Airborne and potentially deadly SARS-CoV-2, that causes the disease COVID-19, was discovered in late December of 2019. Till now no medications including vaccine, antibody, or any antiviral are found with success. SARS-CoV-2 is very similar to SARS-CoV-1 which was discovered in 2003, and recognizes the same host cell receptor for entry into the cell. MERS–CoV, another lethal HCoV discovered in 2012, belongs to the same group of SARS (β-type), but recognizes a different cell receptor for host cell entry.
All these viruses can only be studied safely under high-level biosafety conditions to protect the laboratory workers and the environment. However, these safety precautions slow down efforts to find drugs and vaccines for COVID-19 since many scientists lack access to the required biosafety facilities.
There are four more common human CoVs, such as, HCoV-229E, HCoV-OC43, HCoV-HKU1, and HCoV-NL63, which were known from many years back. They cause self-limiting respiratory infections, such as common cold, in humans, and can be studied safely in BSL2 lab.
In this review, a comparison of SARS and Common cold virus were done in order to search for a better surrogate virus those can be used in BSL2 lab for identifying the disease mechanism and therapeutic intervention of SAR-CoV-2. We focused on the following key questions, like, virus-host cell interaction mechanism; the differential cell line susceptibility; species tropism; viral replication efficiency; antigen expression patterns; mechanistic pathway of apoptosis; and structure-function relationship of the virus.
SARS, NL-63, COVID-19, therapeutics, Sars-CoV-2, antiviral
Coronaviruses (CoVs) are a diverse group of enveloped positive-strand RNA viruses in the family Coronaviridae [1,2]. CoVs have been identified in a wide variety of hosts, including mammals and birds, and are shown to cause a number of respiratory and enteric diseases [1,3,4]. Four coronaviruses are continuously circulating in the human population from long time back, causing common cold, and they are HCoV-229E, HCoV-OC43, HCoV-HKU1, HCoV-NL63 [5-8].
The seasonality of HCoV-OC43, HCoV-229E and HCoV-NL63 tend to be mainly in the winter season [9-15], but in Hong Kong, HCoV-NL63 show a spring-summer peak of activity, also [16]. Until recently, it was commonly accepted that the known human coronaviruses (HCoVs), with the exception of SARS-CoV, mainly cause mild upper respiratory tract infections (URTIs) [9]. For this reason, the circulation of HCoVs was not monitored and no attempt to develop vaccines or drugs against these viruses was made [10].
In 2003, the global epidemic of an atypical form of pneumonia named severe acute respiratory syndrome (SARS), outbreak in south China, led to the discovery of SARS-CoV as an etiologic pathogen [17-20]. Since outbreak, SARS caused more than 800 deaths (~10%) worldwide [21]. Patients with SARS-CoV infection developed diffuse alveolar damage with the potential to progress into acute respiratory distress syndrome and eventually death [22].
Almost 10 years later, another previously unknown CoV, Middle East respiratory syndrome coronavirus (MERS-CoV), was found to cause a new epidemic starting in the Arabian Peninsula in 2012 [23-25]. MERS infection led to acute pneumonia and renal failure, with mortality rate as high as 50% in hospitalized patients [26,27]. Very recent, in late December of 2019, another highly infectious corona virus (SARS-CoV-2) was identified which so far infected 32 million people globally and causes death about 1 million, as of Sep 27, 2020. (https://www.worldometers.info/coronavirus/?utm_campaign=homeAdvegas1). We do not have, yet, any antiviral or vaccines against this virus to protect from the disease, COVID-19.
Here we tried to draw a comparison between low pathogenic common cold viruses and high pathogenic SARS viruses to find the similarities as well as dissimilarities in the mechanism of infection and replication inside the cells. This will help us to find a better and safe surrogate virus to work in a BSL-2 type laboratories.
Comparison of SARS and common cold virus
Classifications: CoVs are ssRNA viruses that infect humans and animals. According to their genomic sequences, these seven HCoVs are further classified into alpha-coronavirus genus (HCoV-229E and HCoV-NL63) and beta-coronavirus genus (HCoV-OC43, HCoV-HKU1, SARS-CoV-1, MERS-CoV, and SARS-CoV-2) [24,28].
Zoonosis: The emergence of CoV infection in human beings are believed to begin with zoonotic transmission from animal reservoirs [21]. For example, high degree of genomic sequence similarity was shown between bovine CoV and HCoV-OC43, suggesting an animal-to-human transmission [23,29]. In the case of human SARS-CoV, over 95% genomic sequence identity was found with bat CoVs, suggesting bats as the potential zoonotic for the SARS viruses.
Host cell receptor recognition: Angiotensin-converting enzyme 2 is the cellular receptor for SARS-CoV-1 and SARS-CoV-2 [30-33], while MERS-CoV, though belongs to the same Group of SARS (β type), recognizes Dipeptidyl peptidase 4 (DPP4) instead of ACE-2 receptor [34]. The cellular receptors for other two α-type CoVs, HCoV-OC43 and HCoV-HKU, have not been confirmed yet, but thought to be a 9-O-Acetylated sialic acid [31,35].
HCoV-229E and HCoV-NL63, both belongs to group 1 (α type) coronaviruses, but the first one while utilizes aminopeptidase N (CD13) as its cellular receptor [36, 37], the second one (HCoV-NL63) utilizes the ACE2 receptor like SARS-CoV to infect cells [34]. A comparison of some properties of common cold viruses and other severe acute respiratory syndrome causing viruses (SARS) were depicted in Table-1.
Table 1. Human Coronavirus and it’s Different types [38]
|
Common Human Coronaviruses
(Less Pathogenic) |
Other Human Coronaviruses
(Severe Disease Causing Pathogens) |
|
HCoV-229E |
HCoV-NL63 |
HCoV-OC43 |
HCoV-HKU1 |
HCoV-SARS-1 |
HCoV-MERS |
HCoV-SARS-2 |
Zoonosis |
Bats à
Camels (?) à
Human |
Bats à
(?) à
Human |
Rodents à
Bovines à
Human |
Rodents à
(?) à
Human |
Batsà
Palm
Civets à
Human |
Batsà
Dromadery
Camels à
Human |
Batsà
Pangulins à
Human |
Classifications |
α-type |
α-type |
β-type |
β-type |
β-type |
β-type |
β-type |
Incubation Period |
2-5 days |
2-5 days |
2-5 days |
2-5 days |
2-11 days |
2-13 days |
3-6 days |
Clinical Symptoms |
Malaise, Headache, Sneezing, Nasal discharge, Sore throat, Fever and Cough |
Cough. Fever, Hypoxia, Croup, Rhinorrhea, Tachypna |
Malaise, Headache, Sneezing, Nasal discharge, Sore throat, Fever and Cough |
Fever, Running Nose, Cough, Dyspnea |
Fever, Myalgia, Headache, Dry Cough, Respiratory Distress, Diarrhea, Dyspnea |
Fever, Myalgia, Headache, Dry Cough, Respiratory Distress, Diarrhea, Dyspnea, Pneumonia |
Fever, Myalgia, Headache, Dry Cough, Respiratory Distress, Diarrhea, Dyspnea, Pneumonia |
Fatality rates |
None |
None |
None |
None |
~10% |
~35% |
~3% |
Host Cell Receptor |
Amino peptidase |
Angiotensin Converting enzyme-2 (ACE2) |
9-O Acetylated Sialic acid |
9-O Acetylated Sialic acid |
Angiotensin Converting enzyme-2 (ACE2) |
Dipeptidyl peptidase 4 (DPP4) |
Angiotensin Converting enzyme-2 (ACE2) |
References |
[8] |
[4] |
[31, 35] |
[31, 35] |
[17] |
[34] |
[39, 40] |
Among the above three high pathogenic CoVs, SARS-CoV-1 and SARS-CoV-2 are phylogenetically much more similar to each other than MERS-CoV [41]. The both SARS-CoVs contain the largest genomes of all RNA viruses [18]. The genomic sequences of SARS-CoV-2 and SARS-CoV-1 have extremely high homology at the nucleotide level, and both requires a highly specific lab to study further with these viruses.
Therefore, we focused here on NL-63, a low-pathogenic common cold virus and allowed to work in a BSL2 lab to study for SARS, as they share the same receptor molecule (ACE2).
Identification of HCoV-NL63
In January 2003, a 7-month-old child appeared in an Amsterdam hospital with coryza, conjunctivitis and fever. Chest radiography showed typical features of bronchiolitis and a nasopharyngeal aspirate specimen was collected 5 days after the onset of disease (sample NL63). Diagnostic tests for all known respiratory viruses were negative. The clinical sample was inoculated onto tertiary monkey kidney cells (tMK; Cynomolgus monkey) and a cytopathic effect was observed. The infectious agent could subsequently be passaged onto LLC-MK2 cells (a monkey kidney cell line), and finally was identified as a novel coronavirus, and named as NL-63 [5]. The initial PCR products showed sequence similarity to the genome sequence of members of the Coronaviridae family, and the complete genome sequence revealed that this virus was not a recombinant one rather a novel member of the group I coronaviruses [42, 43] (Figure 1).
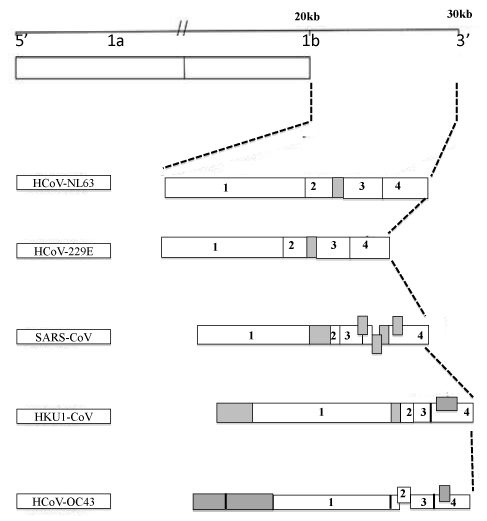
Figure 1. Schematic comparison of the genome organization of human coronaviruses. HCoV-NL63: Human coronavirus HCOV-NL63 (NC_005831); HCoV-229E: Human coronavirus 229E (NC_002645); SARS-CoV: Severe acute respiratory syndrome coronavirus (NC_004718); HKU1-CoV: Human coronavirus HKU1 (NC_006577); HCoV-OC43: Human coronavirus OC43 (NC_005147). ORFs S (1), E (2), M (3) and N (4) are shown, and open reading frames encoding for accessory genes are shown in grey. [6,7]. [Dijkman R, Jebbink MF, Wilbrink B, et al. Human coronavirus 229E encodes a single ORF4 protein between the spike and the envelope genes. Virol J 2006; 3: 106; Abdul-Rasool and Fielding. The Open Virology Journal, 2010, 4, 76-84]
Respiratory clinical findings
Scientific and clinical evidence show that HCoV-NL63 infects both the upper and lower respiratory tract [44], causing symptoms similar to those associated with HCoV-229E and HCoV-OC43.
Nonrespiratory clinical findings: HCoV-NL63 infections have previously been associated with gastrointestinal findings [32,33,45-47]. However, this is not unique to HCoV-NL63 infections, as SARS-CoV and HCoV-HKU1 RNA have been previously detected in patient diarrheic samples [48, 49]. Nontheless, these manifestations appear to be a direct consequence of viral invasion of the intestinal mucosa [10].
Receptor-mediated cell entry of the corona virus
Coronaviruses bind to the cellular receptors via the spike protein to mediate infection of specific target cells. The spike of HCoV-NL63 is a class I fusion protein, similar to the influenza virus haemagglutinin and the HIV-1 Env glycoprotein gp120/gp41. The amino-terminal part of the spike protein (S1) contains the receptor-binding domain, and the carboxy-terminal part (S2) contains a membrane spanning region. The S1 domain mediates an initial high affinity association with the cellular receptor [50- 53], whereas the C-terminal S2 domain mediates fusion of the viral and cellular membranes. The coronavirus fusion peptide is located internally within S2 [54].
Before the discovery of HCoV-NL63, it was generally thought that all group I coronaviruses use CD13 (also known as aminopeptidase N) as receptor, as it was described for HCoV-229E. Unexpectedly, HCoV-NL63 is not able to use CD13 as a receptor for cell entry [34], but uses unusually ACE-2 receptor. Further, the virus replicates in monkey kidney cells [5,14], whereas its closest relative, HCoV-229E, does not.
Intriguingly, SARS-CoV being from a different coronavirus group (group IIβ), is also able to replicate in monkey kidney cells [5]. Huh-7 and 293T cells express SARS-CoV receptor ACE2 [31, 44, 49, 55], and thus are permissive to both NL63- and SARS-CoV-S-driven infection; whereas CEMx174, HeLa, and HOS cells are not permissive [44,56,57]; and do not express ACE2 [44, 49].
Further, Purified antibodies against the ectodomain of ACE1 did not modulate infection of Huh-7 cells by pseudotypes bearing 229E-, NL63-, or SARS-CoV-S [34]. In contrast, purified antibodies against the ectodomain of ACE2 or preincubation of pseudovirions with soluble ACE2 ectodomain potently blocked infection driven by NL63- and SARS-CoV- but not by 229E-S protein, indicating that NL63-S employs ACE2 for infectious cellular entry. Further, CD13 rendered 293T cells highly permissive to infection driven by the S protein of HCoV-229E but not HCoV-NL63 or SARS-CoV. All these information confirms that despite the similarity between 229E- and NL63-S proteins, the latter engages ACE2 and not CD13 like the 229-E for cellular entry [34].
FACS analysis employing soluble S1 domains of NL63- and SARS-CoV-S revealed the binding of NL63-S1 to cells expressing ACE2 but not empty vector, indicating that ACE2 and NL63-S protein directly interact. Of note, SARS-CoV-S bound more efficiently to ACE2- expressing cells than NL63-S, which could be indicative of a higher binding affinity. In brief, these data and the shared cell tropism is suggestive of a shared receptor (ACE2) usage by HCoV-NL63 and SARS-CoV [31]. This ACE2 surface molecule is localized to the ciliated cells of human nasal and tracheobronchial airway epithelia, thus supporting the presence of virus infection in the upper airways [35].
The reduced pathogenicity of HCoV-NL63 suggests that ACE2 binding by the virus is not the only factor that determines the severity of viral pathogenicity. Further research on this subject is warranted, especially on the modulation of the ACE2 expression levels during HCoV-NL63 infection, to understand the difference in lung pathogenicity of SARS-CoV and HCoV-NL63.
Receptor engagement is conferred by the N-terminal S1 subunit; consequently, animal and human CoVs exhibit the same functional organization, particularly the S1 subunits differ in amino acid sequence, resulting in interaction with specific cellular receptors. However, cells expressing the SARS-CoV receptor protein ACE2 were susceptible to NL63-S driven infection [14].
This was unexpected as NL63-S has no striking homology to either the whole S1 subunit of SARS-CoV or the already identified ACE2 interaction domain in SARS-CoV-S [15], suggesting that both proteins either form a common three-dimensional structure that allows ACE2 engagement in a similar fashion or that both S-proteins evolved different strategies to target ACE2.
Direct interaction between ACE2 and NL63-S: The interaction between NL63-S and ACE2 was specific. The closely related ACE1 protein did not react with NL63-S, and on the other hand, ACE2 was not able to confer 229E-S-mediated infection, suggesting that ACE2 is not a functional equivalent of fAPN in class I CoV entry.
HCoV-NL63 entry is sensitive to some changes in ACE2 that interfere with, or are adjacent to residues critical for SARS-CoV entry. In addition, the SARS-CoV RBD can inhibit HCoV-NL63 S-protein-mediated infection [39]. These information indicate that the SARS-CoV and HCoV-NL63 binding sites on ACE2 overlap. In contrast, Hoffman et al., using a different panel of ACE2 variants, tentatively concluded that these viruses (SARS-CoV and HCoV-NL63) have distinct binding sites because they did not identify variants that interfered their entry [58].
Analysis of hCoV-NL63-S1 deletion mutants and chimeric hCoVNL63-229E-S variants: In order to map which region in the NL63 S1-protein is responsible for targeting ACE2, an analysis of a panel of N-terminal S1-deletion mutants were done, suggesting that the central region in the hCoV-229E-S and possibly NL63-S proteins might determine the correct folding or orientation of a C-terminal receptor binding domain, as has been suggested previously for hCoV-229E-S [53,59].
Taken together, a detailed point mutagenesis of NL63-S1 will be required to identify residues with a critical function in ACE2 interaction.
pH-dependent cell entry: The internalization of coronaviruses into the host cell occurs either by direct fusion with the plasma membrane or by endocytosis and subsequent fusion with the endosomal membrane. Viruses that use the latter entry route can be inhibited by lysosomotropic agents that lower the endosomal pH such as bafilomycin A, chloroquine and NH4Cl.
Treatment of Huh-7 cells with bafilomycin A or NH4Cl revealed that entry driven by NL63-S protein depends on the low-pH environment in intracellular vesicles [34]. The same has been described for HCoV-229E infection and SARS-CoV-S mediated infection [60, 44, 56]. However, there is also one opposite result that NL63-S mediated infection of ACE2 expressing-HEK293T cells was not greatly influenced by NH4Cl [61].
Cathepsin helps virus infection: Cathepsins are a diverse group of endosomal and lysosomal proteases that include aspartyl, serine, and cysteine proteases with both endo- and exopeptidase activities [62]. The role of cathepsins in reovirus infection is well established [63-67]. Following receptor-mediated endocytosis, the reovirus virion is converted to an infectious subvirion particle by partial proteolysis, mediated by cathepsins B, L, or S. One or more of these enzymes degrades the reovirus outer capsid protein σ3, exposing the underlying µ1 protein, which mediates penetration of endosomal membranes [68-70]. These indicate that two coronaviruses that utilize a common receptor nonetheless enter cells through distinct mechanisms.
Because cathepsin inhibitors can cross-react, an investigation was done to find out the consequences of exogenous cathepsin B, L, and S expression in 293T cells that express human ACE2. Exogenous cathepsin L markedly increased infection by SARS/MLV but had no effect on NL63/MLV or VSV-G/MLV. Similarly, exogenous cathepsin S also modestly enhanced SARS/MLV infection but, surprisingly, inhibited NL63/MLV infection. This indicate that overexpressed cathepsin S, which is secreted and active at neutral pH, may digest and inactivate the HCoV-NL63 S protein but not that of SARS-CoV.
Collectively, these data show that introduction of cathepsin L into cells where this enzyme is limiting or absent can substantially boost infection mediated by the SARS-CoV but not the HCoV-NL63 S protein [71] (Table 2). Cathepsin S also appears to contribute modestly to SARS-CoV infection and may partially compensate for the absence of cathepsin L in some cells [71]. It remains to be investigated whether other cellular proteases contribute to HCoV-NL63 infection through a mechanism analogous to the role played by cathepsin L in SARS-CoV infection or whether HCoV-NL63, like HIV-1 and VSV, infects cells independently of target-cell proteases.
Table 2. SARS Coronavirus, but Not NL63, Utilizes Cathepsin L to Infect ACE2- expressing Cells [71]
Angiotensin-converting enzyme 2 (ACE2) |
SARS-CoV |
HCoV-NL63 |
Lysosomal cysteine proteases |
Utilizes the enzymatic activity of the cysteine protease |
Doesn’t utilize the enzymatic activity of the cysteine protease |
Cathepsin L |
Cathepsin L to infect ACE2-expressing cells. |
Cathepsin L to infect ACE2-expressing cells. |
Inhibitors of
Cathepsin L |
Blocked infection by SARS-CoV |
No infection by HCoV-NL63 |
Expression of exogenous Cathepsin L |
Substantially enhanced infection mediated by the SARS-CoV S protein |
No infection by the HCoV-NL63 S protein |
Inhibitor of endosomal acidification |
Has much effect on infection mediated by the SARS-CoV S protein |
Less effect on infection mediated by the HCoV-NL63 S protein |
NL-63 and SARS-CoV: Differential Down regulation of ACE2 by the spike proteins
Several lines of evidence suggest that ACE2 plays a key role in SARS-CoV spread: (i) ACE2 expression in cell lines correlates with susceptibility to SARS-CoV spike protein (SARS-S)-driven entry [34,49], and (ii) knockout of ACE2 in mice abrogates permissiveness to SARS-CoV infection [30].
Notably, it has recently been proposed that binding of SARS-CoV but not NL63 to ACE2 induces ACE2 shedding from the cell surface, and evidence has been presented that this process is required for cellular uptake of SARS-CoV [72]. Whether ACE2 shedding is indeed a pre-requisite to infectious entry and contributes to the previously observed ACE2 down regulation by SARS-S remains to be determined.
In addition, it is unclear if SARS-S and NL63-S differentially interfere with ACE2 expression, which might contribute to the differential pathogenicity of these viruses. Binding studies in an enzyme-linked immunosorbent assay (ELISA) format revealed highly efficient capture of recombinant ACE2 by SARS-S, while binding of ACE2 to NL63-S was barely detectable. Finally, preincubation of ACE2-transfected cells with recombinant SARS-S blocked subsequent infection with lentiviruses pseudotyped with SARS-S and NL63-S, while inhibition with recombinant NL63-S was much less pronounced. In summary, it indicates that SARS-S binds to ACE2 with higher efficiency than NL63-S [31,73].
A direct comparison of ACE2 down regulation in SARS-CoV and NL63infected cultures was not possible due to the differential replication efficiencies. However, it can be speculated that relatively inefficient ACE2 engagement by NL63 might contribute to the reduced replication and ACE2 down modulation compared to SARS-CoV. However, ACE2 shedding is a mere byproduct of SARS-CoV and NL63 infection and is not required for infectious entry.
Crystal structure
NL63 and SARS-CoV receptor-binding domain complexed with its host cell receptor: NL63-CoV and SARS-CoV have no structural homology in RBD cores or RBMs; yet the 2 viruses recognize common ACE2 regions, largely because of a ‘‘virus-binding hotspot’’ on ACE2. Among group I coronaviruses, RBD cores are conserved but RBMs are variable, explaining how these viruses recognize different receptors. These results provide a structural basis for understanding viral evolution and virus–receptor interactions.
A fundamental yet unresolved puzzle in virology is how viruses evolve to recognize their receptor proteins [74]. Specifically, how do different viruses recognize the same receptor protein, and how do similar viruses recognize different receptor proteins?
To date, the crystal structure of SARS-CoV RBD complexed with ACE2 is the only atomic structure available for any coronavirus S1 [75]. SARS-CoV RBD contains two subdomains, a core and a receptor-binding motif (RBM), which exclusively contacts ACE2. ACE2 contains a claw-like peptidase domain, with 2 lobes encircling the active site. SARS-CoV binds to the outer surface of the N-terminal lobe of the ACE2 peptidase domain. Structural information has been lacking for group I coronavirus S1, either alone or in complex with its receptor.
The most Important Qs is, despite of having no obvious sequence homology in their S1 Subunits, how do NL63-CoV and SARS-CoV both use ACE2 as their receptor? One hypothesis is that NL63-CoV and SARS-CoV share homologous RBMs, and that through RNA recombination, SARS-CoV acquired its RBM from NL63-CoV or an NL63-CoV–related group I coronavirus, gaining binding affinity for ACE2 and infectivity for human cells [39,76]. The recent analysis of crystal structure revealed that lack of structural homology in RBD cores and in RBMs, indicate 2 independent ways in which NL63-CoV and SARS-CoV recognize their common receptor protein [77].
A common virus-binding hotspot on ACE2: A virus-binding hotspot on ACE2 lies at the center of the NL63-CoV–receptor interface. In the structure of unbound ACE2, Lys-353 projects into solution [78]. Upon NL63-CoV binding, Lys-353 becomes embedded in a hydrophobic tunnel surrounded by 2 aromatic rings of ACE2 Tyr-41 and NL63-CoV Tyr-498 and by 2 alkyl chains of ACE2 Asp-37 and NL63-CoV Ser-535 [77]. At the end of the tunnel, ACE2 Asp-38 forms a salt bridge with Lys-353, neutralizing its charge. Because of the hydrophobic environment, this salt bridge is energetically stabilizing [78], and critical for virus–receptor interactions. Alanine substitutions for Lys- 353 or any other residues involved in the hotspot structure abolish NL63-CoV binding [39,79]. The same virus-binding hotspot on ACE2 is also key to the binding of SARS-CoV. The structures of the hotspots at the 2 different virus–receptor interfaces are strikingly similar [77]. The receptor parts are nearly identical, with subtle changes in protein side chain conformations. In the viral parts, Thr-487 and Tyr-491 on SARS-CoV replace Ser-535 and Tyr-498 on NL63-CoV, respectively, as 2 of the 4 tunnel walls. Adaptation to the hotspot on ACE2 is critical for SARS-CoV pathogenesis.
Therefore, virus-binding hotspots on receptor proteins likely dictate virus–receptor interactions, viral pathogenesis, and viral transmissibility and thus are potentially major binding targets for viruses.
Binding characteristics of ACE2 with NL63 and SARS
ACE2 bears an aspartic acid at this position, whereas the ACE2 of most animals, including human, rat, mouse, cat, and dog express a glycine [80,81]. Similarly, SARS-CoV, HCoV-NL63 S1-Ig (301–749) did not bind rat ACE2 efficiently, but bound the D354G form of palm civet ACE2 more efficiently than human ACE2. However, in striking contrast to the SARS-CoV S1, HCoV-NL63 S1-Ig (301–749) did not bind palm civet ACE2 with the native aspartic acid at residue 354. Therefore, residues in the immediate vicinity of glycine 354 likely contribute to HCoV-NL63 association.
Further, introduction of a glycosylated region of rat ACE2 (residues 82–84; denoted MYP/NFS) into the human protein modestly decreased binding of the SARS-CoV S1 [81]. This modification similarly decreased HCoV-NL63 S1 binding. Again, the HCoV-NL63 S protein followed the same pattern. However, introduction of an aspartic acid at human ACE2 residue 354 had only a modest effect on SARS-CoV S1 association, whereas it completely abolished association with HCoV-NL63 S1. Thus residue 354 modulates HCoV-NL63 S-protein association with both human and palm civet ACE2. The ability of those human ACE2 variants to bind HCoV-NL63 and SARS-CoV S1 was reflected in their ability to support infection mediated by the S proteins of these viruses [81]. Several variants that less efficiently bound the SARS-CoV S1 domain also less efficiently bound HCoV-NL63 S1-Ig [81].
Collectively, these data indicate that the SARS-CoV and HCoV-NL63 S1 domains bind regions of ACE2 that largely overlap. Further, the SARS-CoV RBD inhibits infection mediated by the SARS-CoV and HCoV-NL63 S proteins [77].
Replication of NL-63
Coronaviruses employ posttranslational proteolytic processing as a key regulatory mechanism in the expression of their replicative proteins. The HCoV-NL63 1a and 1ab polyproteins are potentially cleaved by viral proteases to facilitate the assembly of a multi-subunit protein complex that is responsible for viral replication and transcription. The genome of HCoV-NL63 is predicted to encode two proteinases in the 50 region of the 1a polyprotein [82]. First, a papain-like proteinase (PLpro) is expressed by the nonstructural protein (nsp) 3 gene situated near the 5’ end of the genome. This putative papain-like proteinase of HCoV-NL63 consists of two domains, PL1pro and PL2pro, and both are expected to have catalytic activities.
The enzyme is predicted to cleave the 1a/1b protein at three sites between nsp1|nsp2, nsp2|nsp3, and nsp3|nsp4, releasing the functional papain-like proteinase protein (nsp3) molecule by auto-cleavage. Analysis of the predicted cleavage sites of PLpro indicates that this HCoV-NL63 enzyme has the specificity to cut between two small amino acids with short uncharged side chains, similar to homologous enzymes in other coronaviruses [83].
Therapeutic aspects
Antiviral agents: Several inhibitors are known to reduce replication of at least some coronaviruses including HCoV-NL63 [84,85]. These inhibitors act at various steps of the coronavirus replication cycle, e.g. receptor binding, membrane fusion, transcription and posttranslational processing. An interesting HCoV-NL63 inhibitor is intravenous immunoglobulin [83], which is already approved as an intravenously delivered drug by the Food and Drug Administration. Intravenous immunoglobulin has been used successfully to treat several diseases, mostly primary immune deficiencies and autoimmune neuromuscular disorders, but also respiratory diseases (e.g. RSV) [86], and Kawasaki disease [84].
Inhibition of viral replication can also occur at the level of fusion of the viral and cellular membranes. The spike of HCoV-NL63 contains two heptad repeat regions, HR1 and HR2, situated in the S2 part of the spike protein close to the transmembrane domain. After binding of virus to the receptor, a conformational change leads to the formation of a six-helix bundle containing three HR1s and three HR2s and subsequent exposure of the fusion peptide mediates membrane fusion between the virus and the host cell. For retroviruses, paramyxoviruses and coronaviruses [85,87], peptides derived from the HR2 domain can inhibit virus infection, most likely by interacting with HR1. The peptide thus blocks formation of the natural HR1–HR2 interaction, prevents membrane fusion and as a consequence reduces infection.
Another novel means to inhibit replication is RNA interference (RNAi) [88]. Pyrc et al. (2006a) explored the antiviral potential of small interfering RNA (siRNA) targeting HCoV-NL63 [85]. The inhibitory properties of two siRNAs targeting conserved sequences within the spike protein gene were analyzed in cell culture infections. Transfection of a relatively low amount of siRNA into HCoV-NL63-susceptible cells made them resistant to virus infection.
HCoV-NL63 can also be inhibited at the transcriptional level by pyrimidine nucleoside analogues: α-D-N4-hydroxycytidine and 6-azauridine [85]: The exact mechanism by which these agents inhibit HCoV-NL63 transcription is unclear. Generally, protease inhibitors act at the level of posttranslational processing. The Mpro of coronaviruses has a highly conserved substrate recognition pocket, thus providing the opportunity to design broad-spectrum antiviral drugs against several coronavirus species. One potent inhibitor, N3, showed wide-spectrum inhibition of various Mpro enzymes, including the one encoded by HCoV-NL63 [89]. Through comparative study with Mpros from other human CoVs (including the deadly SARSCoV and MERS-CoV) and their related zoonotic CoVs, HCoV-NL63 Mpro structure may provides critical insight into rational development of wide spectrum antiviral therapeutics to treat infections caused by human CoVs.
The carboxypeptidase ACE2 is an important component of the renin–angiotensin system, which controls blood pressure [90,91]. ACE2 expression in lung and intestine explains important aspects of SARS-CoV tropism [13], and the protein likely plays a central role in SARS-CoV spread [80].
SARS-CoV and NL63 though belong to different groups β-type and α-type, respectively, use the same host-cell receptor ACE-2. However, the consequence of entry is very different. Severe respiratory distress in the case of SARS-CoV but frequently only a mild respiratory infection for NL63 are observed. Using a whole recombinant system, it was found that the NL63 S protein has a weaker interaction with ACE-2 than the SARS-CoV S protein, though the residues required for contact are similar. It was also confirmed that the ACE-2-binding site of NL63 S lies between residues 190 and 739, a lower-affinity binding site, and that may explain the different pathological consequences of infection by SARS-CoV and NL63 [5,18,20,31,92,93].
Further, the virus-mediated down regulation of ACE-2 has been suggested the underlie pathology of SARS-CoV infection [30,94], but it is unclear why this should not also be the case for NL63. Following engagement with ACE-2, the cellular pathways of internalization of the two viruses also appear to be different. SARS-CoV requires the presence of the lysosomal cysteine protease cathepsin L to infect susceptible cells, while NL63 has no such requirement [71].
It is not yet clear how cathepsin L facilitates SARS-CoV infection. Several compatible possibilities exist. Cathepsin L may serve to nonspecifically degrade the S1 domain of SARS-CoV S protein, thereby permitting conformational transitions in the S2 domain necessary for fusion. ACE2 could also be a cathepsin target, thereby facilitating its dissociation with the S protein. SARS-CoV infection can be limited by low cathepsin L expression in mature endothelial cells [95]. Studies determining whether HCoV-NL63 more efficiently infects these cells in vivo than SARS-CoV are warranted.
The above information suggest that although both viruses utilize ACE-2 as the receptor, the consequences of receptor binding differ, although the reasons for this remain unclear. Li et al. (2007) showed that incubation of a tagged form of the RBD with cell lines expressing a number of natural and synthetic ACE-2 variants indicated that the ACE-2 contact residues critical for binding both SARS-CoV and NL63 S overlap [39].
The binding site for both viruses is distinct from the active site of the enzyme [78,96], consistent with the fact that treatment of ACE-2-bearing cells with MLN-4760, a potent ACE-2 inhibitor, has no effect on S–RBD interaction or virus entry [97]. Marzi et al. (2004), showed that the soluble forms of both SARS-CoV and NL63-S protein bind soluble ACE-2 in vitro with substantially different affinity [98]. They also confirmed that ACE-2 residues shown to be critical for SARS-CoV S binding also can abolish NL63 S binding, and that the binding of NL63 S does not involve its unique amino-terminal sequence. A secreted form of ACE-2 fused to green fluorescent protein (GFP) to provide a ligand with an alternate tag for the detection of ligand binding revealed CoV S protein binding to ACE-2 in a unified format. SARS-CoV S and S1 while can pulled down ACE-2 effectively, NL63 S proteins pulled down between 10- and 100-fold less ACE-2 on a weight for weight basis. Differences in the affinity of ACE-2 interaction with the different CoV-S proteins are therefore independent of assay format and may underlie the different pathological outcomes of infection.
ACE-2 interaction was apparent for NL63 S, NL63 S15–739 and NL63 S196–739 but not for NL63 S15–195 (subscript numbers indicate the residues contained within each fragment) confirming that the unique 180 residue amino terminus of NL63 does not bind ACE-2 [58,39]. Li et al. (2007) show that NL63 S and SARS-CoV S bind an overlapping ACE-2 sequence [39]. However, this study demonstrated that the role of ACE-2 residue 353 was noticeably different. In addition, incubation of infected cells with an excess of ACE-2–GFP resulted in a two- to threefold difference in ACE-2 binding between SARS-CoV and NL63 S1 [39].
It can be therefore concluded that SARS-CoV S protein has a significantly higher affinity for ACE-2 than NL63 S protein, but that multi-valency partly reduces the factor of difference. We speculate that while NL63 can use ACE-2 as a receptor for virus entry almost as effectively as SARS-CoV, the consequence of binding on events downstream of ACE-2 binding may be different.
In addition, it was confirmed that ACE-2 residues key to SARS-CoV S binding are also involved in NL63 binding but that the contribution of individual residues, exemplified here by K353A, may differ. This will relate to the molecular contact between S protein and the receptor, which has been described for SARS-CoV but remains unknown in NL63 [75], despite the identification of residues critical for contact [79].
It remains to be determined exactly what difference in ACE-2 signaling, if any, follows SARS-CoV and NL63 S protein binding and whether this relates to the pathology of infection. It will be especially interesting to investigate whether the mode of ACE2 engagement by the viral S proteins impacts viral replication and pathogenesis. The establishment of reverse genetics systems and animal models for HCoV-NL63 replication are indispensable for these studies.
Researchers at Washington University School of Medicine in St. Louis have developed a hybrid virus by genetically modifying a mild virus by swapping one of its genes for one from SARS-CoV-2. The resulting hybrid virus infects cells and is recognized by antibodies just like SARS-CoV-2, but can be handled under ordinary laboratory safety conditions However, this safe model virus lacks the other genes that should account for their pathogenicity and/or antiviral therapies.
The characterization of NL63 and SARS-CoV-S interactions with ACE2 might also have important implications for inhibitor development, because the S–ACE2 interface is a major target for therapeutic intervention. Finally, the apparent similarities between HCoV-NL63 and SARS-CoV replication and the frequent HCoV-NL63 infection of humans suggest that pathogenic HCoVs can evolve, highlighting the need for efficient vaccines against HCoVs.
Researchers at Washington University School of Medicine in St. Louis have developed a hybrid virus by genetically modifying a mild virus by swapping one of its genes for one from SARS-CoV-2 [99]. The resulting hybrid virus infects cells and is recognized by antibodies just like SARS-CoV-2, but can be handled under ordinary laboratory safety conditions However, this safe model virus lacks the other genes that should account for their pathogenicity and/or antiviral therapies.
The detection of HCoV-NL63 in samples collected in 1981 and 1988 shows that the virus has been circulating and causing disease in the human population for a long time. HCoV-NL63 causes LRTIs and URTIs in 1.0–9.3% of children, the elderly and the immune-compromised, with symptoms ranging from mild to severe.
Current data clearly show that HCoV-NL63 is clinically more important that previously suspected as it shares the same cellular receptor, ACE-2, similar to most pathogenic BSL-2-incompatible SARS virus. Some similarities along with some dissimilarities of NL-63 and SARS in host cell entry mechanism and pathogenicity may open up a new strategy to find a proper therapeutics for the most virulent SARS virus.
The authors declare no competing interests.
This work was supported by Institutional grant from Nanoviricides (Shelton, CT, USA). Both the authors contributed equally, and we would like to thank to our all colleagues for helping during the preparation of the manuscript.
- Weiss SR, Navas-Martin S (2005) Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol Mol Biol Rev 69: 635-664. [Crossref]
- Masters PS (2006) The molecular biology of coronaviruses. Adv Virus Res 66: 193-292. [Crossref]
- Siddell S, Wege H, Ter Meulen V (1983) The biology of coronaviruses. J Gen Virol 64: 761-776.
- Weiss SR & Leibowitz JL (2011) Coronavirus pathogenesis. Adv Virus Res 81: 85-164.
- van der Hoek L, Pyrc K, Jebbink MF, Vermeulen-Oost W, Berkhout R, Wolthers KC, et al. (2004) Identification of a new human coronavirus. Nat Med 10: 368-373.
- Woo PC, Lau SK, Chu CM, Chan KH, Tsoi HW, et. al. (2005) Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J Virol 79: 884-895.
- Tyrrell DAJ, Bynoe ML (1965) Cultivation of a novel type of common cold virus in organ culture. Br Med J 1: 1467-1470. [Crossref]
- Hamre D, Procknow JJ (1966) A new virus isolated from the human respiratory tract. Proc Soc Exp Biol Med 121:190-193.
- Arden KE, Nissen MD, Sloots TP, Mackay IM (2005) New human coronavirus, HCoV‐NL63, associated with severe lower respiratory tract disease in Australia. J Med Virol 75: 455-462. [Crossref]
- Ebihara T, Endo R, Ma X, Ishiguro N, Kikuta H (2005) Detection of human coronavirus NL63 in young children with bronchiolitis. J Med Virol 75: 463-465.
- Moe¨s E, Vijgen L, Keyaerts E, Zlateva K, Li S, Maes P, et. al. (2005) A novel pan-coronavirus RT-PCR assay: frequent detection of human coronavirus NL63 in children hospitalized with respiratory tract infections in Belgium. BMC Infect Dis 5: 6-16. [Crossref]
- Vabret A, Mourez T, Dina J, van der Hoek L, Gouarin S, et. al. (2005) Human coronavirus NL63, France. Emerg Infect Dis 11: 1225-1229.
- Bastien N, Anderson K, Hart L, Van Caeseele P, Brandt K, et. al. (2005) Human coronavirus NL63 infection in Canada. J Infect Dis 191: 503-506. [Crossref]
- van der Hoek L, Sure K, Ihorst G, Stang A, Pyrc K, et. al. (2005) Croup is associated with the novel coronavirus NL63. PLoS Med. 2: E240. [Crossref]
- Hendley JO, Fishburne HB, Gwaltney JM Jr. (1972) Coronavirus infections in working adults. Eight-year study with 229 E and OC 43. Am Rev Respir Dis 105: 805-11.
- Chiu SS, Chan KH, Chu KW, Kwan SW, Guan Y, et al. (2005) Human coronavirus NL63 infection and other coronavirus infections in children hospitalized with acute respiratory disease in Hong Kong, China. Clin Infect Dis 40: 1721-1729. [Crossref]
- Peiris JS, Lai ST, Poon LL, Guan Y, Yam LYC, et al. (2003) Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 361: 1319-1325.
- Drosten C, Gunther S, Preiser W, van der Werf S, Brodt HR, et al. (2003) Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med 348: 1967-1976.
- Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S, et. al. (2003) A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med 348: 1953-1966.
- Kuiken T, Fouchier RA, Schutten M, Rimmelzwaan GF, van Amerongen G, et. al. (2003) Newly discovered coronavirus as the primary cause of severe acute respiratory syndrome. Lancet 362: 263-270.
- Graham RL, Donaldson EF, Baric RS (2013) A decade after SARS strategies for controlling emerging coronaviruses. Nat Rev Microbiol 11: 836-848. [Crossref]
- Peiris, JS, Guan Y, Yuen KY (2004) Severe acute respiratory syndrome. Nat Med 10: S88-97.
- Coleman CM, Frieman MB (2014) Coronaviruses: important emerging human pathogens. J Virol 88: 5209-5212.
- de Groot RJ, Baker SC, Baric RS, Brown CS, Drosten C, et al. (2013) Middle east respiratory syndrome coronavirus (MERS-CoV): announcement of the Coronavirus Study Group. J Virol 87: 7790-7792.
- Raj VS, Osterhaus AD, Fouchier RA, Haagmans BL (2014) MERS: emergence of a novel human coronavirus. Curr Opin Virol 5: 58-62.
- Memish ZA, Zumla AI, Al-Hakeem RF, Al-Rabeeah, Stephens GM (2013) Family cluster of Middle East respiratory syndrome coronavirus infections. N Engl J Med 368: 2487-2494. [Crossref]
- Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA (2012) Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 367: 1814-1820.
- Woo PC, Huang Y, Lau SK, Yuen KY (2010) Coronavirus genomics and bioinformatics analysis. Viruses 2: 1804-1820.
- Vijgen L, Keyaerts E, Moës E, Thoelen I, Wollants E, et. al. (2005) Complete genomic sequence of human coronavirus OC43: molecular clock analysis suggests a relatively recent zoonotic coronavirus transmission event. J Virol 79: 1595-1604.
- Kuba K, Imai Y, Rao S, Gao H, Guo F, et al. (2005) A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med 11: 875-879. [Crossref]
- Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, et. al. (2003) Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426: 450-454.
- Hamming I, Timens W, Bulthuis ML, Lely AT, Navis GL, et al. (2004) Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus: A first step in understanding SARS pathogenesis. J Pathol 203: 631-637.
- Ding Y, He L, Zhang Q, Huang Z, Che X, et. al. (2004) Organ distribution of severe acute respiratory syndrome (SARS) associated coronavirus (SARS-CoV) in SARS patients: implications for pathogenesis and virus transmission pathways. J Pathol 203: 622-630. [Crossref]
- Hofmann H, Pyrc K, van der Hoek L, Geier M, Berkhout B, et al. (2005) Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc Natl Acad Sci USA 102: 7988-7993.
- Sims AC, Baric RS, Yount B, Burkett SE, Collins PL, et al. (2005) Severe acute respiratory syndrome coronavirus infection of human ciliated airway epithelia: role of ciliated cells in viral spread in the conducting airways of the lungs. J Virol 79: 15511-15524.
- Yeager CL, Ashmun RA, Williams RK, Cardellichio CB, Shapiro LH, et al. (1992) Human aminopeptidase N is a receptor for human coronavirus 229E. Nature 357: 420-422.
- Tresnan DB, Holmes KV (1998) Feline aminopeptidase N is a receptor for all group I coronaviruses. Adv Exp Med Biol 440: 69-75.
- Ye ZW, Yuan S, Yuen KS, Fung SY, Chan CP, et al. (2020) Zoonotic origins of human coronaviruses. Int J Biol Sci 16: 1686-1697. [Crossref]
- Li W, Sui J, Huang IC, Kuhn JH, Radoshitzky SR, et al. (2007) The S proteins of human coronavirus NL63 and severe acute respiratory syndrome coronavirus bind overlapping regions of ACE2. Virology 367: 367-374.
- Huang C, Wang Y, Li X, Ren L, Zhao J, et. al. (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395: 497-506.
- Nakagawa S, Miyazawa T (2020) Genome evolution of SARS-CoV-2 and its virological characteristics. Inflamm Regener 40: 17-24.
- Dijkman R, Jebbink MF, Wilbrink B, Pyrc K, Zaaijer HL, et. al. (2006) Human coronavirus 229E encodes a single ORF4 protein between the spike and the envelope genes. Virol J 3: 106-114.
- Abdul-Rasool, Fielding BC (2010) Understanding Human Coronavirus HCoV-NL63. The Open Virology Journal 4: 76-84. [Crossref]
- Hofmann H, Geier M, Marzi A, Krumbiegel M, Peipp M, et. al. (2004) Susceptibility to SARS coronavirus S protein-driven infection correlates with expression of angiotensin converting enzyme 2 and infection can be blocked by soluble receptor. Biochem Biophys Res. Commun 319: 1216-1221.
- Lai MMC, Holmes KV (2001) Coronaviridae: The viruses and their replication. Fields’ Virology, Knipe DM, Howley PM, eds (Lippincott, Williams & Wilkins, Philadelphia), 1163-1186.
- McIntosh K (1996) Coronaviruses. Fields virology (Fields BN Knipe DM & Howley PM, et al., eds), 1095 pp. Lippincott-Raven Publishers, Philadelphia.
- Mossel EC, Wang J, Jeffers S, Edeen KE, Wang S, et al. (2008) SARS-CoV replicates in primary human alveolar type II cell cultures but not in type I-like cells. Virology 372: 127-135. [Crossref]
- To KF, Lo AW (2004) Exploring the pathogenesis of severe acute respiratory syndrome (SARS): the tissue distribution of the coronavirus (SARS-CoV) and its putative receptor, angiotensin-converting enzyme 2 (ACE2). J Pathol 203: 740-743.
- Nie Y, Wang P, Shi X, Wang G, Chen J, et. al. (2004) Highly infectious SARS-CoV pseudotyped virus reveals the cell tropism and its correlation with receptor expression. Biochem Biophys Res Commun 321: 994-1000.
- Tsai JC, Zelus BD, Holmes KV, Weiss SR (2003) The N-terminal domain of the murine coronavirus spike glycoprotein determines the CEACAM1 receptor specificity of the virus strain. J Virol 77: 841-850.
- Kubo H, Yamada YK, Taguchi F (1994) Localization of neutralizing epitopes and the receptor-binding site within the amino-terminal 330 aminoacids of the murine coronavirus spike protein. J Virol 68: 5403-5410.
- Wong SK, Li WH, Moore MJ, Choe H, Farzan M (2004) A193-amino acid fragment of the coronavirus S protein efficiently binds angiotensin-converting enzyme 2. J Biol Chem 279: 3197-3201. [Crossref]
- Bonavia A, Zelus BD, Wentworth DE, Talbot PJ, Holmes KV (2003) Identification of a receptor-binding domain of the spike glycoprotein of human coronavirus HCoV-229E. J Virol 77: 2530-2538.
- Xiao X, Dimitrov DS (2004) The SARS-CoV S glycoprotein. The SARS-CoV S glycoprotein. Cell Mol Life Sci 61: 2428-2430.
- Wang P, Chen J, Zheng A, Nie Y, Shi X, et. al. (2004) Expression cloning of functional receptor used by SARS coronavirus. Biochem Biophys Res Commun 315: 439-444.
- Simmons G, Reeves JD, Rennekamp AJ, Amberg SM, Piefer AJ, et al. (2004) Characterization of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) spike glycoprotein-mediated viral entry. Proc Natl Acad Sci USA 101: 4240-4245. [Crossref]
- Yang ZY, Huang Y, Ganesh L, Leung K, Kong WP, et. al. (2004) pH-dependent entry of severe acute respiratory syndrome coronavirus is mediated by the spike glycoprotein and enhanced by dendritic cell transfer through DC-SIGN. J Virol 78: 5642-5650.
- Hofmann H, Simmons G, Rennekamp AJ, Chaipan C, Gramberg T, et. al. (2006) Highly conserved regions within the spike proteins of human coronaviruses 229E and NL63 determine recognition of their respective cellular receptors. J Virol 80: 8639-8652.
- Breslin JJ, Mørk I, Smith MK, Vogel LK, Hemmila EM, et. al. (2003) Human coronavirus 229E: receptor binding domain and neutralization by soluble receptor at 37 degrees. J Virol 77: 4435-4438.
- Blau DM, Holmes KV (2001) Human coronavirus HCoV-229E enters susceptible cells via the endocytic pathway. Adv Exp Med Biol 494: 193-198. [Crossref]
- Huang IC, Bosch BJ, Li F, Li W, Lee KH, et. al. (2005) SARS coronavirus, but not human coronavirus NL63, utilizes cathepsin L to infect ACE2-expressing cells. J Biol Chem 281: 3198-3203.
- Turk V, Turk B, Turk D (2001) Lysosomal cysteine proteases: facts and opportunities. EMBO J 20: 4629-4633.
- Golden JW, Bahe JA, Lucas WT, Nibert ML, Schiff LA (2004) Cathepsin S supports acid-independent infection by some reoviruses. J Biol Chem 279: 8547-8557.
- Golden JW, Linke J, Schmechel S, Thoemke K, Schiff LA (2002) Addition of exogenous protease facilitates reovirus infection in many restrictive cells. J Virol 76: 7430-7443.
- Ebert DH, Deussing J, Peters C, Dermody TS (2002) Cathepsin L and cathepsin B mediate reovirus disassembly in murine fibroblast cells. J Biol Chem 277: 24609-24617.
- Jane-Valbuena J, Breun LA, Schiff LA, Nibert ML (2002) Sites and determinants of early cleavages in the proteolytic processing pathway of reovirus surface protein σ3. J Virol 76: 5184-5197.
- Baer GS, Ebert DH, Chung CJ, Erickson AH, and Dermody TS (1999) Mutant cells selected during persistent reovirus infection do not express mature cathepsin L and do not support reovirus disassembly. J Virol 73: 9532-9543.
- Chandran K, Farsetta DL, Nibert ML (2002) Strategy for nonenveloped virus entry: a hydrophobic conformer of the reovirus membrane penetration protein μ1 mediates membrane disruption. J Virol 76: 9920-9933. [Crossref]
- Chandran K, Parker JS, Ehrlich M, Kirchhausen T, Nibert ML (2003) The delta region of outer-capsid protein μ1 undergoes conformational change and release from reovirus particles during cell entry. J Virol 77: 13361-13375.
- Odegard AL, Chandran K, Liemann S, Harrison SC, Nibert ML (2003) Disulfide bonding among μ1 trimers in mammalian reovirus outer capsid: a late and reversible step in virion morphogenesis. J Virol 77: 5389-5400.
- Huang IC, Bosch BJ, Li F, Li W, Lee KH, et. al. (2006) SARS coronavirus, but not human coronavirus NL63, utilizes cathepsin L to infect ACE2-expressing cells. J Biol Chem 281: 3198-3203. [Crossref]
- Haga S, Yamamoto N, Nakai-Murakami C, Osawa Y, Tokunaga K, et. al. (2008) Modulation of TNF-alpha-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-alpha production and facilitates viral entry. Proc Natl Acad Sci USA 105: 7809-7814.
- Mathewson AC, Bishop A, Yao Y, Kemp F, Ren J, et al. (2008) Interaction of severe acute respiratory syndrome-coronavirus and NL63 coronavirus spike proteins with angiotensin converting enzyme-2. J Gen Virol 89: 2741-2745.
- Baranowski E, Ruiz-Jarabo CM, Domingo E (2001) Evolution of cell recognition by viruses. Science 292:1102-1105. [Crossref]
- Li F, Li W, Farzan M, Harrison SC (2005a) Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 309: 1864-1868.
- Li W, Wong SK, Li F, Kuhn JH, Huang IC, et al. (2006) Animal origins of the severe acute respiratory syndrome coronavirus: insight from ACE2-S-protein interactions. J Virol 80: 4211-4219. [Crossref]
- Li W, Zhang C, Sui J, Kuhn JH, Moore MJ, et. al. (2005) Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J 24: 1634-1643.
- Towler P, Staker B, Prasad SG, Menon S, Tang J, et. al. (2004) ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J Biol Chem 279: 17996-18007.
- Lin HX, Feng Y, Wong G, Li B, Zhao X, et. al. (2008) Identification of residues in the receptor-binding domain (RBD) of the spike protein of human coronavirus NL63 that are critical for the RBD-ACE2 receptor interaction. J Gen Virol 89: 1015-1024.
- Hofmann H, Po¨hlmann S (2004) Cellular entry of the SARS coronavirus. Trends Microbiol 12: 466-472. [Crossref]
- Li W, Zhang C, Sui J, Kuhn JH, Moore MJ, et. al. (2005c) Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J 24: 1634-1643.
- Pyrc K, Berkhout B, Van Der Hoek L (2005) Molecular characterization of human coronavirus NL63. Recent Research Developments in Infection & Immunity (SG Pandelai, ed.), pp. 25- 48. Transworld Research Network, Kerala, India.
- Lim KP, Liu DX (1998) Characterization of the two overlapping papain‐like proteinase domains encoded in gene 1 of the coronavirus infectious bronchitis virus and determination of the C‐terminal cleavage site of an 87‐kDa protein. Virology 245: 303-312. [Crossref]
- Stiehm ER, Ashida E, Kim KS, Winston DJ, Haas A (1987) Intravenous immunoglobulins as therapeutic agents. Ann Intern Med 107: 367- 382.
- Pyrc K, Bosch BJ, Berkhout B, Jebbink MF, Dijkman R, et al. (2006a) Inhibition of HCoV‐NL63 infection at early stages of the replication cycle. Antimicrob Agents Chemother 50: 2000-2008.
- Hemming VG, Rodriguez W, Kim HW, Brandt CD, Parrott RH, et. al. (1987) Intravenous immunoglobulin treatment of respiratory syncytial virus infections in infants and young children. Antimicrob Agents Chemother 31: 1882-1886.
- Bosch BJ, Martina BE, Van Der Zee R, Lepault J, Haijema BJ, et. al. (2004) Severe acute respiratory syndrome coronavirus (SARS‐CoV) infection inhibition using spike protein heptad repeat‐derived peptides. Proc Natl Acad Sci USA 101: 8455- 8460. [Crossref]
- Haasnoot PCJ, Cupac D, Berkhout B (2003) Inhibition of virus replication by RNA interference. J Biomed Sci 10: 607- 616.
- Yang H, Xie W, Xue X, Yang K, Ma J, et. al. (2005) Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol 3: e324.
- Rice GI, Thomas DA, Grant PJ, Turner AJ, Hooper NM (2004) Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem J 383: 45-51. [Crossref]
- Danilczyk U, Eriksson U, Crackower MA, Penninger JM (2003) A story of two ACEs. J Mol Med 81: 227-234.
- Pyrc K, Berkhout B, van der Hoek L (2007) Identification of new human coronaviruses. Expert Rev Anti Infect Ther 5: 245-53.
- Sloots TP, Whiley DM, Lambert SB, Nissen MD (2008) Emerging respiratory agents: new viruses for old diseases? J Clin Virol 42: 233-243.
- Imai Y, Kuba K, Rao S, Huan Y, Guo F, et. al. (2005) Angiotensin converting enzyme 2 protects from severe acute lung failure. Nature 436: 112-116.
- Urbich C, Heeschen C, Aicher A, Sasaki K, Bruhl T, et. al. (2005) Cathepsin L is required for endothelial progenitor cell-induced neo-vascularization. Nat Med 11: 206-213. [Crossref]
- Guy JL, Jackson RM, Jensen HA, Hooper NM, Turner AJ (2005) Identification of critical active-site residues in angiotensin converting enzyme-2 (ACE2) by site-directed mutagenesis. FEBS J 272: 3512-3520.
- Li W, Shi Z, Yu M, Ren W, Smith C, et. al. (2005b) Bats are natural reservoirs of SARS-like coronaviruses. Science 310: 676-679.
- Marzi A, Gramberg T, Simmons G, Moller P, Rennekamp AJ, et. al. (2004). DC-SIGN and DC-SIGNR interact with the glycoprotein of Marburg virus and the S protein of severe acute respiratory syndrome coronavirus. J Virol 78:12090-12095.
- Case JB, Rothlauf PW, Chen RE, Liu Z, Zhao H, et. al. (2020) Neutralizing antibody and soluble ACE2 inhibition of a replication-competent VSV-SARS-CoV-2 and a clinical isolate of SARS-CoV-2. Cell Host and Microbe 28: 475-485.