and GCLM) and diminished GSH levels [1,3,5,14] . Expression of GCLC and GCLM is regulated by Nrf2. The CRF rats in the experimental studies showed significant elevation of lipid peroxidation products and marked reductions of GCLC and GCLM, and the GSH-to-GSSG ratio in the renal tissue [9,10,15]. Accordingly, impaired Nrf2 activation shows contribution to the severity of oxidative stress and inflammation and the progression of tissue damage in the remnant kidney in CRF.
Anti-oxidant and anti-atherogenic actions of HDL in CKD are well known [8,11,14] and are shown in the next scheme:
A- HDL acts as reverse cholesterol on lipid transport
B- HDL influences endothelial cells migration and endothelial repair (estimated via scavenger receptor B-1 (SRB-1)
C- HDL exhibits antioxidant and anti-inflammatory actions
a. HDL has an ApoA-I mediated extraction of oxidized phospholipids from lipoproteins and cell membrane.
b. HDL forces the lecithin-cholesterol acyltransferase (LCAT) - mediated hydrolysis of pro-inflammatory oxidized phospholipids (AA at sn-2).
c. HDL prevents LDL oxidation and destruction of oxidized phospholipids by paraoxonase-1 and glutathione peroxidase (GPX) (Studies show that in CKD the LDL is more susceptible to “in vitro” oxidation than in the healthy).
D- HDL inactivates PAF and PAF-like phospholipids by PAF acetyl hydrolase (anti-inflammatory and anti-thrombotic).
ROS production is markedly increased in the diseased kidney, accompanied by impaired Nrf2 (Erythroid-derived Nuclear factor2) activation and consequent down-regulation of the antioxidant and cyto-protective molecules. All usual therapies effective in retarding CKD progression (i.e. RAS blockade, Glycemia and HTN control) reduce oxidative stress and inflammation [1,3,5,14,16].
Therapies against OS, some of them currently in clinical trial, are as follow:
- The Nrf2 (Erythroid-derived Nuclear factor2) activator Bardoxolone, which can lower oxidative stress and inflammation by raising expression of endogenous antioxidant enzymes and related molecules.
- AST-120, a specially formulated activated charcoal which limits absorption of the pro-oxidant gut–derived uremic toxins.
- Some trace elements as selenium (Se), zinc (Zn), cuprum (Cu) are co-factors in a number of antioxidant enzymes and their deficit requires adequate supplementation, helping to overcome OS development. High doses of anti-oxidant vitamins are not accepted for treatment of OS without controversy17,18, and some authors suggest they may increase the risk of cardiovascular diseases and to provoke other serious complications.
Identification of biochemical and/or functional biomarkers that could be used in clinical practice to monitor oxidative imbalance in CKD may allow the development of an optimal interventional strategy to reduce oxidative stress in CKD.
- Vaziri ND, Yuan J, Rahimi A, Ni Z, Said H, et al. (2012) Disintegration of colonic epithelial tight junction in uremia: a likely cause of CKD-associated inflammation. Nephrol Dial Transplant 27: 2686-2693. [Crossref]
- Duni A, Liakopoulos V, Rapsomanikis K-P, Dounousi E (2017) Chronic Kidney Disease and Disproportionally Increased Cardiovascular Damage: Does Oxidative Stress Explain the Burden? Oxid Med Cell Longev 2017: 1-15.
- Meenakshi Sundaram SP, Nagarajan S, Manjula Devi AJ (2014) Chronic Kidney Disease—Effect of Oxidative Stress. Chinese J of Biology 2014: 1-6.
- Kakey MIS, Abdoulrahman KK (2017) Estimation of liver parameters and oxidative stress in chronic renal failure patients on hemodialysis in Erbil governorate. AIP Conference Proceedings p. 1888.
- Liakopoulos V, Roumeliotis S, Gorny X, Dounousi E, Mertens P (2017) Oxidative Stress in hemodialysis patients: A review of the literature. Oxid Med Cell Longev 2017: 1- 22.
- Xie LM, Ge YY, Huang X, Zhang YQ, Li JX (2015) Effects of fermentable dietary fiber supplementation on oxidative and inflammatory status in hemodialysis patients. Int J Clin Exp Med 8: 1363-1369.
- Kuo K-L, Tarng D-C (2010) Oxidative stress in chronic kidney disease. Adaptive Medicine 2: 87-94.
- Cho KH, Kim HJ, Rodriguez-Iturbe B, Vaziri ND (2009) Niacin ameliorates oxidative stress, inflammation, proteinuria, and hypertension in rats with chronic renal failure. Am J Physiol Renal Physiol 297: F106–F113.
- Kim HJ, Vaziri ND (2010) Contribution of impaired Nrf2-K1 pathway to OS and inflammation in CRF. Am J Physiol Renal Physiol 298: F662-F671.
- Aminzadeh MA, Nicholas SB, Norris KC, Vaziri ND (2013) Role of impaired Nrf2 activation in the pathogenesis of oxidative stress and inflammation in chronic tubulo-interstitial nephropathy. Nephrol Dial Transplant 28: 2038-2045.
- SHARP Collaborative Group (2010) Study of Heart and Renal Protection (SHARP): randomized trial to assess the effects of lowering low-density lipoprotein cholesterol among 9, 438 patients with chronic kidney disease. Am Heart J 160: 785-794.
- Liu M, Grigoryev DN, Crow MT, Haas M, Yamamoto M, et al. (2009) Transcription factor Nrf2 is protective during ischemic and nephrotoxic AKI in mice. Kidney Int 76: 277-285.
- Yoon JW, Pahl MV, Vaziri ND (2007) Spontaneous leukocyte activation and oxygen-free radical generation in end stage renal disease. Kidney Int 71: 167-172.
- Vaziri ND, Moradi H, Pahl MV, Fogelman AM, Navab M (2009) In vitro stimulation of HDL anti-inflammatory activity and inhibition of LDL pro-inflammatory activity in the plasma of patients with end-stage renal disease by an apoA-1 mimetic peptide. Kidney Int 76: 437-444.
- Fujihara CK, Antunes GR, Mattar AL, Malheiros DM, Vieira JM Jr, et al. (2007) Chronic inhibition of nuclear factor-kB attenuates renal injury in the 5/6 renal ablation model. Am J Physiol Renal Physiol 292: F92-F99.
- Ling CL, Kuo K (2018) Oxidative stress in chronic kidney disease. Renal Replacement Therapy 4: 53-59.
- Schmidt HW, Stocker R, Vollbracht C, Paulsen G, Riley D, et al. (2015) Antioxidants in translational medicine. Antioxid Redox Signal 23: 1130-1143.
- Kooman JP, Kotanko P, Schols AMWJ, Shiels PG, Stenvinkel P (2014) Chronic kidney disease and premature ageing. Nature Reviews Nephrology 10: 732-742.