Abstract
Transforming growth factor-β (TGF-β) is known to be a critical mediator of the cancer associated fibroblast (CAF) phenotype, and osteopontin (OPN) expression in tumors is associated with more aggressive phenotypes and poor patient outcomes. The potential link between these two pathways has not been well characterized. In human breast cancer and hepatocellular cancer models (HCC), OPN induces mesenchymal stem cell (MSC)-to-CAF transformation. OPN binds to cell-surface integrin receptors to activate the transcription factor, myeloid zinc finger 1 (MZF1) to induce MSC production of TGF-β1. OPN induces MZF1 protein to positively feedback regulate its own transcription via binding to the MZF1 promoter and further enhance TGF-β1 expression. The adoption of the CAF phenotype is associated with increased local tumor growth and metastases. Blockade of OPN abolishes this MZF1-TGF-β1 mediated MSC-to-CAF transformation. This suggests interrupting the MZF1-dependent elaboration of TGF-β by blockade of OPN may be an effective clinical strategy for tumor growth inhibition. This review will describe the regulatory pathways by which OPN and MZF1 mediated TGF-β expression induces the CAF phenotype to potentiate cancer growth and metastasis.
Introduction
Interactions between tumor cells and cancer-associated fibroblasts (CAFs) in the tumor microenvironment (TMEN) significantly influence cancer growth and metastasis. Tumor growth and metastasis is highly dependent on complex dynamic interactions between cancer cells and tumor stroma, mediated by direct cell-cell contact and secreted growth factors and cytokines.
CAFs, a tumor stromal element, are involved in tumor growth potentiation, extracellular matrix degradation, tumor cell motility, inhibition of host anti-tumor response, promotion of angiogenesis and metastasis. CAF origin is likely multifactorial, derived from local fibroblasts, bone marrow-mesenchymal stem cells (MSCs), pericytes and tumor cells. Specific molecular markers and signaling pathways that mediate CAF activation are poorly understood. Nevertheless, it is generally accepted that α-smooth muscle actin (SMA), tenascin-C, vimentin and fibroblast specific protein-1 (FSP-1), among others, describe the CAF phenotype [1,2]. Transforming growth factor-β (TGF-β) is known to be a critical mediator of the CAF phenotype. TGF-β is critical for CAF activation and elaboration of a pro-tumorigenic microenvironment [3-6]. Signaling by TGF-β regulates tumor initiation, progression and metastasis through tumor cell-autonomous and host–tumor interactions. TGF-β mediates fibroblast differentiation, tumor stroma formation and regulates all stages of tumor development via tumor cell-autonomous and host-tumor interactions [3-6]. Resident fibroblasts and bone marrow-derived MSCs convert into CAFs during tumor progression. A TGF-β autocrine/paracrine signaling loop acts to initiate/maintain this CAF phenotype and CAFs increase tumor invasion [7-10]. The loss of TGF-β1 reverses this growth advantage imparted by CAFs [11].
The process wherein epithelial cells differentiate back into ECM-secreting mesenchymal stem cells is termed epithelial mesenchymal transition (EMT). Epithelial cells lose their cell-cell adhesions, polarity, and epithelial cell markers and gain mobility and mesenchymal cell markers. As such, EMT is vital to the physiologic process of wound healing and the pathologic process of metastasis. Various transcription factors and signaling pathways act to induce EMT and promote either the epithelial or mesenchymal phenotype, including TGF-β, an important suppressor of epithelial cell proliferation and inducer of tumor progression and metastasis. TGF-β is secreted from the tumor stroma in many malignancies, largely sourced from CAFs [7]. CAF-derived TGF-β can induce EMT in several tumors via a multitude of mechanisms. Breast cancer cell lines undergo EMT in response to treatment with conditioned medium from CAFs isolated from invasive breast specimens. In this model, neutralizing antibody and small-molecule inhibition of TGF-β signaling prevents EMT in these cell lines [12]. TGF-β secreted by CAFs can also lead to EMT in bladder cancer cells, a process regulated by the long-coding RNA ZEB2NAT. Following the depletion of lncRNA-ZEB2NAT, conditioned media from CAFs fail to induce EMT in human bladder cancer cell lines (5637, T24, and J82) [13].
TGF-β can also result in the formation of cancer stem cell populations within tumors—a process attributable to EMT. For example, TGF-β treatment of patient-derived colorectal tumor specimens results in upregulation of CD44+ stem cell populations and expression of N-cadherin. This suggests that TGF-β induces the formation of cancer stem cells through EMT [14]. In non-small cell lung cancer, TGF-β has a similar effect on cancer stemness [15]. Another intermediate for TGF-β-related acquisition of cancer stemness is miR-155 which, when overexpressed, results in EMT in liver cancer [16].
These studies demonstrated the role of TGF-β signaling in CAF initiation and maintenance, leading to EMT. However, the initiating steps for TGF-β synthesis in this context have never been addressed. Osteopontin (OPN), a phosphoprotein secreted by malignant cells and tumor stromal cells, is a key mediator of tumor cell migration and metastasis and a marker of breast cancer progression and metastasis [17-23]. Recent findings suggest that tumor-derived OPN instigates bone marrow-derived MSC trafficking to the TMEN [24-26], which is characterized by the outgrowth of a desmoplastic stroma rich in CAFs that promotes cancer growth and metastasis [24,25,27]. Serendipitously, OPN induces TGF-β expression and fibroblast activation in models of inflammation and fibrosis [28,35].
In our previous work with in vitro and in vivo human cancer models, we found that OPN induces MSC-to-CAF transformation. OPN binds to cell-surface integrin receptors to activate the transcription factor, myeloid zinc finger 1 (MZF1), and induce MSC production of TGF-β1 with increased CAF phenotype markers. The adoption of the CAF phenotype is associated with increased local tumor growth and metastases. Extracellular blockade of OPN abolishes this MZF1-TGF-β1 mediated MSC-to-CAF transformation. However, the relationship between OPN, TGF-β and the CAF phenotype remains poorly understood. This review will describe the regulatory pathways by which OPN and MZF1 mediated TGF-β expression induces the CAF phenotype to potentiate cancer growth and metastasis.
Osteopontin
Osteopontin (OPN) was initially characterized in 1979 as a phosphoprotein secreted by transformed, malignant epithelial cells [36]. Investigators have since independently detected this molecule as secreted phosphoprotein I (Spp1), 2ar, uropontin and early T-lymphocyte activation-1 (Eta-1) [37-42]. OPN is a 34 kDa protein that is extensively modified post-translationally; it presents as a 60-kDa phosphoprotein initially labeled as transformation-associated gene and a major sialoprotein in the extracellular matrix of bone [41,43]. Post-translational modifications leading to cell-type and condition-specific variations result in a measured molecular weight of 41–75 kda for OPN and its associated isoforms [42-47]. OPN is classically described as a ligand for αvβ3 integrin and CD44 receptors [48-50]. It has multiple Ser and Thr phosphorylation sites, sites for N- and O-linked glycosylation, as well as a thrombin cleavage site. A variety of additional phosphorylation, glycosylation and sulfation can generate different functional forms of OPN which provide tissue specific and function specific versatility [51].
OPN is a member of the small integrin-binding ligand N-linked glycoprotein (SIBLING) family of proteins which include bone sialoprotein (BSP), dentin matrix protein 1 (DMP1), dentin sialoprotein (DSPP), and matrix extracellular phoshoglycoprotein (MEPE) [52]. Elevated OPN expression has been implicated as an important mediator of tumor metastasis and has been investigated for use as a biomarker for advanced disease and as a potential therapeutic target in the regulation of cancer metastasis. The molecular structure of OPN is rich in aspartate and sialic-acid residues and contains unique functional domains [45]. These structural motifs mediate critical cell-matrix and cell–cell signaling through the αvβintegrin and CD44 receptors in a variety of normal and pathologic processes. Interestingly, the role of OPN appears to be maintained across species with similar expression and functions detected in humans and rodents [41,45,53-66]. Cell types which express OPN include osteoclasts, osteoblasts, kidney, breast and skin epithelial cells, nerve cells, vascular smooth muscle cells and enodothelial cells [41,45,50,67]. Activated immune cells such as T-cells, NK cells, macrophages and Kupffer cells also express OPN. The secreted OPN protein is widely distributed in plasma, urine, milk and bile [68-70]. The induced expression of OPN has been detected in T lymphocytes, epidermal cells, bone cells, macrophages and tumor cells in remodeling processes such as inflammation, ischemia–reperfusion, bone resorption and tumor progression [50,67]. OPN gene has an alternative translation start site and thus there are two major variants of OPN, intracellular and extracellular [71,72]. OPN is subject to alternative splicing, as well as post-translational modifications such as phosphorylation, glycosylation and proteolytic cleavage.
An important area of investigation involves the transcriptional regulation of OPN expression during tumorigenesis and metastasis and the identification of trans-elements that could potentially affect the metastatic phenotype. A variety of stimuli including phorbol 12-myristate 13-acetate (PMA), 1,25-dihydroxyvitamin D, basic fibroblast growth factor (bFGF), tumor necrosis factor-α (TNF-α), IL-1, interferon gamma (IFN-γ) and lipopolysaccharide (LPS) upregulate OPN expression [50,66,67].
The gene (SPP1; Secreted Phosphoprotein 1) maps to chromosome 4 (4q13) and contains 7 exons. Human, mouse, rat, pig, bovine and chicken OPN cDNA samples share a high degree of sequence homology with human and mouse genes consisting of seven exons and extending over 8 and 7 kb, respectively [39,45,73-75]. Cloning analysis of OPN reveals a hydrophilic and negatively charged sialo-phosphoprotein of ×298 amino acids in mice and ×314 amino acids in humans [39,43,46,50]. Alternative RNA splicing of human OPN occurs with at least three OPN cDNAs identified [50]. These cancer specific splice variants are OPN-a, OPN-b and OPN-c. OPN-b and -c in the blood are biomarkers for distinct cancers [76].
OPN is rich in aspartate, glutamate and serine residues and contains functional domains for calcium-binding, glycosylation, phosphorylation and extra-cellular matrix adhesion. Several domains include (1) the OPN NH2 and COOH-terminal regions, (2) the thrombin cleavage site (RSK) and (3) the glycine–arginine–glycine–aspartate–serine (GRGDS) sequence [67,70]. The GRGDS sequence contains a functional arginine–glycine–asparate (RGD) binding motif that ligates cell surface αvβ3, αvβ1, αvβ5, and α5β1 integrins [66,77]. Six amino acids away from the RGD motif resides a thrombin cleavage site [50,77].
Efficient engagement of the integrin receptor by OPN requires thrombin cleavage and phosphorylation of the N-terminal fragment, and this improves cell attachment and migration when compared to uncleaved OPN [49,53,78]. Both melanoma cells and K562 erythroleukemia cells bind exclusively to thrombin cleaved OPN but not to intact OPN [79,80]. Thrombin cleaves OPN to reveal the integrin and CD44 binding domains and produce two fragments of approximately equivalent size resulting in OPN-R and exposing the cryptic C-terminal α4β1, α9β1 and α9β4 integrin receptors integrin-binding motif (SVVYGLR) [77,81-85]. The SVVYGLR sequence may alsp play a role in angiogenesis [86].
A correlation between elevated OPN expression and malignant invasion was initially established when OPN was observed within tumor cells and in the surrounding stroma of numerous human cancers [68,87-93]. Further clinical studies detected a significant increase in OPN plasma concentration in patients with metastatic disease compared with normal sera [68,90,94]. Histologic analysis of a wide variety of tumor specimens have shown that increased OPN expression is associated with invasion, progression or metastasis in cancers of the breast, stomach, lung, prostate, liver, and colon [91,93,95-102]. Elevated plasma OPN correlates with increased breast cancer tumor burden, metastasis and prognosis [21,91,94,103]. OPN inhibition decreases local breast cancer growth and metastasis [27,104-108]. In colon cancer, OPN has been identified as a lead marker for tumor progression in a screen of 12,000 human genes with a significant association between the degree of OPN expression and advancing Astler Collier (AC) stage [102,109]. Almost 10–20-fold OPN induction exists in samples with liver metastases over normal mucosa. In breast cancer, similar models have been established. Tuck et al. analyzed a series of 154 lymph node negative breast cancer patients and demonstrated that elevated OPN expression is associated with poorer clinical outcome [91]. Rudland et al. determined that prognostic significance could be assigned to tumor–OPN expression by demonstrating that OPN-negative specimens correlate with a median survival of >228 months compared with 68 months for OPN-positive specimens taken from 333 patients with operable stage I and II breast cancer. Several signaling pathways when mis-regulated can result in activation of OPN expression. Noteworthy among them are oncogenic, tumor promoting pathways such as receptor tyrosine pathway, G-protein coupled pathways, Wnt/β-catenin, Hedgehog (Hh), NFκβ and estrogen signaling pathways. OPN is not only a hypoxia-responsive gene but also transcriptionally upregulates HIF1α expression under normoxia and hypoxia [111,112]. Events such as upregulation of transcription by promoter polymorphism, abnormal activation, lack of repressor function and aberrant epigenetic regulation are of specific importance in the expression patterns of OPN in cancers [76]. Cook et al. identified gene expression changes differentially regulated by OPN in a model of human breast cancer. These changes reflect the six “hallmarks of cancer” in a model of breast cancer progression [22].
Studies utilizing high throughput gene profiling arrays have identified OPN as a target for use as a serum biomarker in predicting tumor metastasis. The practical application of plasma OPN biomarkers in clinical medicine is currently the subject of intense investigation. Multiple pilot studies examining the plasma expression of OPN in patients with advanced disease in comparison with patients with no disease or local/regional disease have demonstrated that elevated OPN levels can be sensitive and specific in predicting disease progression in head and neck, renal, gastric, hepatocellular, lung, pancreatic cancers, and uveal melanoma. Wu et al. used analyses of nonparametric receiver-operating characteristics to calculate cutoff values for OPN values associated with specific clinicopathological features in 132 gastric cancer patients. In this study, a cutoff OPN level of 129.8 ng/ml was associated with 80% sensitivity, 88.2 specificity and 87.7 accuracy in predicting the presence of liver metastasis. Logistic regression analyses confirmed that elevated plasma OPN level is associated with advanced stage, serosal invasion, venous invasion, and liver metastasis. Further, these levels correlated with significantly lower survival rates. In similar multivariate analyses, Chang and colleagues determined that stage is the only independent factor influencing circulating levels of OPN. Patients with stage IV non-small cell lung cancer had median OPN levels measured at 158.2 ng/ml compared with 86.2 ng/ml in stage I patients. Chang et al. also showed that patients with bone metastasis had higher OPN levels than patients without bony metastasis (136.6 vs 112.3 ng/ml). In other studies, Bramwell et al. demonstrated that measuring OPN levels over time during the course of breast metastasis enhanced the prognostic value, with an increase in OPN level > 250 ng/ml being the strongest prognostic indicator for overall survival [19]. Developing such biomarkers to identify subgroups of patients that carry an occult invasive or metastatic phenotype will be helpful in stratifying approaches to clinical management.
Immunohistochemical studies demonstrate that OPN localizes to the leading edge of invading tumors, which are enriched in MSC-like cells, suggesting that OPN+ cells interact in a paracrine fashion with TMEN constituents [87,115,116]. In animal models, OPN produced by tumor and stromal cells stimulates the incorporation of activated BM-derived cells into breast tumors, instigating local growth and distant metastases [24,117]. Large numbers of BM-MSCs were found in explanted tumors and metastases as shown by GFP+ BM transplantation studies. By description, this is similar to our findings [27]. In xenograft studies with 4T1(OPN+) murine breast cancer cells (spontaneous metastatic model), MSCs injected into circulating blood selectively localized and proliferated in subcutaneous and metastatic sites.
Additionally, in murine breast cancer models, OPN has been demonstrated to facilitate the migration of MSC from the primary tumor site to the bone marrow (BM), contributing to the formation of a premetastatic niche. In both MCF7(OPN-) and MB231(OPN+) human breast cancer cell lines, blockade or absence of OPN prohibited the migration of MSC to the BM, the transformation of MSC to CAF within the BM premetastatic niche and reduced the stem cell profile of the cancer cells within the niche. Relative CAF profiles were quantified using rt-PCR to measure SMA and vimentin mRNA levels and cancer stem cell profile was measured using Nanog, Oct4, and Sox2 mRNA levels. These results indicate OPN modulates not only the transformation of MSC to CAF within the primary tumor site but also the formation of a premetastatic niche and the relative stem cell profile of the cancer cells within that niche. Increased cancer stem cell profile has been associated with more aggressive phenotypes. Additionally, the presence of BM micrometastases in breast cancer patients correlates closely with prognosis and survival. These results indicate OPN may be a potent option by which therapeutic interventions may act to disrupt the formation of the bone marrow premetastatic niche in breast cancer.
There exists a relative wealth of information regarding the role of OPN in cancer progression. Furthermore, the link between OPN and TGF-β1 has been described extensively in numerous settings including inflammation, fibrosis, and cancer. However, little is known of the relationship between OPN and TGF-β. Previous studies have shown that when lentiviral expression vectors of OPN transcripts in the MCF-7 human breast cancer cell line were injected into nude mice, all OPN transcripts significantly upregulated TGF-beta1 and MCP-1 production by tumor cells. In hepatocellular cancer, hepatocyte OPN promotes TGF-β1 mediated hepatic stellate cell activation. Our lab was the first to identify MZF1 as the primary transcription factor activated by OPN which then induces TGF-β1 expression. In a separate study, OPN was found to induce MZF1 to stimulate its own transcription via a positive feedback mechanism. OPN also induces MZF1 phosphorylation. In addition, we demonstrate that protein kinase A (PKA) signaling enhances TGF-β1 protein expression and MZF1 promoter activation and phosphorylation in the presence of OPN. These results indicate that OPN and MZF1 help to regulate TGF-β expression.
Myeloid zinc finger-1
MZF1, originally isolated from a myelogenous leukemia patient, is a Kruppel transcription factor containing 13 C2H2 zinc fingers and belongs to the SCAN-ZF family. MZF1 forms a peptide loop that binds to G-rich DNA consensus sequences [124-126]. MZF1 is preferentially expressed in primitive hematopoietic cells and plays an important role in regulating myelopoiesis. Reports identify MZF1 as a tumor suppressor, namely an in vitro cervical cancer model and an in vivo knockout model; however the majority of recent literature supports the oncogenic nature of MZF1 in a variety of cancers [125,128-132]. Expression of MZF1 is associated with the anti-apoptotic and oncogenic transformation of cervical (SiHA, HeLa) and colorectal (Rko, SW480) cancer cell lines. Wu et al. showed that MZF1 was a key transcription factor that binds to the promoter of cytokeratin 17 which then, induced by the TGF-β1-ERK1/2-MZF1 signaling pathway, facilitates metastasis by promoting the acquisition of stemness in cervical cancer.
Retroviral transduction and subsequent overexpression of MZF1 results in loss of contact inhibition, loss of substrate dependence, and more rapid cell cycling in NIH 3T3 cells. Disruption of the tight lineage- and stage-specific regulation of MZF1 can result in neoplastic transformation of embryonic fibroblasts. These MZF1-transformed 3T3 cells form aggressive fibrosarcomas in athymic mice. MZF1 is essential for YAP1 transcription; YAP1 and Sox2 each reinforce the expression of the other to maintain stemness and transcription in osteosarcoma.
MZF1 antisense oligonucleotide inhibits PKCα expression and decreases cell migration and invasion in HCC (SK-Hep1). This mechanism has, more recently, been elucidated; PKCα is regulated by the cooperative interaction of the transcription MZF1 and Ets-like protein-1 (Elk-1) in human cancer cells. The acidic domain of MZF1 and the heparin-binding domain of Elk-1 facilitate the heterodimeric interaction between the two genes before the complex binds to the PKCα promoter. Ye et al. demonstrated that the expression of MZF1/Elk-1 is correlated with that of PKCα in HCC. Blocking the interaction between MZF1 and Elk-1 through the transfection of their binding domain MZF160-72 decreased PKCα expression and decreased the epithelial-mesenchymal transition potential of the HCC cells [136,137]. In triple negative breast cancers (TNBC), the same group showed that blocking the formation of the heterodimer by transfection of MZF160-72 or Elk-1145-157 peptide fragments at the MZF1 / Elk-1 interface decreases DNA-binding activity of the MZF1 / Elk-1 complex at the PKCα promoter region, decreasing expression, migration, tumorigenicity, and the EMT-potential of TNBC [138,139].
MZF1 is also known as an ErbB2-responsive transcription factor that promotes invasion of breast cancer cells via upregulation of lysosomal cathepsins B and L. Tvingsholm et al. identified let-7 microRNA, a breast cancer tumor suppressor, as a direct negative regulator of MZF1. Analysis of primary breast cancer tissues showed a gradual upregulation of MZF1 from normal breast epithelium to invasive ductal carcinoma and a negative correlation between several let-7 family members and MZF1 mRNA, suggesting that the inverse regulatory relationship between let-7 and MZF1 may play a role in the development of invasive breast cancer.
MZF1 was found to enhance induction of apoptosis by interacting with LDOC1, a gene encoding a leucine-zipper protein whose expression was decreased in pancreatic and gastric cancer cell lines. Overexpression of LDOC1 caused externalization of the cell membrane phosphatidylserine for early-phase apoptotic events, and reduced cell viability in human cell lines.
MZF1 has also been identified to bind to the MMP-14 promoter to facilitate its nascent transcription and expression in gastric cancer cell lines. MMP-14 is a membrane-anchored proteinase that promotes tumorigenesis and aggressiveness of gastric cancer.
In endometrial cancer, PAX2 stimulates cell growth and cell motility. The overexpression of PAX2 is regulated by MZF1 and promoter hypermethylation.
Additional genes regulated by MZF1 include: CD34, lactoferrin, myeloperoxidase, PADI1 and N-cadherin.
As described above, MZF1 has been the recent focus of increased interest as the result of its association with a wide variety of cancers and signaling pathways. However, the role of MZF1 in TGF-β1 expression has not been extensively characterized.
Osteopontin, MZF1 and TGF-β
The TMEN significantly influences tumor behavior [6,144-147]. CAFs are a critical constituent, residing within the tumor or at its margins. However, there is no single accepted biomarker that defines a CAF and there exists controversy over the identity and location of the CAF precursor cell [1,145,148,149]. Utilizing in vitro and in vivo models of human breast and liver cancer, we have demonstrated that: 1) OPN induces MZF1-dependent TGF-β1 production in resident MSCs in the TMEN to promote local tumor growth and metastasis, 2) OPN creates a positive feedback loop in which MZF1 protein induces its own transcription via binding to the MZF1 promoter, 3) MSCs adopt a CAF phenotype as a result of autocrine TGF-β1 signaling, and 4) inactivation of extracellular OPN by an RNA aptamer (APT) blocks TGF-β1-mediated MSC-to-CAF transformation and tumor growth and metastasis. APT is a short ssRNA oligonucleotide that assumes a stable 3-dimensional shape to bind and inactivate extracellular OPN [123,150].
OPN elicits TGF-β expression in MSCs
MSCs were exposed to OPN at increasing concentrations (0 to 320 ng/ml), and active TGF-β protein expression was measured at 48 h (Figure 1A). The mean concentration of TGF-β1 after exposure to 80 ng/ml was 227±7 pg/ml; 619±14 pg/ml after 160 ng/ml; and 872±4 pg/ml after 320 ng/ml (ANOVA p=0.0001). For TGF-β2 and TGF-β3, after exposure to 80 ng/ml, 160 ng/ml, and 320 ng/ml, mean concentrations of each were ~155 pg/ml, 260 pg/ml, and 425 pg/ml, respectively (ANOVA p=0.0001). Based on this data, in conjunction with the reported median plasma OPN level of 177 ng/ml in metastatic breast cancer patients, all subsequent studies used an OPN dose of 180 ng/ml [19]. MSCs were exposed to OPN at increasing time intervals (0 to 96 h) (Figure 1B). Time-dependent expression of TGF-β was observed. All isoforms peaked at 12 h. At all time intervals, except TGF-β3 at 96 hours, the increase was significant from baseline (p<0.05). Further studies used the 48 h time point.
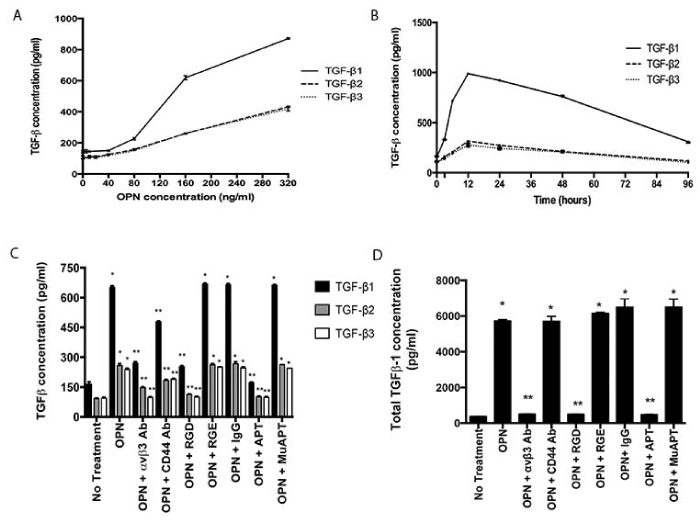
Figure 1. OPN induces TGF-β1, 2 and 3 protein expression in MSCs in vitro. Data presented as mean±SEM of three experiments. A. Concentration-dependent expression in MSCs following exposure to OPN for 48 h (ANOVA p=0.0001). B. Time-dependent expression in MSCs following exposure to OPN (180 ng/ml). C. Active TGF-β1, 2 and 3 protein expression in MSCs exposed to OPN (180 ng/ml for 48 h) and one of the following inhibitors: RGD, αvβ3 integrin Ab, CD44 Ab, or APT. Control inhibitors: RGE, IgG, and MuAPT (*p<0.01 vs. No Treatment; **p<0.01 vs. OPN). D. Total TGF-β1 protein expression in MSCs exposed to OPN (180 ng/ml for 48h) (*p<0.01 vs. No Treatment; **p<0.01 vs. OPN) [123].
OPN binds to avβ3 integrin and CD44 receptors to mediate important cell-cell and cell-matrix signaling pathways [48]. To examine the role of these cell-surface receptor pathways in OPN-mediated TGF-β expression, MSCs were exposed to OPN and the following inhibitors: RGD, an integrin competitive ligand inhibitor, avβ3 integrin Ab, CD44 Ab, or OPN neutralizing RNA aptamer (APT); control inhibitors: RGE, IgG, and mutant aptamer (MuAPT) (Figure 1C). Extracellular binding of OPN by APT and blockade of integrin and CD44 receptors ablated OPN-mediated active TGF-β1, 2, and 3 expressions (p<0.01 vs. OPN). The control inhibitors did not alter TGF-β expression. These results indicate that OPN-mediated TGF-β expression in MSCs requires OPN binding to integrin receptors, and to a lesser extent, CD44 receptors.
As TGF-β1 expression is up-regulated to a larger degree than other isoforms, remaining studies focus on TGF-β1. A similar expression pattern was seen in total TGF-β1 after OPN exposure (Figure 1D). APT blockade of OPN and integrin receptor blockade resulted in a significant decrease in total TGF-β1 expression (p<0.01). However, CD44 receptor blockade had no effect. These results indicate that the primary receptor involved in OPN-mediated TGF-β1 expression in MSCs is likely integrin.
MSC CAF marker expression is OPN-mediated
MSCs were exposed to OPN once or daily for one week to determine the duration of CAF marker (SMA, tenascin-C, vimentin and FSP-1) and TGF-β1 expression. At 2 days, all CAF markers and TGF-β1 were elevated by 8- to 12-fold in both groups (p<0.01). In the group exposed once, all CAF markers and TGF-β1 were decreased by day 7 (p<0.05). In the daily exposure group, by day 5, TGF-β1 was elevated even further (p<0.05). By day 7, tenascin-C and TGF-β1 were elevated compared to day 2 (p<0.05) (Figure 2D). These results indicate that under continuous OPN stimulation, as would likely occur an OPN-producing tumor TMEN, a robust and durable CAF marker profile persists and TGF-β1 expression is up-regulated to a similar/larger degree.
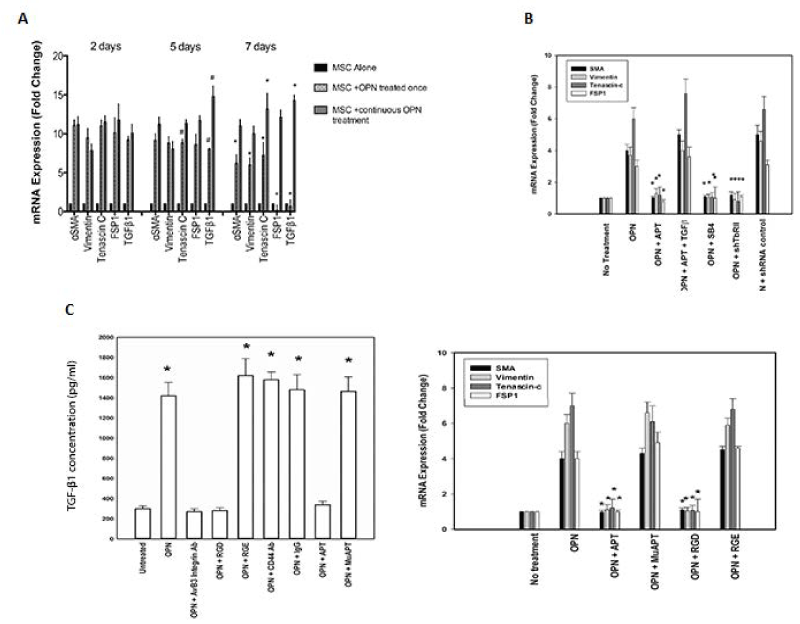
Figure 2. OPN induces a CAF phenotype in MSCs in vitro. Data presented as mean±SEM of three experiments. D. CAF marker and TGF-β1 mRNA expression in MSCs exposed to OPN either once or daily for one week. (* and # p<0.01 vs. Day 2). E. CAF marker mRNA expression after TGF-β1 signaling blockade (SB4 or shTGFβR2) (*p<0.01 vs. OPN) or exogenous recombinant TGF-β (400 pg/ml) (*p<0.01 vs. OPN+APT). F. TGF-β1 protein and CAF marker mRNA expression in primary human MSCs (TGF-β1: *p<0.01 vs. Untreated, OPN+αvβ3 integrin Ab, OPN+RGD. CAF marker mRNA: *p<0.01 vs. OPN, OPN+MuAPT and OPN+RGE) [123]
To determine the functional role of TGF-β1, its signaling was blocked using a TGF-β1 type I receptor inhibitor (SB4) or TGF-β1 type II receptor shRNA. OPN-stimulated CAF marker expression was ablated (p<0.01 vs. OPN) (Figure 2E). In selected instances, exogenous recombinant TGF-β1 (400 pg/ml) was administered [7]. Exogenous TGF-β1 restored CAF marker expression in OPN+APT+TGF-β1 (p<0.01 vs. OPN+APT). BM-MSCs (3000 cells), enriched from normal human bone marrow, were exposed to OPN. Active TGF-β1 protein increased ~7-fold (p<0.01), total: active TGF-β1 ratio was ~8-10:1 (Data not shown). OPN significantly increased CAF marker mRNA expression, which was blocked by RGD and APT (p<0.01 vs. OPN) (Figure 2F). Human skin, mammary and lung fibroblasts and murine embryonic fibroblasts were exposed to human OPN. Active TGF-β1 protein and CAF marker expression was not altered (Data not shown). In summary, OPN-integrin binding induces TGF-β1 and CAF marker expression in MSCs.
OPN activates TGF-β1 transcription via MZF1
The role of OPN in TGF-β1 transcriptional regulation was determined by measuring recruitment of RNA polymerase II to a consensus TATA box (ATTA located at -1195bp) and the final exon. TGF-β1 transcription increased ~20-fold after OPN exposure (p<0.01) (Figure 3A). A full-length human TGF-β1 promoter (−3360/+124) was amplified (Figure 3B). Stimulation with OPN resulted in >35-fold transactivation with lesser activation of the first two deletion constructs (Figure 3C). The remaining three deletion constructs showed no activation after OPN stimulation. The 67bp region (-691 to -624) contained a canonical binding site for MZF1 (GGGGAGGAGGGGGA), as predicted by TFSearch [124,125]. Two MZF1 consensus DNA-binding sequences have been identified: 5'-AGTGGGGA-3' and 5'-CGGGGAGGGGGAA-3' (Figure 3D). This G-rich region is 100% homologous to the mouse TGF-β1 promoter (-653 to -637). Human and mouse MZF1 proteins are 83% homologous. MZF1 is involved in cell differentiation, migration and proliferation [126,128,133].
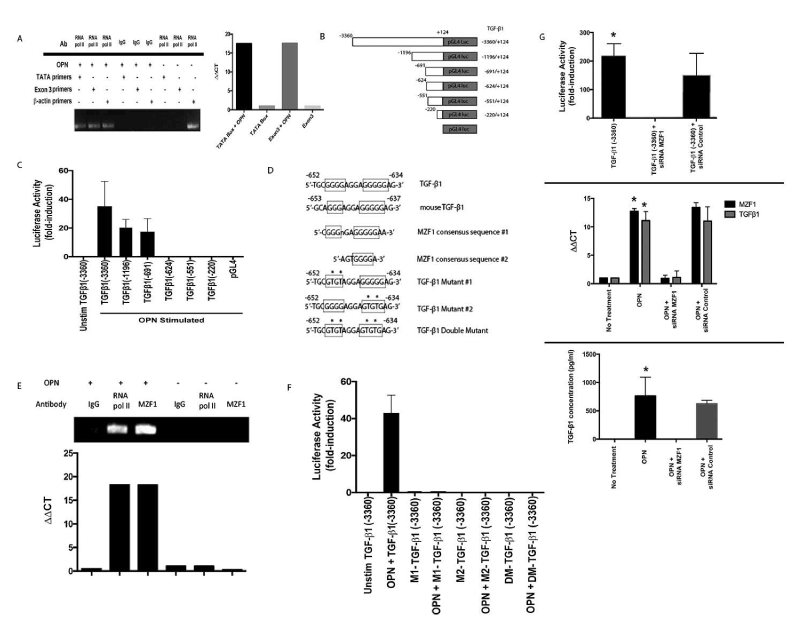
Figure 3. OPN mediates TGF-β transcription. (Data are presented as mean±SEM of at least three experiments. Gels are representative of three experiments.) A. RNA polymerase II binding to TGF-β1 promoter. B. Schematic depiction of TGF-β1 promoter luciferase-reporter constructs. C. Transient transfection analysis of TGF-β1 promoter constructs in MSCs. D. Schematic representation of MZF1 binding sites in human and murine TGF-β1 promoter and associated mutations of putative MZF1 sites. E. ChIP assays for MZF1 and RNA polymerase II binding to the TGF-β1 promoter. F. Transient transfection analysis of mutant and wild-type TGF-β1 promoter constructs in MSCs. G. Effect of siRNA inhibition of MZF1 expression in OPN stimulated MSCs. TGF-β1 promoter luciferase activity was determined by transient transfection analysis (*p<0.01 vs. TGF-β1 (-3360)+siRNA MZF1). TGF-β1 and MZF1 mRNA expression was measured by RT-PCR (*p<0.01 vs. OPN+siRNA MZF1). TGF-β1 protein in MSC media was analyzed with ELISA (*p<0.01 vs. OPN+siRNA MZF1) [123]
Chromatin immunoprecipitation (ChIP) assays targeting -697bp to -600bp confirmed binding of MZF1 and RNA polymerase II to the TGF-β1 promoter in the presence of OPN (Figure 3E). After OPN exposure, MZF1 binding to the G-rich promoter region increased ~18-fold (p<0.02 vs. MSCs). This putative MZF1 binding site was mutated to generate two single mutants and one double mutant (Figure 3D). All mutation constructs demonstrated a significant decrease in luciferase activity (p<0.03) (Figure 3F). siRNA-mediated silencing of MZF1 significantly decreased TGF-β1 full-length promoter luciferase activity (p<0.01), MZF1 and TGF-β1 mRNA (p<0.01) and active TGF-β1 protein expression (p<0.02) in OPN stimulated MSCs compared to controls (Figure 3G). Taken together, this data indicates that MZF1 regulates OPN-mediated TGF-β1 transcription and protein expression.
OPN induces TGF-β1-dependent MSC CAF marker expression in breast cancer co-cultures
MSCs were co-cultured in Boyden chambers with MB231(OPN+) or MCF7(OPN-) human breast cancer cells. All ELISA and qRT-PCR data presented below are results of media from MSC well or MSC cell lysates only. OPN+ cells exhibit aggressive in vitro indices of adhesion and migration/invasion. OPN- cells exhibit the opposite [105,106]. MSCs do not express OPN (Data not shown). After 24 h, in MB231+MSC, active TGF-β1 increased 9-fold compared to baseline; blockade by αvβ3 Ab, RGD and APT abolished the TGF-β1 response (p<0.01). Conversely, in MCF7+MSC, active TGF-β1 expression was minimal. Addition of OPN increased active TGF-β1 ~7-fold; this was blocked by αvβ3 Ab, RGD and APT (p<0.01) (Figure 4A and 4B). In co-culture with MB231, CAF marker and TGF-β1 mRNA expression increased 20- to 30-fold (p<0.01); abolished by APT, avβ3 Ab and RGD (p<0.01); indicating a causative role for OPN-integrin binding. As expected, exogenous OPN significantly increased CAF marker and TGF-β1 mRNA expression in an integrin-dependent fashion (Figures 4C and 4D).
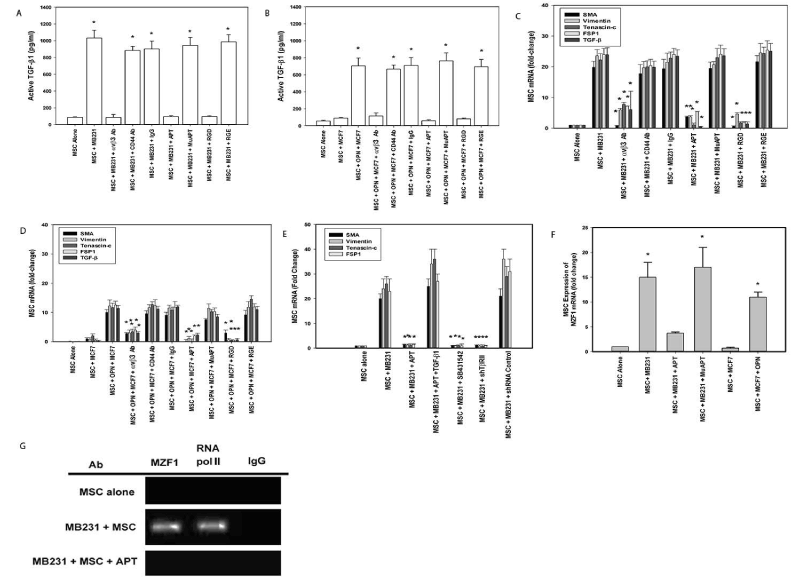
Figure 4. MB231(OPN+), MCF7 (OPN-) and MSC co-culture studies of TGF-β1 expression. (Data are presented as mean±SEM of at least three experiments. Gels are representative of three experiments.) A. Active TGF-β1 expression in MB231-MSC co-culture media. (*p<0.01 vs. MSC alone, MSC+MB231+αvβ3 integrin Ab, MSC+MB231+APT, and MSC+MB231+RGD). B. Active TGF-β1 expression in MCF7+MSC co-culture media with exogenous OPN (*p<0.01 vs. MSC, MSC+MCF7, MSC+OPN+MCF7+αvβ3 integrin Ab, MSC+OPN+MCF7+APT, and MSC+OPN+MCF7+RGD). C. TGF-β1 and CAF marker mRNA expression by qRT-PCR in MSCs co-cultured with MB231. (*p<0.01 vs. MSC+MB231, MSC+MB231+CD44 Ab, MSC+MB231+IgG, MSC+MB231+MuAPT and MSC+MB231+RGE). D. TGF-β1 and CAF marker mRNA expression by qRT-PCR in MSCs co-cultured with MCF7 and exogenous OPN (*p<0.01 vs. MSC+OPN+MCF7, MSC+OPN+MCF7+CD44 Ab, MSC+OPN+MCF7+IgG, MSC+OPN+MCF7+MuAPT and MSC+OPN+MCF7+RGE). E. CAF marker mRNA expression in MSCs co-cultured with MB231 and inhibitors of TGF- β1 signaling (*p<0.01 vs. MSC+MB231, MSC+MB231+APT+TGF-β1, and MSC+MB231+shRNA Control). F. MZF1 mRNA expression by qRT-PCR in MSCs co-cultured with MB231 (OPN+) or MCF7(OPN) (*p<0.01 vs. MSC, MSC+MB231+APT, and MSC+MCF7). G. ChIP assay for MZF1 and RNA polymerase II binding to the TGF-β1 promoter region, -697 to -600, in MSCs co-cultured with MB231 [123]
Co-culture studies were repeated with SB4, shTGFβR2 and exogenous TGF-β1 (Figure 4E) [7]. CAF markers in MB231+MSC were significantly decreased with APT and reversed with exogenous TGF-β1. Similarly, TGF-β1 receptor blockade ablated CAF marker expression (p<0.01). This data indicates that tumor-derived OPN interacts with MSC integrin receptors to induce TGF-β1-dependent CAF marker expression. APT blockade of OPN in MB231 co-culture significantly decreased MZF1 mRNA while ablating MZF1 and RNA polymerase II binding to the TGF-β1 promoter region. Conversely, co-culture with MCF7 did not express MZF1 mRNA without exogenous OPN (Figure 4F and 4G). In parallel with our previous results, this data suggests that OPN mediates MZF1-dependent TGF-β1 expression to induce CAF marker expression in MSCs.
MZF1 and TGF-β1 expression in mouse models
In a MB231+MSC orthotopic xenotransplant model, we previously demonstrated that extracellular OPN blockade and inactivation by APT significantly inhibits: 1) local breast cancer growth and metastasis and 2) MSC expression of CAF markers [27]. This mouse model was repeated to study TGF-β1 and MZF1 expression. Animals were injected via tail vein with APT or MuAPT every 2 days and imaged for 8 weeks. Bioluminescence was greater by 10-fold in MB231+MSC compared with MB231 (p< 0.01) (Figure 5A). At 8 weeks, MSCs were isolated from primary tumors and metastatic sites. In MB231+MSC, MZF1 and TGF-β1 mRNA increased by 4- and 8-fold, respectively, in all locations (p<0.01); expression ablated by APT (Figures 5B and 5C). As expected, CAF markers were readily expressed but ablated in the presence of APT (Data not shown).
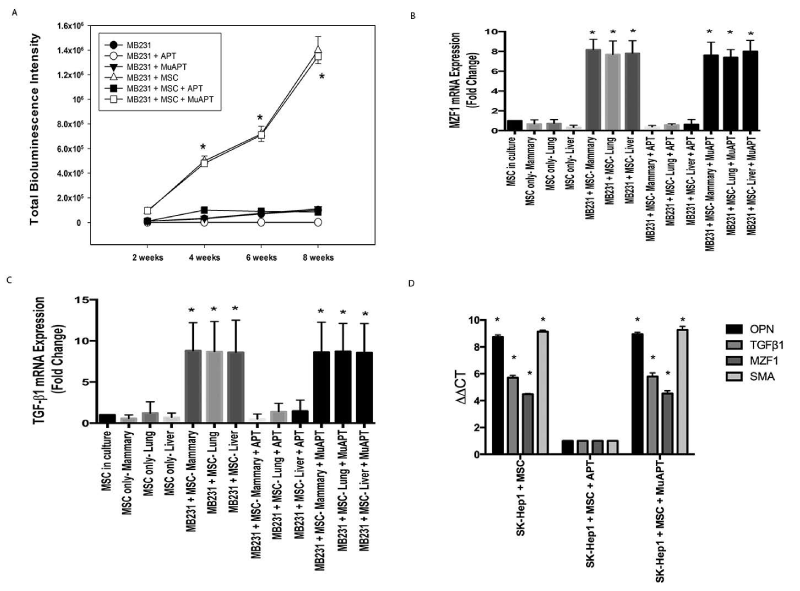
Figure 5. MB231 or SK-Hep1 in vivo co-implantation models with MSCs in NOD-scid mice. Data derived from four animals in each group. A. Bioluminescence intensity in MB231 mouse model (*p<0.01 for MB231+MSC or MB231+MSC+MuAPT vs. MB231, MB231+APT, MB231+MuAPT and MB231+MSC+APT). B. MZF1 mRNA expression by qRT-PCR in MSCs isolated from MB231±MSC mice. (*p<0.01 vs. MSC only and MB231+MSC+APT among the various locations). C. TGF-β1 mRNA expression by qRT-PCR in MSCs isolated from MB231±MSC mice. (*p<0.01 vs. MSC only and MB231+MSC+APT among the various locations). D. OPN, TGF-β1, MZF1, SMA mRNA expression by qRT-PCR in MSCs isolated from SK-Hep1+MSC mice. (*p<0.01 vs. SK-Hep1+MSC+APT) [123]
We have previously reported OPN-mediated EMT (epithelial-mesenchymal transition) associated growth of hepatocellular cancer (HCC) in a mouse xenograft model. To ascertain the role of our newly proposed OPN-MZF1-TGF-β1 axis in HCC, qRT-PCR was performed using cDNA from MSCs isolated by FACS from the primary liver tumors of SK-Hep1+MSC xenografts. APT blockade significantly decreased OPN, MZF1, TGF-β1 and SMA mRNA expression (p<0.01), supporting the likely presence of the OPN-MZF1-TGF-β1 axis in HCC (Figure 5D).
A series of xenotransplant experiments was performed using dual-labeled MCF7 cells that constitutively express OPN (RFP-luc-MCF7-lvOPN) implanted alone or co-implanted with GFP-MSCs. A subset of GFP-MSCs had constitutive MZF1 knockdown (MSC(dMZF1)). When MCF7-lvOPN were implanted alone, increased growth did not occur until 8 weeks. In MCF7-lvOPN+MSC mice, tumor growth was significantly greater. Deletion of MZF1 resulted in abrogation of OPN-associated tumor growth in MCF7-lvOPN. In a similar fashion, APT treatment resulted in ablation of tumor growth in MCF7-lvOPN+MSC mice (Figure 6A). After 8 weeks, FACS was used to isolate GFP-MSCs and RFP-MCF7 cells from primary tumors and liver metastases. MCF7-lvOPN+MSC and MCF7-lvOPN+MSC+MuAPT combinations demonstrated significantly greater fractions of tumor cells (RFP+) and MSCs (GFP+), suggesting enhanced local growth and metastasis. Similarly, only MCF7-lvOPN+MSC and MCF7-lvOPN+MSC+MuAPT expressed CAF marker, MZF1 and TGF-β1 mRNA. When compared to MSCs isolated from MCF7-lvOPN+MSC±MuAPT, the MSC(dαZF1)s expressed minimal CAF marker, MZF1 and TGF-β1 mRNA (p<0.01) (Figures 6B and 6C). This data demonstrates that enhanced OPN expression in MCF7 in the presence of MSCs promotes local tumor growth and metastasis, which is inhibited by MZF1 ablation in MSCs or APT-inactivation of OPN.
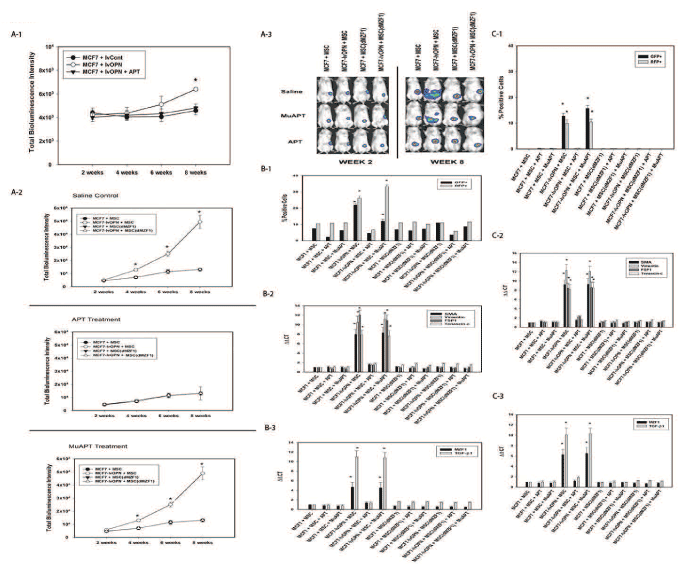
Figure 6. MCF7 in vivo co-implantation models with MSCs in NOD-scid mice. Data derived from four animals in each group. A-1. Bioluminescence intensity in MCF7±lvOPN mouse model (*p<0.01 vs. MCF7-lvCont and MCF-lvOPN+APT). A-2. Bioluminescence intensity in MCF7±lvOPN± MSC(dMZF1) mouse model (*p<0.01 vs. MCF7+MSC, MCF7+MSC(dMZF1) and MCF7-lvOPN+MSC(dMZF1) treated with saline control, APT or MuAPT. A-3. Representative photos of bioluminescence in MCF7 mouse models at Week 2 and Week 8. B-1. FACS analysis of MCF7 cells (RFP+) and MSCs (GFP+) isolated from primary tumor. (*p<0.01 vs. MCF7+MSC, MCF7+MSC+APT, MCF7-lvOPN+MSC+APT, MCF7+MSC(dMZF1) and MCF7-lvOPN+MSC(dMZF1)). B-2. CAF marker mRNA expression by qRT-PCR in MSCs. (*p<0.01 vs. MCF7+MSC, MCF7+MSC+APT, MCF7-lvOPN+MSC+APT, MCF7+MSC(dMZF1) and MCF7-lvOPN+MSC(dMZF1)). B-3. MZF1 and TGF-β1 mRNA expression by qRT-PCR in MSCs. (*p<0.01 vs. MCF7+MSC, MCF7+MSC+APT, MCF7-lvOPN+MSC+APT, MCF7+MSC(dMZF1) and MCF7-lvOPN+MSC(dMZF1)). C-1. FACS analysis of MCF7 cells (RFP+) and MSCs (GFP+) isolated from liver. (*p<0.01 vs. MCF7+MSC, MCF7+MSC+APT, MCF7-lvOPN+MSC+APT, MCF7+MSC(dMZF1) and MCF7-lvOPN+MSC(dMZF1)). C-2. CAF marker mRNA expression by qRT-PCR in MSCs. (*p<0.01 vs. MCF7+MSC, MCF7+MSC+APT, MCF7-lvOPN+MSC+APT, MCF7+MSC(dMZF1) and MCF7-lvOPN+MSC(dMZF1)). C-3. MZF1 and TGF-β1 mRNA expression by qRT-PCR in MSCs. (*p<0.01 vs. MCF7+MSC, MCF7+MSC+APT, MCF7-lvOPN+MSC+APT, MCF7+MSC(dMZF1) and MCF7-lvOPN+MSC(dMZF1)) [123]
To elucidate the role of TGF-β1, exogenous TGF-β1 (1.2 ng/mouse/day via tail vein) was administered to a subgroup of MCF7-lvOPN+MSC(dMZF1). With this gain-of-function study bypassing the inhibition of MSC elaboration of TGF-β1 in MSC(dMZF1)s, exogenous systemic TGF-β1 accelerated growth and metastasis (Figure 7A and 7B). At Week 5, luciferase activity was enhanced ~10-fold with TGF-β1 (p<0.01). Primary tumor MSC(dMZF1)s were isolated by FACS after 6 weeks. CAF markers were strongly expressed with exogenous TGF-β1; MZF1 was not expressed; TGF-β1 was expressed in a positive feedback manner (Figure 7C and 7D) [7]. These results indicate that OPN mediates MZF1-dependent TGF-β1 expression in MSCs to promote local tumor growth and metastases via MSC-to-CAF transformation.
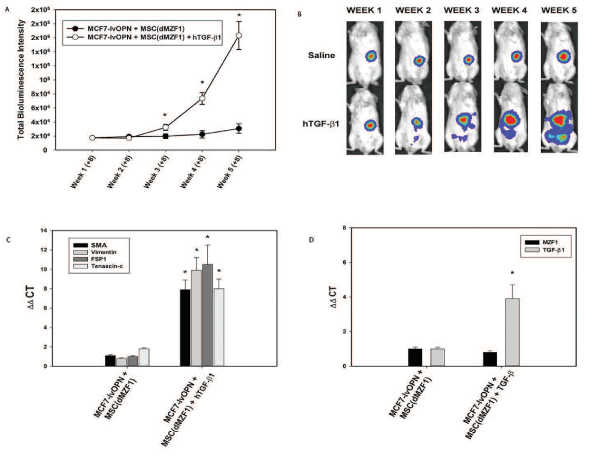
Figure 7. Gain of function study with administration of exogenous TGF-β1 to a subgroup of MCF7-lvOPN+MSC(dMZF1) beginning 8 weeks after co-implantation. Data are presented as mean±SEM of three animals in each group. A. Bioluminescence intensity in MCF7-lvOPN+MSC(dMZF1) mouse model (*p<0.01 vs. MCF7-lvOPN+MSC(dMZF1)) B. Representative images of luciferase activity in mice at Weeks 1 through 5. (*p<0.01 vs. MCF7-lvOPN+MSC(dMZF1)). C. CAF mRNA expression by qRT-PCR in MSCs isolated from primary tumors. (*p<0.01 vs. MCF7-lvOPN+MSC(dMZF1)). D. MZF1 and TGF-β1 mRNA expression by qRT-PCR in MSCs isolated from primary tumors. (*p<0.01 vs. MCF7-lvOPN+MSC(dMZF1) [123]
OPN-MZF1 positive feedback loop upregulates MZF1 transcription
To further assess the role of OPN in modulating MZF1 activity, ChIP Real Time-PCR was used to examine transcriptional regulation of the MZF1 gene in MSC. A primer set was designed to include the MZF1 promoter TATA box. OPN was found to significantly induce binding of RNA polymerase II to the MZF1 promoter (1.2% input). (Figure 8A and Figure 8B). The human MZF1 promoter sequence was amplified, and the PCR products (2.2, 1.8, 0.7, 0.6, 0.5, and 0.4 kb) were sequenced and subcloned into the pGL3 luciferase reporter (Figure 9A). Luciferase induction was quantified as relative luciferase activity (RLA) using Renilla signal to control for transfection efficiency. Stimulation with OPN significantly induced luciferase activation of the 2.2 (21,409 RLA), 1.8 (29,664 RLA), and 0.7 kb (20,007 RLA) MZF1 promoter sequences but was not sufficient for induction of the shorter MZF1 promoter sequences (Figure 9B). The MZF1 promoter DNA sequence was then analyzed for known transcription factor binding sequences. A MZF1 binding sequence within the MZF1 gene promoter region was identified and then mutated (Figure 9C). The 2.2-kb fragment of the MZF1 promoter containing the mutated MZF1 DNA binding site was subcloned into the pGL3 luciferase reporter. Luciferase assay demonstrated that mutation of the MZF1 binding site was sufficient to nearly completely ablate the luciferase activity of the 2.2-kb MZF1 promoter fragment when stimulated with OPN (Figure 9D). ChIP Real Time-PCR was used to determine the effect of OPN on the interactions of MZF1 with its binding site located within the MZF1 promoter. ChIP and subsequent pulldown with an anti-MZF1 antibody showed that OPN exposure induced binding of MZF1 to its own promoter at the expected MZF1 binding site. (fold-enrichment, MSC + OPN, 31305). Binding of RNA polymerase to the MZF1 promoter was similarly induced by OPN, (fold-enrichment: MSC + OPN, 82614.6) (Figure 10A and 10B). These results support a positive feedback mechanism in which the MZF1 protein induces its own transcription via binding to the MZF1 promoter.
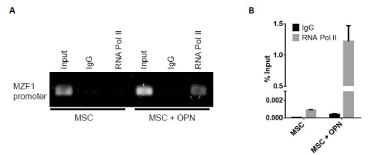
Figure 8. TGF-β1 and MZF1 promoter activation. A. Binding of RNA pol II to the MZF1 promoter. ChIP-qRT-PCR was performed using primers that included the MZF1 TATA box. B. Graphical representation of RNA pol II binding to the MZF1 promoter. The percent input was calculated from ChIP-qRT-PCR CT values using input samples as internal controls for the amount of chromatin [150]
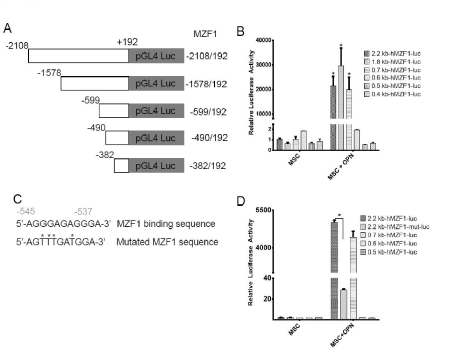
Figure 9. OPN mediated MZF1-luciferase construct activation. A. Schematic depiction of MZF1 promoter-luciferase reporter constructs. B. Graphical representation of RNA pol II and MZF1 binding to the TGF-β1 promoter. The percent input was calculated from ChIP Real Time-PCR CT values using input samples as internal controls for the amount of chromatin. C. Schematic representation of the wild type and mutated MZF1 binding site within the MZF1 promoter sequence. D. Luciferase activity of the wild type and mutant MZF1 promoter constructs in MSC (*p<0.0001) [150]
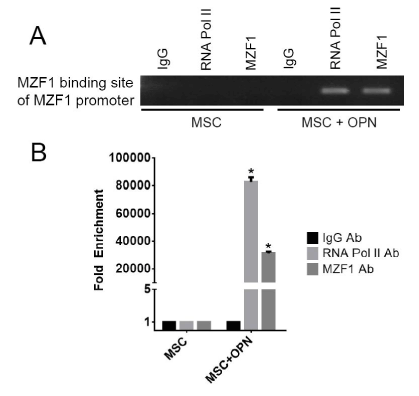
Figure 10. MZF1 binding to the MZF1 promoter. A. Binding of MZF1 to the MZF1 promoter. ChIP-qRT-PCR was done using primers that amplified the promoter region at -545 to -537 base pairs upstream of the MZF1 start codon which contains an MZF1 binding sequence. B. Graphical representation of RNA pol II and MZF1 binding to the MZF1 promoter. Fold enrichment was calculated using non-specific IgG binding as an internal control (*p<0.0001) [150]
Conclusion
OPN interacts with MSCs via integrin receptors to up-regulate MZF1-mediated expression of TGF-β1, which results in conversion of MSCs to a CAF phenotype and promotion of more aggressive local tumor growth and metastasis in human breast and HCC models. In addition, OPN induces MZF1 protein to positively feedback regulate its own transcription via binding to the MZF1 promoter and further enhancing TGF-β1 expression. Blockade and inactivation of extracellular OPN by R3 APT effectively abolishes these pathways. This suggests that OPN blockade may be an effective clinical strategy for tumor growth inhibition.
References
- Mishra PJ, Mishra PJ, Humeniuk R, Medina DJ, Alexe G, et al. (2008) Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res 68: 4331-4339. [Crossref]
- Spaeth EL, Dembinski JL, Sasser AK, Watson K, Klopp A, et al. (2009) Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS One 4: e4992. [Crossref]
- Bierie B, Moses HL (2006) Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer 6: 506-520. [Crossref]
- Worthington JJ, Klementowicz JE, Travis MA (2011) TGFbeta: a sleeping giant awoken by integrins. Trends Biochem Sci 36: 47-54
- Liu Z, Bandyopadhyay A, Nichols RW, Wang L, Hinck AP, et al. (2012) Blockade of Autocrine TGF-beta Signaling Inhibits Stem Cell Phenotype, Survival, and Metastasis of Murine Breast Cancer Cells. J Stem Cell Res Ther 2: 1-8.
- Dumont N, Liu B, Defilippis RA, Chang H, Rabban JT, et al. (2013) Breast Fibroblasts Modulate Early Dissemination, Tumorigenesis, and Metastasis through Alteration of Extracellular Matrix Characteristics. Neoplasia 15: 249-262.
- Kojima Y, Acar A, Eaton EN, Mellody KT, Scheel C, et al. (2010) Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci U S A 107: 20009-20014.
- Casey T, Eneman J, Crocker A, White J, Tessitore J, et al. (2008) Cancer associated fibroblasts stimulated by transforming growth factor beta1 (TGF-β1) increase invasion rate of tumor cells: a population study. Breast Cancer Research and Treatment 110: 39-49.
- Quante M, Tu SP, Tomita H, Gonda T, Wang SS, et al. (2011) Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 19: 257-272.
- Hawinkels LJ, Paauwe M, Verspaget HW, Wiercinska E, van der Zon JM, et al. (2012) Interaction with colon cancer cells hyperactivates TGF-beta signaling in cancer-associated fibroblasts. Oncogene 33: 97-107. [Crossref]
- Su G, Sung KE, Beebe DJ, Friedl A (2012) Functional Screen of Paracrine Signals in Breast Carcinoma Fibroblasts. PLoS One 7: e46685.
- Yu Y, Xiao CH, Tan LD, Wang QS, Li XQ, et al. (2013) Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-[beta] signalling. Br J Cancer 110: 724-732. [Crossref]
- Zhuang J, Lu Q, Shen B, Huang X, Shen L, et al. (2015) TGFbeta1 secreted by cancer-associated fibroblasts induces epithelial-mesenchymal transition of bladder cancer cells through lncRNA-ZEB2NAT. Sci Rep 5: 11924.
- Nakano M, Ariyama H, Tamura S, Isobe T, Miyawaki K, et al. (2015) Abstract 1520: Plasticity of CD44+ colorectal cancer stem cells depend on TGF-beta-induced epithelial mesenchymal transition (EMT): evidences from ex vivo culture system. Cancer Research 75: 1520.
- Pore MM, Buikema L, Hiltermann T, Kruyt F (2012) Abstract 2402: TGF beta-mediated epithelial to mesenchymal transition in non-small cell lung cancer: effects on stemness, invasiveness and chemotherapy sensitivity. Cancer Res 72: 2402.
- Liu F, Kong X, Lv L, Gao J (2015) TGF-β1 acts through miR-155 to down-regulate TP53INP1 in promoting epithelial–mesenchymal transition and cancer stem cell phenotypes. Cancer Lett 359: 288-298. [Crossref]
- Ye QH, Qin LX, Forgues M, He P, Kim JW, et al. (2003) Predicting hepatitis B virus-positive metastatic hepatocellular carcinomas using gene expression profiling and supervised machine learning. Nat Med 9: 416-423.
- Wai PY, Kuo PC (2008) Osteopontin: regulation in tumor metastasis. Cancer Metastasis Rev 27: 103-118. [Crossref]
- Bramwell VH, Doig GS, Tuck AB, Wilson SM, Tonkin KS, et al. (2006) Serial plasma osteopontin levels have prognostic value in metastatic breast cancer. Clin Cancer Res 12: 3337-3343.
- Mi Z, Guo H, Wai PY, Gao C, Wei J, et al. (2004) Differential osteopontin expression in phenotypically distinct subclones of murine breast cancer cells mediates metastatic behavior. J Biol Chem 279: 46659.-46667.
- Singhal H, Bautista DS, Tonkin KS, O'Malley FP, Tuck AB, et al. (1997) Elevated plasma osteopontin in metastatic breast cancer associated with increased tumor burden and decreased survival. Clin Cancer Res 3: 605-611.
- Cook AC, Tuck AB, McCarthy S, Turner JG, Irby RB, et al. (2005) Osteopontin induces multiple changes in gene expression that reflect the six "hallmarks of cancer" in a model of breast cancer progression. Mol Carcinog 43: 225-236.
- Rodrigues LR, Teixeira JA, Schmitt FL, Paulsson M, Lindmark-Mansson H (2007) The Role of Osteopontin in Tumor Progression and Metastasis in Breast Cancer. Cancer Epidemiol. Biomarkers Prev 16: 1087-1097.
- McAllister SS, Gifford AM, Greiner AL, Kelleher SP, Saelzler MP, et al. (2008) Systemic endocrine instigation of indolent tumor growth requires osteopontin. Cell 133: 994-1005.
- Elkabets M, Gifford AM, Scheel C, Nilsson B, Reinhardt F, et al. 2011. Human tumors instigate granulin-expressing hematopoietic cells that promote malignancy by activating stromal fibroblasts in mice. J Clin Invest 121: 784-799.
- Anderberg C, Li H, Fredriksson L, Andrae J, Betsholtz C, et al. (2009) Paracrine Signaling by Platelet-Derived Growth Factor-CC Promotes Tumor Growth by Recruitment of Cancer-Associated Fibroblasts. Cancer Res 69: 369-378.
- Mi Z, Bhattacharya SD, Kim VM, Guo H, Talbot LJ, et al. (2011) Osteopontin promotes CCL5-mesenchymal stromal cell-mediated breast cancer metastasis. Carcinogenesis 32: 477-487.
- Nicholas SB, Liu J, Kim J, Ren Y, Collins AR, et al. (2010) Critical role for osteopontin in diabetic nephropathy. Kidney Int 77: 588-600.
- Vetrone SA, Montecino-Rodriguez E, Kudryashova E, Kramerova I, Hoffman EP, et al. (2009) Osteopontin promotes fibrosis in dystrophic mouse muscle by modulating immune cell subsets and intramuscular TGF-beta. J Clin Invest 119: 1583-1594.
- Wolak T, Kim H, Ren Y, Kim J, Vaziri ND, et al. (2009) Osteopontin modulates angiotensin II-induced inflammation, oxidative stress, and fibrosis of the kidney. Kidney Int 76: 32-43.
- Szalay G, Sauter M, Haberland M, Zuegel U, Steinmeyer A, et al. (2009) Osteopontin: a fibrosis-related marker molecule in cardiac remodeling of enterovirus myocarditis in the susceptible host. Circ Res 104: 851-859.
- Kohan M, Breuer R, Berkman N (2009) Osteopontin induces airway remodeling and lung fibroblast activation in a murine model of asthma. Am J Respir Cell Mol Biol 41: 290-296.
- Miyazaki K, Okada Y, Yamanaka O, Kitano A, Ikeda K, et al. (2008) Corneal wound healing in an osteopontin-deficient mouse. Invest Ophthalmol Vis Sci 49: 1367-1375.
- Syn WK, Choi SS, Liaskou E, Karaca GF, Agboola KM, et al. (2011) Osteopontin is induced by hedgehog pathway activation and promotes fibrosis progression in nonalcoholic steatohepatitis. Hepatology 53: 106-115.
- Lenga Y, Koh A, Perera AS, McCulloch CA, Sodek J, et al. (2008) Osteopontin expression is required for myofibroblast differentiation. Circ Res 102: 319-327.
- Senger DR, Wirth DF, Hynes RO (1979) Transformed mammalian cells secrete specific proteins and phosphoproteins. Cell 16: 885-93
- Fisher LW, Hawkins GR, Tuross N, Termine JD (1987) Purification and partial characterization of small proteoglycans I and II, bone sialoproteins I and II, and osteonectin from the mineral compartment of developing human bone. J Biol Chem 262: 9702-9708.
- Kiefer MC, Bauer DM, Barr PJ. 1989. The cDNA and derived amino acid sequence for human osteopontin. Nucleic Acids Res 17: 3306.
- Young MF, Kerr JM, Termine JD, Wewer UM, Wang MG, et al. (1990) cDNA cloning, mRNA distribution and heterogeneity, chromosomal location, and RFLP analysis of human osteopontin (OPN). Genomics 7: 491-502.
- Zhang Q, Wrana JL, Sodek J (1992) Characterization of the promoter region of the porcine opn (osteopontin, secreted phosphoprotein 1) gene. Identification of positive and negative regulatory elements and a 'silent' second promoter. Eur J Biochem 207: 649-659.
- Craig AM, Denhardt DT (1991) The murine gene encoding secreted phosphoprotein 1 (osteopontin): promoter structure, activity, and induction in vivo by estrogen and progesterone. Gene 100: 163-171. [Crossref]
- Patarca R, Freeman GJ, Singh RP, Wei FY, Durfee T, et al. (1989) Structural and functional studies of the early T lymphocyte activation 1 (Eta-1) gene. Definition of a novel T cell-dependent response associated with genetic resistance to bacterial infection. J Exp Med 170: 145-161.
- Franzen A, Heinegard D (1985) Isolation and characterization of two sialoproteins present only in bone calcified matrix. Biochem J 232: 715-724
- Smith JH, Denhardt DT (1987) Molecular cloning of a tumor promoter-inducible mRNA found in JB6 mouse epidermal cells: induction is stable at high, but not at low, cell densities. J Cell Biochem 34: 13-22.
- Denhardt DT, Guo X (1993) Osteopontin: a protein with diverse functions. FASEB J 7: 1475-1482. [Crossref]
- Oldberg A, Franzen A, Heinegard D (1986) Cloning and sequence analysis of rat bone sialoprotein (osteopontin) cDNA reveals an Arg-Gly-Asp cell-binding sequence. Proc Natl Acad Sci USA 83: 8819-8823.
- Senger DR, Asch BB, Smith BD, Perruzzi CA, Dvorak HF (1983) A secreted phosphoprotein marker for neoplastic transformation of both epithelial and fibroblastic cells. Nature 302: 714-715
- Denhardt DT, Guo X (1993) Osteopontin: a protein with diverse functions. FASEB J 7: 1475-1482. [Crossref]
- Helluin O, Chan C, Vilaire G, Mousa S, DeGrado WF, et al. (2000) The activation state of alphavbeta 3 regulates platelet and lymphocyte adhesion to intact and thrombin-cleaved osteopontin. J Biol Chem 275: 18337-18343.
- O'Regan A, Berman JS (2000) Osteopontin: a key cytokine in cell-mediated and granulomatous inflammation. Int J Exp Pathol 81: 373-390.
- Sodek J, Ganss B, McKee MD (2000) Osteopontin. Crit Rev Oral Biol Med 11: 279-303. [Crossref]
- Fedarko NS, Jain A,2021 Copyright OAT. All rights reservall integrin binding ligand N-linked glycoproteins (SIBLINGs) bind and activate specific matrix metalloproteinases. FASEB J 18: 734-736.
- Senger DR, Perruzzi CA, Papadopoulos-Sergiou A, Van de WL (1994) Adhesive properties of osteopontin: regulation by a naturally occurring thrombin-cleavage in close proximity to the GRGDS cell-binding domain. Mol Biol Cell 5: 565-574.
- Attur MG, Dave MN, Stuchin S, Kowalski AJ, Steiner G, et al. (2001) Osteopontin: an intrinsic inhibitor of inflammation in cartilage. Arthritis Rheum 44: 578-584.
- Guo H, Cai CQ, Schroeder RA, Kuo PC (2001) Osteopontin is a negative feedback regulator of nitric oxide synthesis in murine macrophages. J Immunol 166: 1079-1086.
- Johnson RJ, Gordon KL, Giachelli C, Kurth T, Skelton MM, et al. (2000) Tubulointerstitial injury and loss of nitric oxide synthases parallel the development of hypertension in the Dahl-SS rat. J Hypertens 18: 1497-1505.
- Noble BS, Reeve J (2000) Osteocyte function, osteocyte death and bone fracture resistance. Mol Cell Endocrinol 159: 7-13. [Crossref]
- Scott JA, Weir ML, Wilson SM, Xuan JW, Chambers AF, et al. (1998) Osteopontin inhibits inducible nitric oxide synthase activity in rat vascular tissue. Am J Physiol 275: H2258-H2265.
- Thomas SE, Lombardi D, Giachelli C, Bohle A, Johnson RJ (1998) Osteopontin expression, tubulointerstitial disease, and essential hypertension. Am J Hypertens 11: 954-961.
- Rollo EE, Laskin DL, Denhardt DT (1996) Osteopontin inhibits nitric oxide production and cytotoxicity by activated RAW264.7 macrophages. J Leukoc Biol 60: 397-404. [Crossref]
- Singh K, Balligand JL, Fischer TA, Smith TW, Kelly RA (1995) Glucocorticoids increase osteopontin expression in cardiac myocytes and microvascular endothelial cells. Role in regulation of inducible nitric oxide synthase. J Bio Chem 270: 28471-28478.
- Feng B, Rollo EE, Denhardt DT (1995) Osteopontin (OPN) may facilitate metastasis by protecting cells from macrophage NO-mediated cytotoxicity: evidence from cell lines down-regulated for OPN expression by a targeted ribozyme. Clin Exp Metastasis 13: 453-462.
- Denhardt DT, Lopez CA, Rollo EE, Hwang SM, An XR, et al. (1995) Osteopontin-induced modifications of cellular functions. Ann N YAcad Sci 760: 127-142.
- Hwang SM, Wilson PD, Laskin JD, Denhardt DT (1994) Age and development-related changes in osteopontin and nitric oxide synthase mRNA levels in human kidney proximal tubule epithelial cells: contrasting responses to hypoxia and reoxygenation. J Cell Physiol 160: 61-68.
- Hwang SM, Lopez CA, Heck DE, Gardner CR, Laskin DL, et al. (1994) Osteopontin inhibits induction of nitric oxide synthase gene expression by inflammatory mediators in mouse kidney epithelial cells. J Biol Chem 269: 711-715.
- Hijiya N, Setoguchi M, Matsuura K, Higuchi Y, Akizuki S, et al. (1994) Cloning and characterization of the human osteopontin gene and its promoter. Biochem J 303: 255-262. [Crossref]
- Weber GF (2001) The metastasis gene osteopontin: a candidate target for cancer therapy. Biochim Biophys Acta 1552: 61-85.
- Senger DR, Perruzzi CA, Gracey CF, Papadopoulos A, Tenen DG (1988) Secreted phosphoproteins associated with neoplastic transformation: close homology with plasma proteins cleaved during blood coagulation. Cancer Res 48: 5770-5774.
- Senger DR, Perruzzi CA, Papadopoulos A, Tenen DG (1989) Purification of a human milk protein closely similar to tumor-secreted phosphoproteins and osteopontin. Biochim Biophys Acta 996: 43-48.
- Bautista DS, Denstedt J, Chambers AF, Harris JF (1996) Low-molecular-weight variants of osteopontin generated by serine proteinases in urine of patients with kidney stones. J Cell Biochem 61: 402-409.
- Suzuki K, Zhu B, Rittling SR, Denhardt DT, Goldberg HA, et al. (2002) Colocalization of Intracellular Osteopontin With CD44 Is Associated with Migration, Cell Fusion, and Resorption in Osteoclasts. J Bone Miner Res 17: 1486-1497. [Crossref]
- Shinohara ML, Kim HJ, Kim JH, Garcia VA, Cantor H (2008) Alternative translation of osteopontin generates intracellular and secreted isoforms that mediate distinct biological activities in dendritic cells. Proc Natl Acad Sci U S A 105: 7235-7239. [Crossref]
- Patarca R, Saavedra RA, Cantor H (1993) Molecular and cellular basis of genetic resistance to bacterial infection: the role of the early T-lymphocyte activation-1/osteopontin gene. Crit Rev Immunol 13: 225-246. [Crossref]
- Behrend EI, Chambers AF, Wilson SM, Denhardt DT (1993) Comparative analysis of two alternative first exons reported for the mouse osteopontin gene. J Biol Chem 268: 11172-11175.
- Yamamoto S, Hijiya N, Setoguchi M, Matsuura K, Ishida T, et al. (1995) Structure of the osteopontin gene and its promoter. Ann N Y Acad Sci 760: 44-58. [Crossref]
- Shevde LA, Samant RS (2014) Role of osteopontin in the pathophysiology of cancer. Matrix Biol 37: 131-141. [Crossref]
- Senger DR, Perruzzi CA (1996) Cell migration promoted by a potent GRGDS-containing thrombin-cleavage fragment of osteopontin. Biochi Biophys Acta 1314: 13-24.
- Ashkar S, Weber GF, Panoutsakopoulou V, Sanchirico ME, Jansson M, et al. (2000) Eta-1 (osteopontin): an early component of type-1 (cell-mediated) immunity. Science 287: 860-864. [Crossref]
- Denhardt DT, Noda M, O'Regan AW, Pavlin D, Berman JS (2001) Osteopontin as a means to cope with environmental insults: regulation of inflammation, tissue remodeling, and cell survival. J Clin.Invest 107: 1055-1061.
- Smith LL, Giachelli CM (1998) Structural requirements for alpha 9 beta 1-mediated adhesion and migration to thrombin-cleaved osteopontin. Exp Cell Res 242: 351-360.
- Rehman M, Iqbal M, Sharif M, Raza M (2012) Content based image retrieval: survey. World Applied Sciences Journal 19: 404-412.
- Senger DR, Perruzzi CA (1985) Secreted phosphoprotein markers for neoplastic transformation of human epithelial and fibroblastic cells. Cancer Res 45: 5818-5823.
- Laffón A, García-Vicuña R, Humbría A, Postigo AA, Corbí AL, et al. (1991) Upregulated expression and function of VLA-4 fibronectin receptors on human activated T cells in rheumatoid arthritis. J Clin Invest 88: 546-552. [Crossref]
- Smith LL, Cheung HK, Ling LE, Chen J, Sheppard D, et al. (1996) Osteopontin N-terminal domain contains a cryptic adhesive sequence recognized by alpha9beta1 integrin. J Biol Chem 271: 28485-28491.
- Yokosaki Y, Matsuura N, Sasaki T, Murakami I, Schneider H, et al. (1999) The integrin alpha(9)beta(1) binds to a novel recognition sequence (SVVYGLR) in the thrombin-cleaved amino-terminal fragment of osteopontin. J Biol Chem 274: 36328-36334.
- Hamada Y, Nokihara K, Okazaki M, Fujitani W, Matsumoto T, et al. (2003) Angiogenic activity of osteopontin-derived peptide SVVYGLR. Biochem Biophys Res Commun 310: 153-157.
- Brown LF, Papadopoulos-Sergiou A, Berse B, Manseau EJ, Tognazzi K, et al. (1994) Osteopontin expression and distribution in human carcinomas. Am J Pathol 145: 610-623.
- Bellahcene A, Castronovo V (1995) Increased expression of osteonectin and osteopontin, two bone matrix proteins, in human breast cancer. Am J Pathol 146: 95-100.
- Hirota S, Ito A, Nagoshi J, Takeda M, Kurata A, et al. (1995) Expression of bone matrix protein messenger ribonucleic acids in human breast cancers. Possible involvement of osteopontin in development of calcifying foci. Lab Invest 72: 64-69.
- Singhal H, Bautista DS, Tonkin KS, O'Malley FP, Tuck AB, et al. (1997) Elevated plasma osteopontin in metastatic breast cancer associated. Clin Cancer Res 3: 605-611.
- Tuck AB, O'Malley FP, Singhal H, Harris JF, Tonkin KS, et al. (1998) Osteopontin expression in a group of lymph node negative breast cancer patients. Int J Cancer 79: 502-508
- Casson AG, Wilson SM, McCart JA, O'Malley FP, Ozcelik H, et al. (1997) ras mutation and expression of the ras-regulated genes osteopontin and cathepsin L in human esophageal cancer. Int J Cancer 72: 739-745. [Crossref]
- Gotoh M, Sakamoto M, Kanetaka K, Chuuma M, Hirohashi S (2002) Overexpression of osteopontin in hepatocellular carcinoma. Pathol Int 52: 19-24. [Crossref]
- Fedarko NS, Jain A, Karadag A, Van Eman MR, Fisher LW (2001) Elevated serum bone sialoprotein and osteopontin in colon, breast, prostate, and lung cancer. Clin Cancer Res 7: 4060-4066.
- Tuck AB, O'Malley FP, Singhal H, Tonkin KS, Harris JF, et al. (1997) Osteopontin and p53 expression are associated with tumor progression in a case of synchronous, bilateral, invasive mammary carcinomas. Arch Pathol Lab Med 121: 578-584.
- Tuck AB, Arsenault DM, O'Malley FP, Hota C, Ling MC, et al. (1999) Osteopontin induces increased invasiveness and plasminogen activator expression of human mammary epithelial cells. Oncogene 18: 4237-4246.
- Ue T, Yokozaki H, Kitadai Y, Yamamoto S, Yasui W, et al. (1998) Co-expression of osteopontin and CD44v9 in gastric cancer. Int J Cancer 79: 127-132.
- Shijubo N, Uede T, Kon S, Maeda M, Segawa T, et al. (1999) Vascular endothelial growth factor and osteopontin in stage I lung adenocarcinoma. Am J Respir Crit Care Med 160: 1269-1273. [Crossref]
- Chambers AF, Wilson SM, Kerkvliet N, O'Malley FP, Harris JF, et al. (1996) Osteopontin expression in lung cancer. Lung Cancer 15: 311-323. [Crossref]
- Thalmann GN, Sikes RA, Devoll RE, Kiefer JA, Markwalder R, et al. (1999) Osteopontin: possible role in prostate cancer progression. Clin Cancer Res 5: 2271-2277.
- Pan HW, Ou YH, Peng SY, Liu SH, Lai PL, et al. (2003) Overexpression of osteopontin is associated with intrahepatic metastasis, early recurrence, and poorer prognosis of surgically resected hepatocellular carcinoma. Cancer 98: 119-127.
- Agrawal D, Chen T, Irby R, Quackenbush J, Chambers AF, et al. (2002) Osteopontin identified as lead marker of colon cancer progression, using pooled sample expression profiling. J Natl Cancer Inst 94: 513-521.
- Tuck AB, Chambers AF (2001) The role of osteopontin in breast cancer: clinical and experimental studies. J Mammary Gland Biol Neoplasia 6: 419-429.
- Yu KN, Minai-Tehrani A, Chang SH, Hwang SK, Hong SH, et al. (2010) Aerosol Delivery of Small Hairpin Osteopontin Blocks Pulmonary Metastasis of Breast Cancer in Mice. PLoS One 5: e15623.
- Mi Z, Guo H, Russell MB, Liu Y, Sullenger BA, et al. (2009) RNA Aptamer Blockade of Osteopontin Inhibits Growth and Metastasis of MDA-MB231 Breast Cancer Cells. Mol Ther 17: 153-161.
- Mi Z, Guo H, Kuo PC (2009) Identification of osteopontin-dependent signaling pathways in a mouse model of human breast cancer. BMC Res Notes 2: 119.
- Tuck AB, Chambers AF, Allan AL (2007) Osteopontin overexpression in breast cancer: knowledge gained and possible implications for clinical management. J Cell Biochem 102: 859-868.
- Shevde LA, Samant RS, Paik JC, Metge BJ, Chambers AF, et al. (2006) Osteopontin knockdown suppresses tumorigenicity of human metastatic breast carcinoma, MDA-MB-435. Clin Exp Metastasis 23: 123-133.
- Yeatman TJ, Chambers AF (2003) Osteopontin and colon cancer progression. Clin Exp Metastasis 20: 85-90. [Crossref]
- Rudland PS, Platt-Higgins A, El-Tanani M, De Silva RS, Barraclough R, et al. (2002) Prognostic significance of the metastasis-associated protein osteopontin in human breast cancer. Cancer Res 62: 3417-3427.
- Kale S, Raja R, Thorat D, Soundararajan G, Patil TV, et al. (2014) Osteopontin signaling upregulates cyclooxygenase-2 expression in tumor-associated macrophages leading to enhanced angiogenesis and melanoma growth via alpha9beta1 integrin. Oncogene 33: 2295-2306.
- Raja R, Kale S, Thorat D, Soundararajan G, Lohite K, et al. (2014) Hypoxia-driven osteopontin contributes to breast tumor growth through modulation of HIF1alpha-mediated VEGF-dependent angiogenesis. Oncogene 33: 2053-2064.
- Wu CY, Wu MS, Chiang EP, Wu CC, Chen YJ, et al. (2007) Elevated plasma osteopontin associated with gastric cancer development, invasion and survival. Gut 56: 782-789.
- Chang YS, Kim HJ, Chang J, Ahn CM, Kim SK, et al. (2007) Elevated circulating level of osteopontin is associated with advanced disease state of non-small cell lung cancer. Lung Cancer 57: 373-380.
- Matsuzaki H, Shima K, Muramatsu T, Ro Y, Hashimoto S, et al. (2007) Osteopontin as biomarker in early invasion by squamous cell carcinoma in tongue. J Oral Pathol Med 36: 30-34. [Crossref]
- Gao MQ, Kim BG, Kang S, Choi YP, Park H, et al. (2010) Stromal fibroblasts from the interface zone of human breast carcinomas induce an epithelial-mesenchymal transition-like state in breast cancer cells in vitro. J Cell Sci 123: 3507-3514.
- Udagawa T, Puder M, Wood M, Schaefer BC, D'Amato RJ (2006) Analysis of tumor-associated stromal cells using SCID GFP transgenic mice: contribution of local and bone marrow-derived host cells. FASEB J 20: 95-102.
- Wang H, Cao F, De A, Cao Y, Contag C, et al. (2009) Trafficking mesenchymal stem cell engraftment and differentiation in tumor-bearing mice by bioluminescence imaging. Stem Cells 27: 1548-1558.
- Kuo MC, Kothari AN, Kuo PC, Mi Z (2018) Cancer stemness in bone marrow micrometastases of human breast cancer. Surgery 163: 330-335.
- Bao B, Ahmad A, Azmi AS, Ali S, Sarkar FH (2013) Overview of cancer stem cells (CSCs) and mechanisms of their regulation: implications for cancer therapy. Current protocols in pharmacology Chapter 14: Unit 14.
- Sun J, Feng A, Chen S, Zhang Y, Xie Q, et al. (2013) Osteopontin splice variants expressed by breast tumors regulate monocyte activation via MCP-1 and TGF-beta1. Cell Mol Immunol 10: 176-182. [Crossref]
- Xiao X, Gang Y, Gu Y, Zhao L, Chu J, et al. (2012) Osteopontin Contributes to TGF-β1 Mediated Hepatic Stellate Cell Activation. Dig Dis Sci 57: 2883-2891. [Crossref]
- Weber CE, Kothari AN, Wai PY, Li NY, Driver J, et al. (2015) Osteopontin mediates an MZF1-TGF-beta1-dependent transformation of mesenchymal stem cells into cancer-associated fibroblasts in breast cancer. Oncogene 34: 4821-4833.
- Morris JF, Hromas R, Rauscher FJ 3rd. (1994) Characterization of the DNA-binding properties of the myeloid zinc finger protein MZF1: two independent DNA-binding domains recognize two DNA consensus sequences with a common G-rich core. Mol Cell Biol 14: 1786-1795.
- Hromas R, Morris J, Cornetta K, Berebitsky D, Davidson A, et al. (1995) Aberrant expression of the myeloid zinc finger gene, MZF-1, is oncogenic. Cancer Res 55: 3610-3614.
- Gaboli M, Kotsi PA, Gurrieri C, Cattoretti G, Ronchetti S, et al. (2001) Mzf1 controls cell proliferation and tumorigenesis. Genes Dev 15: 1625-1630. [Crossref]
- Tsai SJ, Hwang JM, Hsieh SC, Ying TH, Hsieh YH (2012) Overexpression of myeloid zinc finger 1 suppresses matrix metalloproteinase-2 expression and reduces invasiveness of SiHa human cervical cancer cells. Biochem Biophys Res Commun 425: 462-467. [Crossref]
- Hsieh YH, Wu TT, Tsai JH, Huang CY, Hsieh YS, et al. (2006) PKCα expression regulated by Elk-1 and MZF-1 in human HCC cells. Biochem Biophys Res Commun 339: 217-225.
- Rafn B, Nielsen Christian F, Andersen Sofie H, Szyniarowski P, Corcelle-Termeau E, et al. (2012) ErbB2-Driven Breast Cancer Cell Invasion Depends on a Complex Signaling Network Activating Myeloid Zinc Finger-1-Dependent Cathepsin B Expression. Molecular Cell 45: 764-776.
- Mudduluru G, Vajkoczy P, Allgayer H (2010) Myeloid Zinc Finger 1 Induces Migration, Invasion, and In vivo Metastasis through Axl Gene Expression in Solid Cancer. Mol Cancer Res 8: 159-169. [Crossref]
- Hsieh YH, Wu TT, Huang CY, Hsieh YS, Liu JY (2007) Suppression of tumorigenicity of human hepatocellular carcinoma cells by antisense oligonucleotide MZF-1. Chin J Physiol 50: 9-15.
- Eguchi T, Prince T, Wegiel B, Calderwood SK (2015) Role and Regulation of Myeloid Zinc Finger Protein 1 in Cancer. J Cell Biochem 116: 2146-2154. [Crossref]
- Tsai SJ, Hwang JM, Hsieh SC, Ying TH, Hsieh YH (2012) Overexpression of myeloid zinc finger 1 suppresses matrix metalloproteinase-2 expression and reduces invasiveness of SiHa human cervical cancer cells. Biochem Biophys Res Commun 425: 462-467.
- Wu L, Han L, Zhou C, Wei W, Chen X, et al. (2017) TGF-beta1-induced CK17 enhances cancer stem cell-like properties rather than EMT in promoting cervical cancer metastasis via the ERK1/2-MZF1 signaling pathway. FEBS J 284: 3000-3017. [Crossref]
- Verma NK, Gadi A, Maurizi G, Roy UB, Mansukhani A, et al. (2017) Myeloid Zinc Finger 1 and GA Binding Protein Co-Operate with Sox2 in Regulating the Expression of Yes-Associated Protein 1 in Cancer Cells. Stem Cells 35: 2340-2350.
- Ye JC, Hsu LS, Tsai JH, Yang HL, Hsiao MW, et al. (2017) MZF-1/Elk-1/PKCalpha is Associated with Poor Prognosis in Patients with Hepatocellular Carcinoma. J Cancer 8: 3028-3036. [Crossref]
- Yue CH, Huang CY, Tsai JH, Hsu CW, Hsieh YH, et al. (2015) MZF-1/Elk-1 Complex Binds to Protein Kinase Calpha Promoter and Is Involved in Hepatocellular Carcinoma. PLoS One 10: e0127420.
- Lee CJ, Hsu LS, Yue CH, Lin H, Chiu YW, et al. (2016) MZF-1/Elk-1 interaction domain as therapeutic target for protein kinase Calpha-based triple-negative breast cancer cells. Oncotarget 7: 59845-59859.
- Yue CH, Liu LC, Kao ES, Lin H, Hsu LS, et al. (2016) Protein kinase C alpha is involved in the regulation of AXL receptor tyrosine kinase expression in triple-negative breast cancer cells. Mol Med Rep 14: 1636-1642. [Crossref]
- Tvingsholm SA, Hansen MB, Clemmensen KKB, Brix DM, Rafn B, et al. (2018) Let-7 microRNA controls invasion-promoting lysosomal changes via the oncogenic transcription factor myeloid zinc finger-1. Oncogenesis 7: 14.
- Inoue M, Takahashi K, Niide O, Shibata M, Fukuzawa M, et al. (2005) LDOC1, a novel MZF-1-interacting protein, induces apoptosis. FEBS Lett 579: 604-608. [Crossref]
- Zheng L, Jiao W, Mei H, Song H, Li D, et al. (2016) miRNA-337-3p inhibits gastric cancer progression through repressing myeloid zinc finger 1-facilitated expression of matrix metalloproteinase 14. Oncotarget 7: 40314-40328.
- Jia N, Wang J, Li Q, Tao X, Chang K, et al. (2016) DNA methylation promotes paired box 2 expression via myeloid zinc finger 1 in endometrial cancer. Oncotarget 7: 84785-84797.
- Xu LN, Xu BN, Cai J, Yang JB, Lin N (2013) Tumor-associated fibroblast-conditioned medium promotes tumor cell proliferation and angiogenesis. Genet Mol Res 12: 5863-58671.
- Mao Y, Keller E, Garfield D, Shen K, Wang J (2012) Stromal cells in tumor microenvironment and breast cancer. Cancer Metastasis Rev 32: 303-315. [Crossref]
- Huang WH, Chang MC, Tsai KS, Hung MC, Chen HL, et al. (2013) Mesenchymal stem cells promote growth and angiogenesis of tumors in mice. Oncogene 32: 4343-4354. [Crossref]
- Brauer HA, Makowski L, Hoadley KA, Casbas-Hernandez P, Lang LJ, et al. (2012) Impact of tumor microenvironment and epithelial phenotypes on metabolism in breast cancer. Clin Cancer Res 19: 571-585. [Crossref]
- Markers for Characterization of Bone Marrow Multipotential Stromal Cells. Stem Cells International 2012: 12.
- Quante M, Tu SP, Tomita H, Gonda T, Wang SSW, et al. (2011) Bone Marrow-Derived Myofibroblasts Contribute to the Mesenchymal Stem Cell Niche and Promote Tumor Growth. Cancer Cell 19: 257-272.
- Driver J, Weber CE, Callaci JJ, Kothari AN, Zapf MA, et al. (2015) Alcohol inhibits osteopontin-dependent transforming growth factor-beta1 expression in human mesenchymal stem cells. J Biol Chem 290: 9959-9973. [Crossref]
- Kim SJ, Glick A, Sporn MB, Roberts AB (1989) Characterization of the promoter region of the human transforming growth factor-beta 1 gene. J Biol Chem 264: 402-408. [Crossref]
- Qi W, Gao S, Wang Z (2008) Transcriptional regulation of the TGF-beta1 promoter by androgen receptor. Biochem J 416: 453-462. [Crossref]
- Rajaram M, Li J, Egeblad M, Powers RS (2013) System-Wide Analysis Reveals a Complex Network of Tumor-Fibroblast Interactions Involved in Tumorigenicity. PLoS Genet 9: e1003789.
- Korpal M, Yan J, Lu X, Xu S, Lerit DA, et al. (2009) Imaging transforming growth factor-[beta] signaling dynamics and therapeutic response in breast cancer bone metastasis. Nat Med 15: 960-966.