Hyperglycemia is linked to many inflammatory, metabolic and malignant diseases. High glucose provides inflammatory and cancer cells with more abundant “fuel” that promotes the cell motility, proliferation and production of pro-inflammatory mediators. The “malicious behavior” of activated inflammatory cells and cancer cells is further exacerbated by over-expression of chemoattractant receptors, notably FPRs (mouse Fprs) and tyrosine kinase receptors (TKRs) that are traditionally discovered as mediators of cell migration in response to a number of pathogen and host-derived chemotactic molecular patterns (PMAPs and DAMPs) existing at the diseased sites. In addition, the M1 macrophage polarizing capacity of one of FPRs, Fpr2, acts as a double-edged sword that exacerbates the insulin resistance and obesity in high-fat diet-fed mice. Therefore, while controlling glucose to a physiological level is important, targeting cell surface FPRs and TKRs should also be critical to manage hyperglycemia-associated disease conditions.
Hyperglycemia (or high glucose, HG, as is often referred to), in addition to be a characteristic feature of diabetes mellitus, occurs when human body system fails to maintain the normal serum glucose under 6 mmol. Hyperglycemia initiates the release of cortisol, glucagon, and growth hormones leading to increased insulin resistance, hyperinsulinemia, lipolysis, gluconeogenesis, and glycogenolysis that, in turn, exacerbating hyperglycemia as a vicious circle. The insulin resistance, needing higher-than-normal concentrations of insulin to promote the transport of circulating glucose into the cells [1], triggers body responses detrimental to overall body metabolism. In the presence of hyperglycemia, excessive reactive oxygen species (ROS) are generated, resulting in oxidative stress. Hyperglycemia also stimulates calcium flux into the cell, causing mitochondrial fragmentation, further generating high levels of ROS. These events disrupt normal cell metabolism, signaling, and cell-to-cell interactions in association with activation of downstream transcription factors, including the nuclear factor-kappa B (NF-kB), which induces the overexpression of inflammatory cytokines (IL-6, TNFα, and IFNγ, as a few), chemokines (such as CXCL8 and CCL2), and prostaglandins [2]. These inflammatory molecules perturb the function of immune system with excessive cell adherence, chemotaxis, phagocytosis, and apoptosis, preventing normal detection and elimination of infectious pathogens and damaged tissue. Moreover, hyperglycemia-associated inflammatory responses are a major contributor to the progression of cancer [3-5].
The hallmark of inflammation is leukocyte infiltration at disease sites in response to pathogen- or damage-associated chemotactic molecular patterns (PAMPs and DAMPs), which are recognized by a superfamily of seven transmembrane, Gi-protein-coupled receptors (GPCRs) on cell surface. Chemotactic GPCRs consist of two major subfamilies: the classical GPCRs and chemokine GPCRs. Formyl-peptide receptors (FPRs) belong to the classical chemotactic GPCR subfamily with unique properties that are increasingly appreciated for their expression on diverse host cell types and the capacity to interact with a plethora of chemotactic PAMPs and DAMPs. Three members of FPRs have been identified in human: FPR1–FPR3, with putative corresponding mouse counterparts (Fprs) [6,7]. FPR expression was initially described in myeloid cells but subsequently in many non-hematopoietic cells, including cancer cells. Accumulating evidence demonstrates that FPRs possess multiple functions in addition to controlling inflammation and participate in the processes of many pathophysiologic conditions. They are not only critical mediators of myeloid cell trafficking, but are also implicated in tissue repair, angiogenesis, and inflammation-associated tumorigenesis. Recent discoveries have verified and expanded the scope of FPRs in pathophysiological conditions by using genetically engineered mouse strains, indicating the essential roles of FPRs in step-wise trafficking of myeloid cells, including neutrophils and dendritic cells (DCs), in host responses to bacterial infection, tissue injury and wound healing. Also of great interest is that FPRs are exploited by cancer cells for their growth, invasion and metastasis [7,8].
The cellular expression of FPRs is subject to regulation by various stimulants, including bacterial molecular patterns and pro-inflammatory cytokines. The ability of high glucose (HG) to increase the transcription and translation of FPRs (mouse Fprs) in retinal Müller cells [9] and human glioblastoma multiforme (GBM) [10] raises much interest that posting FPRs in the loop of HG-induced inflammatory responses and cancer progression. In this mini-review, we list examples of HG regulation of FPR expression by retinal Müller glial cells (MGCs) and human GBM cells, as well as one of the FPRs, FPR2 (mouse Fpr2), in sustaining M1 polarization of macrophages that normally are involved in positive host response to pathogen and cancer [11] but as a dark side, increases host insulin tolerance of mice on high fat diet, therefore promoting obesity (Figure 1) [12].
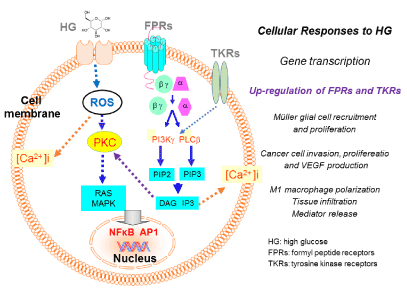
Figure 1. The effect of high glucose (HG) on Müller glial, glioblastoma cell and macrophage function.
HG activates intracellular ROS, which through a PKC-MAPK-mediated signaling cascade triggers translocation of the transcription factors NFκB and/or AP1 that increases the expression of formylpeptide receptors (FPRs). In Müller glial cells, an FPR variant FPR2 (Mouse Fpr2) mediates increased cell chemotaxis, thus recruitment, and proliferation in the retina, in response to an endogenous Fpr2 agonist peptide CRAMP. This process exacerbates the inflammatory conditions and the progression of diabetic retinopacy. In human glioblastoma cells, HG increases the expression and function of the prototype formylpeptide receptor FPR1, which promotes tumor cell invasion, proliferation and production of the angiogenic factor vascular endothelial cell growth factor (VEGF), by interaction with an agonist Annexin 1 (Anx A1) released by necrotic tumor cells. HG also elevates the expression and function of EGFR on glioblastoma cells and bFGFR on Müller cells. Both TKRs cooperation with FPRs to promote cell chemotaxis and proliferation.
Diabetic retinopathy (DR) is a severe complication of diabetes and the leading cause of blindness. Many abnormalities seen in the retina of diabetes patients are associated with inflammation [13]. Consequently, anti-inflammatory therapies significantly reduce the progression of DR in animal models. DR is staged based on levels of severity to mild, moderate, and severe non-proliferative DR, followed by advanced proliferative DR (PDR). In PDR, proliferative neo-vasculature and fibrovascular tissues extend from the retina into the vitreous. Tractional forces originating within these tissues cause vitreous hemorrhage, retinal scars, and retinal detachment, leading to irreversible vision loss. Although the origin and the process of fibrovascular membrane formation remain unclear, it is believed that inflammation linked to hyperglycemia is the basis for DR [14].
In PDR, activated MGCs exhibit increased motility and a fibroblast-like proliferation phenotype that contribute to the formation of fibrovascular membrane. Our investigation of the capacity of HG to regulate the expression of cell surface receptors that may participate in the proinflammatory responses of MGCs revealed that moue MGCs express Fpr2 and fibroblast growth factor receptor 1 (FGFR1), which mediate MGC migration and proliferation in response to their respective ligands. HG upregulates Fpr2 in MGCs through an NF-κB mediated pathway, increased the activation of MAPKs coupled to Fpr2 and FGFR1, which also further enhanced the production of vascular endothelial growth factor (VEGF) by MGCs. In vivo, Fpr2 was more highly expressed by retina MGCs of diabetic mice and the human counterpart FPR2 was detected in the retina MGCs in fibrovascular membrane of PDR patients. To support the potential pathological relevance of Fpr2, an endogenous Fpr2 agonist cathelin-related antimicrobial peptide (CRAMP) was detected in mouse MGCs and the retinal tissues. CRAMP was further upregulated by HG [9]. It is also reported that FPRs mediate inflammation and angiogenesis in PDR vitreous, which paves the way for targeting FPRs for anti-inflammatory/anti-angiogenic therapy of PDR [15].
GBM is the most common and aggressive subtype of gliomas with poor patient survival despite the treatment [16]. Putative risk factors for gliomas include aging, male gender and white ethnicity, without effective means of prevention [17]. Studies suggest that elevated glucose is associated with increased malignancy and the rate of recurrence of gliomas [18]. HG triggers intracellular pathways in tumor cells, which promote cancer progression, such as increased leptin levels and pro-cell survival AKT/mTOR, enhancement of WNT/β-catenin signaling, induction of epithelial mesenchymal transition (EMT), and up-regulation of inflammatory cytokine levels in circulation [19].
Our studies of the effect of HG on the growth of human GBM cells demonstrate that HG promotes the proliferation and inhibited the apoptosis of a human GBM cell line. Mechanistically, HG upregulates the expression and function of FPR1 and EGFR on GBM cells, by triggering a signaling cascade that involves MAPKs and NFκB. FPR1 and EGFR, upon activation by their agonists, promote GBM cell migration, proliferation and colony formation. In addition, HG increases the invasiveness and the production of VEGF by GBM cells, which are further augmented by FPR1 and EGFR agonists. The tumor promoting activity of HG was substantiated by increased tumorigenicity and growth of xenograft tumors formed by GBM cells in nude mice with chemically induced diabetes mellitus [10]. Thus, HG is clearly demonstrated to promote GBM progression via enhancement of the function of chemoattractant receptor FPR1 and the growth factor receptor EGFR.
Obesity and related inflammation are critical for the pathogenesis of insulin resistance. As discussed earlier, FPR2 (Mouse Fpr2) plays important roles in host immune responses and inflammation-associated diseases by mediating myeloid recruitment and M1 polarization of macrophages [11]. Studies of the contribution of Fpr2 to macrophage accumulation and polarization in white adipose tissue of high-diet (HFD) fed obese mice [12] show that systemic deletion of Fpr2 alleviated HFD-induced obesity, insulin resistance, hyperglycemia, hyperlipidemia, and hepatic steatosis. Furthermore, Fpr2 deletion in HFD-fed mice elevated body temperature, reduced fat mass, and inhibited inflammation by reducing macrophage infiltration and M1 polarization in metabolic tissues. Bone marrow transplantations between wild-type and Fpr2-/- mice and myeloid-specific Fpr2 deletion demonstrate the capacity of Fpr2-expressing myeloid cells to exacerbate HFD-induced obese syndrome. Mechanistically, HFD-fed Fpr2 deletion mice manifest enhanced energy expenditure probably through increasing thermogenesis in skeletal muscle. Serum amyloid A3 and other factors secreted by adipocytes induce macrophage chemotaxis via Fpr2, and Fpr2 deletion attenuates macrophage chemotaxis and lipopolysaccharide-, palmitate-, and interferon-γ-induced M1 polarization by inhibiting the signaling pathways coupled to signaling pathways of these molecules. Altogether, these results demonstrate Fpr2 in myeloid cells being critical for promoting obesity and related metabolic disorders.
Hyperglycemia has been linked to many inflammatory, metabolic and malignant diseases. High glucose per se provides inflammatory and cancer cells with more abundant “fuel” that promote the cell motility, proliferation and production of pro-inflammatory mediators. Meanwhile, the more active function of inflammatory cells and increased malignant behavior of cancer cells are further exacerbated by over-expression of chemoattractant GPCRs, notably FPRs, that are traditionally discovered as mediators of cell migration in response to a number of pathogen and host-derived chemotactic molecular patterns (PMAPs and DAMPs) existing at the diseased sites. In addition, HG also up-regulates the expression of functional TKRs (FGFR and EGFR) that act cooperatively with FPRs (Fprs) to enhance cell motility and growth. Furthermore, the M1 macrophage polarizing capacity of Fpr2 clearly indicates the receptors as a double-edged sword that requires careful tuning of its function in anti-tumor responses versus obesity promoting function. Therefore, the biological consequences of hyperglycemia are multi-facet with several layers of consideration in therapeutic development. To boil down, while controlling glucose to a physiological level is important, targeting cell surface FPRs and TKRs should also be critical to keep the inflammation and tumor progression in check.
The authors thanks Ms. Cheri Rhoderick for secretarial assistance. This project has been funded in part by Federal funds from the National Cancer Institute (NCI), National Institutes of Health (NIH), under Contract No. HSN261200800001E, and is also supported in part by the Intramural Research Program of NCI, NIH. Zhiyao Bao was also supported in part by grant No. 81600004 from the National Nature Science Foundation of China and grant No. YG2016QN63 from Shanghai Jiaotong University. Yingying Le was supported by the National Natural Science Foundation of China (31671232). YY was supported in part by fundings from National Natural Science Foundation of China 81700853 and China Postdoctoral Science Foundation 2019M651927.
- Park SE, Park CY, Sweeney G (2015) Biomarkers of insulin sensitivity and insulin resistance: Past, present and future. Crit Rev Clin Lab Sci 52:180-190. [Crossref]
- Esposito K, Nappo F, Marfella R, Giugliano G, Giugliano F, et al. (2002) Inflammatory cytokine concentrations are acutely increased by hyperglycemia in humans: role of oxidative stress. Circulation 106: 2067-2072. [Crossref]
- Tsalamandris S, Antonopoulos AS, Oikonomou E, Papamikroulis GA, Vogiatzi G, et al. (2019) The Role of Inflammation in Diabetes: Current Concepts and Future Perspectives. Eur Cardiol 14: 50-59. [Crossref]
- Chang SC, Yang WV (2016) Hyperglycemia, tumorigenesis, and chronic inflammation. Crit Rev Oncol Hematol 108: 146-153. [Crossref]
- Hammer M, Storey S, Hershey DS, Brady VJ, Davis E, et al. (2019) Hyperglycemia and Cancer: A State-of-the-Science. Review. Oncol Nurs Forum 46: 459-472. [Crossref]
- Krepel SA, Wang JM (2019) Chemotactic ligands that activate G-protein-coupled formylpeptide receptors. Review Int J Mol Sci 20: 3426.
- Chen K, Bao Z, Gong W, Tang P, Yoshimura ,et al. (2017) Regulation of inflammation by members of the formyl-peptide receptor family. J Autoimmun 85: 64-77. [Crossref]
- Zhou J, Xiang Y, Yoshimura T, Chen K, Gong W, et al. (2014) The role of chemoattractant receptors in shaping the tumor microenvironment. Biomed Res Int 2014: 751392. [Crossref]
- Yu Y, Bao Z, Wang X, Gong W, Chen H, et al. (2017) The G-protein-coupled chemoattractant receptor Fpr2 exacerbates high glucose-mediated proinflammatory responses of Müller glial cells. Front Immunol 8: 1852. [Crossref]
- Bao Z, Chen K, Krepel S, Tang P, Gong W, et al. (2019) High glucose promotes human glioblastoma cell growth by increasing the expression and function of chemoattractant and growth factor receptors. Transl Oncol 12: 1155-1163. [Crossref]
- Liu Y, Chen K, Wang C, Gong W, Yoshimura T, et al. (2013) Cell surface receptor FPR2 promotes antitumor host defense by limiting M2 polarization of macrophages. Cancer Res 73: 550-560. [Crossref]
- Chen X, Zhuo S, Zhu T, Yao P, Yang M, et al. (2019) Fpr2 Deficiency Alleviates Diet-Induced Insulin Resistance Through Reducing Body Weight Gain and Inhibiting Inflammation Mediated by Macrophage Chemotaxis and M1 polarization. Diabetes 68: 1130-1142. [Crossref]
- Roodhooft JM (2002) Leading causes of blindness worldwide. Bull Soc Belge Ophtalmol 283: 19-25. [Crossref]
- Ting DS, Cheung GC, Wong TY (2016) Diabetic retinopathy: global prevalence, major risk factors, screening practices and public health challenges: a review. Clin Exp Ophthalmol 44: 260-277. [Crossref]
- Rezzola S, Corsini M, Chiodelli P, Cancarini A, Nawaz IM, et al. (2017) Inflammation and N-formyl peptide receptors mediate the angiogenic activity of human vitreous humour in proliferative diabetic retinopathy. Diabetologia 60:719-728. [Crossref]
- Ostrom QT, Gittleman H, Fulop J, Liu M, Blanda R, et al. (2015) CBTRUS Statistical Report: Primary brain and central nervous system tumors diagnosed in the United States in 2008-2012. Neuro Oncol 17: 1-62
- Chaichana KL, McGirt MJ, Woodworth GF, Datoo G, Tamargo RJ, et al. (2010) Persistent outpatient hyperglycemia is independently associated with survival, recurrence and malignant degeneration following surgery for hemispheric low grade gliomas. Neurol Res 32: 442-448. [Crossref]
- Chambless LB, Parker SL, Hassam-Malani L, McGirt MJ, Thompson RC (2012) Type 2 diabetes mellitus and obesity are independent risk factors for poor outcome in patients with high-grade glioma. J Neurooncol 106: 383-389. [Crossref]
- Scappaticcio L, Maiorino MI, Bellastella G, Giugliano D, Esposito K (2017) Insights into the relationships between diabetes, prediabetes, and cancer. Endocrine 56: 231-239. [Crossref]