Abstract
MET pathway dysregulation has been identified in several solid tumors including lung cancers. MET- deregulated NSCLC (non-small cell lung cancer) represents a molecularly defined lung cancer. METexon14 (METex14) mutations and high-level MET amplification represent primary, targetable oncogenic drivers. Type I MET tirosine kinase inhibitors (crizotinib, capmatinib, tepotinib, savolitinib, AMG-337) and type II (cabozantinib, glesatinib, merestinib) showed promising results in preclinical and clinical trials. The main challenge of targeted therapies is the development of acquired resistance. Resistance to MET tirosine kinase inhibitors (MET TKIs) depends on target mutations or upregulation of MET ligand expression. Several strategies are under investigation to overcome acquired resistance.
Key words
MET exon 14 skipping alterations, MET amplification, Non-Small Cell Lung Cancer, MET tirosine kinase inhibitors, Acquired resistance
Introduction
The treatment paradigm of NSCLC radically changed after the discovery that the inhibition of driver oncogenes by targeted agents could reduce tumor burden and improve patient survival. Based on data showing efficacy of targeted therapies in molecularly defined subgroups of patients, testing for EGFR mutations, BRAF mutations, ALK rearrangements and ROS1 rearrangements is strongly advised. Further emerging biomarkers in NSCLC, for which target agents are currently approved for other indications, include HER-2 mutations, RET gene rearrangements and MET deregulation: MET-exon 14 mutations and high-level MET-amplification [1]. Knowledge about clinical and molecular characteristics of MET-deregulated patients is rising as preclinical and clinical evidence of activity and efficacy of different agents targeting MET.
HGF and c-MET signaling pathway
The proto-oncogene MET (mesenchymal epithelial transition factor), located in the long (q) arm of chromosome 7 at position 31.2 (7q31.2), encodes for the receptor tyrosine kinase c-MET or hepatocyte growth factor receptor (HGFR), a transmembrane tyrosine kinase receptor which consists of alpha and beta subunits linked via disulfide bonds [2]. Its ligand, hepatocyte growth factor (HGF), also known as scatter factor, binding the receptor, induces dimerization and autophosphorylation of MET on its intracellular domain. This process activates several downstream signaling pathways including mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K), v-src avian sarcoma (Schmidt-Ruppin A-2) viral oncogene homolog (SRC), and signal transducer and activator of transcription (STAT) signaling pathways [2,3]. Physiologically, during embryogenesis, MET-signaling is involved in gastrulation, development and migration of muscles and neuronal precursors, angiogenesis and kidney formation. In adults plays a role in wound healing, organ regeneration, tissue remodelling, differentiation and proliferation of hematopoietic cells and may regulate cortical bone osteogenesis [2,3]. MET and HGF are found in low levels in normal adult tissues. Their expression is frequently dysregulated in a broad spectrum of human tumors [4] where the excessive activation of MET signaling enhances the malignant properties resulting in the up-regulation of cell proliferation, motility, migration, and invasion [5]. MET-deregulation is described in a variety of human cancers including non-small cell lung cancer (NSCLC), breast cancer, ovarian cancer, papillary renal cell carcinoma, hepatocellular carcinoma, gastric cancer, colon cancer, thyroid cancer, head-and-neck squamous cell carcinomas, and human rhabdomyosarcomas [2,4,6].
MET and Non-small-cell lung cancer
MET is an emerging target in NSCLC and represents a new chance of treatment. MET deregulation may occur by different mechanisms including gene amplification, activating mutations, protein overexpression as a consequence of trascritional upregulation, increased autocrine or paracrine ligand-mediated stimulation [6,8]. High-level MET amplifications and MET exon 14 skipping mutation (MET ex14) have been recognized as potentially targetable oncogenic drivers in NSCLC. MET gene amplification is a target-independent mechanism of acquired resistance to EGFR TKI therapy. Initially it was identified in 20% [9,10] of patients [1,7,11] subsequently a lower incidence was noted, and it was described in 5% of cases [12].
METex14 splicing mutations
MET alterations occur in 3-4% of lung adenocarcinomas and in 2% of squamous cell lung cancers [5]. MET mutations, causing exon 14 skipping (METΔex14), produce c-Met receptors lacking a negative regulatory site. Deletion of the juxtamembrane domain (exon 14) is one mechanism for MET activation. Tyrosine 1003 (Y1003), located in the juxtamembrane domain of MET is encoded by exon 14 and is a binding site for c-Cbl, a ubiquitin protein ligase (E3) which causes ubiquitination, receptor endocytosis, and degradation of MET. Normally, introns flanking METex14 in pre-mRNA are spliced out, resulting in mRNA containing METex14. Somatic intronic mutations lead to aberrant splicing, disrupt splice sites and result in METex14 skipping. This produce a mutant MET receptor lacking the Y1003 c-Cbl binding site which results in decreased ubiquitination and degradation Figure 1 [7,13,14]. In a comprehensive genomic profiling conducted on a large series of lung cancer, METex14 was identified in squamous cell carcinoma with a frequency of 2.1% and in adenocarcinoma with a frequency of 2.9%. Most of patients harbouring METex14 was elderly, more than two-thirds of them age 65 years or older [15]. Next-generation sequencing (NGS) performed on 933 no squamous non–small-cell lung cancer (NSCLC), detected MET exon 14 mutations in 3.0% of cases. None of these patients had an activating mutation in KRAS, EGFR, or ERBB2 or a chromosomal rearrangement in ALK, ROS1, or RET. The median age at disease onset in patients with MET exon 14 mutations was 72.5 years, 68% of patients were women, 36% were never-smokers. At the time of diagnosis 46% had stage I NSCLC, 7% stage II disease, 14% stage III disease, and 32% stage IV disease [16]. Demographic characteristics of patients with MET exon 14 mutations were compared with those of patients harbouring other molecular drivers. MET exon 14 mutations were identified in older population than ALK and ROS1 rearrangements and EGFR, KRAS, BRAF mutations which are mostly found respectively at 50 to 60 years and 61 to 66 years. 64% of patients with MET exon 14 mutations had a history of tobacco use while EGFR-, ALK-, and ROS1- driven NSCLC tend to occur in light or never-smokers [16]. In the study by Awad et al. stage IV MET exon 14–mutated NSCLCs were significantly more likely to have concurrent MET genomic amplification and strong c-Met immunohistochemical expression than stage IA to IIIB [16]. However, in a larger series of METex14 NSCLC, Schrock et al. identified concurrent METamp in 15% of METex14 cases and did not demonstrated a significant association between METamp and stage IV disease [15]. A retrospective analysis of 687 Asian patients with resected NSCLC showed that MET exon 14 mutation is mutually exclusive with known driver mutations but tends to coexist with MET amplification or copy number gain. Both METex14 and high-level amplification were negative prognostic factors that predicted poorer survival [8]. Whole-exome sequencing (WES) analyses, conducted on a large collection of Pulmonary Sarcomatoid Carcinoma (PSC), highlighted high frequency of MET exon 14 skipping mutation in this subtype of lung cancer. Pulmonary sarcomatoid carcinoma is a rare (0.1-0.4 of all pulmonary malignancies), highly aggressive and poorly differentiated non–small-cell lung carcinoma which is characterized by poor prognosis and resistance to chemotherapy [17]. Five subtypes are recognized: pleomorphic carcinoma, spindle cell carcinoma, giant cell carcinoma, carcinosarcoma, and pulmonary blastoma [8,17]. The biology of sarcomatoid carcinoma is poorly understood. MET is implicated in the epithelial mesenchymal transition process, therefore MET activation might affect the differentiation state of the tumor cells [8,17]. Approximately 20% to 30% of sarcomatoid carcinomas harbor METex14 alterations [7,8]. This alteration is mutually exclusive with known driver mutations and therefore should be considered as potentially targetable driver [17].
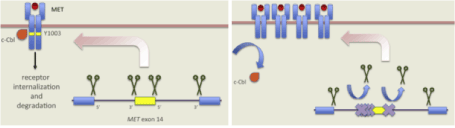
Figure 1. MET exon 14 alteration [7]
MET -Amplification
MET gene amplification occurs in about 4% of lung adenocarcinomas, 1% of squamous cell lung cancers [9,18]. MET-amplification causes rise in gene copy number, protein overexpression and constitutive kinase activation [7]. Gene amplification and polisomy are distinct mechanisms leading to an increase in gene copy number. Gene amplification is a copy number increase of a restricted region of a chromosome arm [19] without a change in copy number for genes located in other regions of the same chromosome [5]. Common chromosomal fragile sites, defects in DNA replication or telomere dysfunction might promote amplification [19]. Preclinical studies highlighted that amplification of drug-selected genes is driven by recurrent breaks within chromosomal common fragile sites (CFSs), via the breakage-fusion-bridge (BFB) mechanism [20]. Polisomy results in an increased in gene copy number because of the presence of extra copies of the entire chromosome without reflecting selection of one gene over another Figure 2 [19]. Only true MET amplification has been identified as an oncogenic driver [7]. PCR essays identify a gain in gene copy number regardless of the underlying mechanisms and are unable to discriminate gene amplification from polysomy. FISH analysis can be performed with two probes: one for the determination of the signal of the target gene (MET) and one for the determination of the signal of the centromere (CEPT7) which directly indicates the number of the corresponding chromosome. In polysomy, each copy of MET is associated with a corresponding centromere, preserving the MET/CEP7 ratio. In true MET amplification, copy number increases without an increase in CEP7 and the MET/CEP7 ratio increases [5,7,11]. MET gene copy number gains, detected by FISH or NGS, is a continuous variable. Agreement about the threshold to define MET positivity has not yet been reached. MET gene copy number gain can be evaluated with two approaches: mean MET copy number per cell (mean MET/cell) and the MET copy number per centromere 7 ratio (MET/CEP7) [11]. According to Cappuzzo scoring system, MET FISH-positive are considered all cases with mean 5 or more copies per cell and negative all cases with mean fewer than 5 copies per cell [7,18]. Considering MET/CEP7 ratio, level of MET amplification is consider low, if MET/CEP7 ratio is in the range 1.8 to 2.2, intermediate, if MET/CEP7 ratio is in the range 2.2 to 5, high if MET/CEP7 ratio is >=5. 14-15 [18,21]. Positive MET FISH result includes, moreover, tumor with MET/CEP7 ratio >= 2 (PathVysion) [22,23]. Since most oncogenic mutations are usually mutually exclusive, oncogene overlap analysis was used to identify a subset of MET-driven NSCLC based on MET copy number [11]. Using a low threshold to define MET copy number gain, overlap with a large number of known dominant oncogenes was detected. Patients with high level amplification with a MET/CEP7 ratio =5 was the only MET positive group associated with zero oncogenic driver overlap. This group was also associated with the highest ORR to crizotinib. Only high level of MET amplification is mutually exclusive with other oncogenic drivers and represent a true MET-driven state whereas lower copy number may represent a coincident event [11]. Polysomy is unlikely a driver event in NSCLC because most of polysomy tumors harbored other driver mutations [8]. Increased MET gene copy number is a negative prognostic factor. In radically resected NSCLC MET FISH-positive patients had a significantly shorter survival than MET FISH-negative patients [18,24,25].
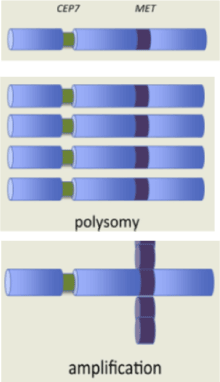
Figure 2. Gene amplification and polisomy [7]
Targeting MET
Several strategies to target MET pathway are in clinical development: small molecules tirosine kinase inhibitors (TKIs) and monoclonal antibodies (mAb) against MET or its ligand HGF. TKIs can be mainly divided into two types. Type I ATP-competitive MET-inhibitors bind to MET unique autoinhibitory conformation through the interaction with Y1230 in the MET activation loop. Type I inhibitors can be further divided into type Ia (crizotinib) and type Ib (capmatinib, tepotinib, savolitinib, AMG-337). Type Ib are highly selective for MET with fewer off target effects as compared with type Ia inhibitors. Type II ATP-competitive inhibitors (cabozantinib, glesatinib, merestinib) bind to the ATP binding site and extend to the hydrophobic back pocket [26,27]. Type II inhibitors often inhibit a broader array of kinase targets, potentially resulting in more off-target side effects. Response to MET TKI was observed both in patients with METamp and in patients without METamp METex14 Table 1 [15].
Table 1. Tyrosine kinase inhibitors targeting MET exon 14 skipping alterations [28]
MET-TKI |
Targets |
Type of inhibitor |
Clinical Trial |
Crizotinib |
ALK,MET,ROS1 |
Type Ia |
NCT00585195 (PROFILE-1001)
NCT02465060 (NCI-MATCH)
NCT02499614 (METROS)
NCT02664935 (Matrix) |
Capmatinib |
MET |
Type Ib |
NCT02750215
NCT01324479(GEOMETRY) |
Tepotinib |
MET |
Type Ib |
NCT02864992/2015-005696-24(VISION) |
Savolitinib |
MET |
Type Ib |
NCT02897479 |
AMG337 |
MET |
Type Ib |
No current clinical trials |
Cabozantinib |
MET, VEGFR2, RET, KIT, TIE-2, AXL |
Type II |
NCT01639508 |
Glesatinib |
MET, VEGFR, RON, TIE-2 |
Type II |
NCT02544633 |
Merestinib |
MET, TIE-1,AXL, ROS1, DDR1/2,FLT3
MERTK, RON, MKNK1/2 |
Type II |
NCT02920996 |
Crizotinib
Crizotinib (PF-02341066) is an orally available, ATP-competitive tirosine-kinase inhibitor of MET, ALK and ROS1. Based on result from PROFILE 1001, PROFILE 1005 [29,30] and subsequently PROFILE 1007 [31] and PROFILE 1014 [32], crizotinib was approved for patients with locally advanced or metastatic ALK-rearranged non–small cell lung cancer both in first and in second line of treatment. Crizotinib was also approved for ROS-1-rearranged advanced NSCLC [33-35]. In the phase 1 trial PROFILE 1001 (NCT00585195), crizotinib, initially developed as MET-inhibitor, was tested in patients with advanced cancers [36]. In a cohort of this study Camidge et al. analyzed efficacy and safety of crizotinib in patients with advanced MET-amplified NSCLC within 3 categories of amplification: MET/CEP7 ratio ≥ 1.8-≤ 2.2 (Low), >2.2-<5 (Intermediate) and ≥ 5 (High). The best tumor shrinkage was seen only in intermediate and high MET cohorts [37,38]. Additionally, in an expansion cohort of PROFILE 1001 study, crizotinib demonstrated antitumor activity in patients with MET exon 14-altered NSCLC [39] NCI-MATCH (Molecular Analysis for Therapy Choice) trial is a phase II basket trial evaluating different targeted therapies directed by genetic testing in patients with advanced refractory solid tumors [40]. The National Lung Matrix Trial consists of a series of parallel single arm phase II trial each testing an experimental targeted drug in population of NSCLC selected by actionable biomarkers. In both of these trial crizotinib is tested in the subpopulation of patients with METex14 alteration [41]. METROS trial is an Italian prospective phase II study designed to assess the efficacy and safety of crizotinib in ROS1-rearranged and MET-positive advanced NSCLC who have failed at least one standard chemotherapy regimen. The best results in terms of response rate, progression free survival and overall survival were registred in ROS-1 population confirming the efficacy of crizotinib in this subgroup of patients and underlining the need of additional and dedicated researches in MET-deregualted NSCLC [42].
Capmatinib
Capmatinib (INC280) is an oral, ATP-competitive, selective MET inhibitor. In preclinical models it suppresses c-MET activation and signaling, blocks cell proliferation, migration and induces apoptosis [43]. In a phase I study efficacy and safety of capmatinib was assessed in patients with solid tumors including an expansion group of cMET-dysregulated NSCLC and an expansion group of patients with EGFR wild type and high cMET-expressing NSCLC. GEOMETRY mono-1 trial is a phase II study (NCT02414139) evaluating capmatinib in patients with METΔex14 mutated or MET amplified advanced NSCLC. The overall response rate (ORR), primary end-point of this trial, was of 72.0% in treatment-naive patients and 39.1% in previously treated patients. GEOMETRY duo-1 is phase Ib/II study investigating the combination of capmatinib plus gefitinib in patients with EGFR-mutated, MET-dysregulated (amplified/overexpressing) NSCLC who experienced disease progression while receiving EGFR-TKI treatment [44].
Tepotinib
Tepotinib (EMD 1214063) is an oral, ATP-competitive, highly selective MET inhibitor. In preclinical studies tepotinib inhibits c-Met phosphorylation and downstream signaling pathways, interferes with tumor cell proliferation and induces tumor regression in xenograft models [45]. In a phase I study (NCT01014936) in patients with advanced solid cancers, tepotinib demonstrated antitumor activity, particularly in c-Met overexpressing/amplified tumors [46]. VISION study (NCT02864992) [47] is a single-arm phase II trial investigating the efficacy and safety of tepotinib in patients with NSCLC. Patients with stage IIIB/IV METΔex14+ NSCLC without EGFR-activating mutations or ALK rearrangements who have received 0–2 lines of prior therapy are eligible. Preliminary data, based on investigator assessment, demonstrated that 60.0% of evaluable patients had a confirmed partial response and 20.0% had stable disease. Recruitment in this trial is ongoing [48].
Savolitinib
Savolitinib (AZD6094, volitinib, HMPL-504) is a potent and selective small molecule inhibitor of MET which showed activity in preclinical models against MET-driven cancer cell lines and in xenograft model of metastatic EGFR- and KRAS-wild type NSCLC disease [49]. In the phase I, dose-escalation study, in patients with advanced solid tumors, volitinib demonstrated anti-tumor activity in MET-deregulated diseases [50]. A phase II, multicenter study is ongoing to evaluate the efficacy and safety of savolitinib in locally advanced/metastatic MET-mutation-positive pulmonary sarcomatoid carcinomas and other NSCLC patients with MET Exon 14 mutation [51]. Interim results from two expansion cohorts of the phase lb clinical trail TATTON, recently presented at the AACR Annual Meeting 2019, showed clinical activity of the combination of Savolitinib plus osimertinib in EGFR-mutant non-small cell lung cancer that had developed resistance to prior EGFR-targeted therapies through MET-gene amplification [52].
AMG-337
AMG-337 is an oral MET kinase inhibitor. In a first in human, sequential dose escalation and expansion study in subjects with advanced solid tumors AMG 337 showed clinical activity in patients with MET-amplified gastroesophageal junction, gastric and esophageal cancer [53]. There are no clinical trials evaluating activity of AMG337 against METex14-positive NSCLC [54].
Glesatinib
GLESATINIB (MGCD265) is an oral, ATP-competitive inhibitor targeting MET, VEGFR1/2/3, Tie-2, Ron and Axl. In NSCLC xenograft models, preclinical antitumor activity of MGCD265 was evaluated in association with taxanes and erlotinib [55]. In a phase I study of patients with advanced solid tumors, glesatinib demonstrated anti-tumor activity in cancer with dysregulation of MET or Axl. Patients with NSCLC and other solid tumors with specific genetic alterations for MET and AXL will be enrolled in the expansion cohort of this study [56]. In a phase 2 trial MGCD265 in being evaluating in pre-treated patients with locally advanced, unresectable or metastatic non-small cell lung cancer with the MET gene deregulation (mutation or amplification) [57].
Merestitinib
Merestitinib (LY2801653) is an oral, potent, type II ATP-competitive inhibitor of MET which also targets RON, AXL, MER receptor tyrosine kinase (MERTK), TIE-2, TIE-1, ROS1, and discoidin domain receptor tyrosine kinase 1 (DDR1). Merestitinib showed anti-tumor activities in multiple mouse xenograft models, anti-angiogenic, and anti-proliferative/cytostatic activities [58]. Merestitinib is being evaluating in a phase II study in patients with advanced NSCLC with MET exon 14 mutation or patients with advanced cancer harboring an NTRK1, 2, or 3 rearrangement [59].
Cabozantinib
Cabozantinib (XL184) is a potent inhibitor of MET, VEGFR2, RET, KIT, AXL, FLT3 involved in tumor pathogenesis [4]. Preclinical studies highlighted that cabozantinib inhibits tumor cell proliferation, angiogenesis and invasive tumor growth [4]. Cabozantinib is currently approved for metastatic medullary thyroid carcinoma harboring RET mutations or chromosomal rearrangements leading to RET gene fusions [59] and for metastatic renal cell carcinoma both in first line treatment, for patients with poor/intermediate risk disease, and as subsequent therapy after progression on a previous TKI [60-62]. In a phase II randomized discontinuation trial, which included patients with prior exposure to anti-EGFR therapy and to anti-VEGF pathway, the activity of cabozantinib was evaluated in a small cohort (60 patients) of metastatic, pre-treated NSCLC. Overall response rate (ORR) was 10% and overall disease control rate (partial response +stable disease) was 40%. 64% of patients experimented objective tumor regression. In contrast to non- responder, some of responders had a known driver mutation at baseline (four EGFR mutations, three KRAS mutations) [63]. ECOG-ACRIN 1512 trial is a three arm, randomised, phase 2 study conducted in metastatic or recurrent non-squamous NSCLC progressing after first line platinum-doublet chemotherapy, and optionally progressed following a second-line chemotherapy regimen. Patients with known EGFR TKI sensitizing mutations and prior erlotinib or MET TKI therapy were excluded. 125 patients were enrolled and randomized (1:1:1) to receive erlotinib monotherapy, cabozantinib monotherapy, and the combination of erlotinib and cabozantinib. Progression free survival (PFS) was significantly improved in the cabozantinib arm (4.3 months) and in the erlotinib plus cabozantinib arm (4.7 months) compared to erlotinib alone (median 1.8 months). Principal limitations of this study are the lack of detailed molecular driver oncogene characterization and the modest sample size. Moreover, erlotinib should not be utilized in control arm since several studies demonstrated that erlotinib is minimally effective in EGFR-wil-type population [64]. The combination of erlotinib and cabozantinib was evaluated in a phase Ib/II study in patients with advanced pre-treated NSCLC, the majority of whom previously received erlotinib. 54 patients received study-drugs in a 3+3 design using combination doses across 5 cohorts in 2 parallel arms. Among the 36 patients assessable for response, six had a ≥ 30% reduction in tumor measurement including 3 with a partial response. In some cases, a prolonged stable disease for more than four months has been observed. Among the responders, one presented MET amplification, among patients with long lasting stabilization one had EGFR T790M mutation [65]. In a phase II trial a combination of cabozantinib and erlotinib was tested in EGFR mutation-positive NSCLC following progression on EGFR TKI. 37 patients were enrolled, ORR was 12.5% in T790M-positive tumors, but DCR and PFS was increased in T790M-negative tumors. MET gene amplification was not detect in post-progression biopsies [66]. A phase II trial is ongoing to evaluate cabozantinib in patients with advanced RET ROS1, or NTRK fusion-positive NSCLC and in patients harbouring increased MET or AXL activity [67]. Although the incidence and management of brain metastases in MET-deregulated NSCLC is still unknown, preliminary data demonstrated intracranical activity of cabozantinib in MET Exon14–positive NSCLC with brain metastases. A patient with METex14-altered NSCLC with intracranial progression and extracranial disease control during crizotinib therapy was treated with cabozantinib reaching a complete intracranial response and maintaining a systemic response. This is the first observation of intracranial penetration and clinical activity of a type II inhibitor (cabozantinib) after exposure to a type I inhibitor (crizotinib) [68].
Acquired resistance to MET TKI
In oncogene addicted cancers, after initial response to targeted therapies, acquired resistance is an inevitable consequence and the principle limitation of this therapeutic strategy. Several mechanisms of acquired resistance have been identified: target modification (gene amplification and second site mutations), activation of bypass tracts, and histologic transformation [69]. Preclinical studies, with random mutagenesis screen in MET-driven tumors cells, highlighted that the most common acquired mutations to type I MET TKI are Y1230 and D1228 which weaken the interactions between type I inhibitors and the MET activation loop [70]. Clinically, two different case reports described the emergence respectively of MET D1228N [71] mutation and MET Y1230C [72] mutation in patients with METex14+ NSCLC after progression during crizotinib treatment. Given that type I MET TKIs bind to the MET unique autoinhibitory conformation through interaction with Y1230 in the MET activation loop, mutations in Y1230 block the binding of type I MET TKIs. Type II MET TKI is less dependent to the interaction with Y1230 thanks to its additional interaction with the hydrophobic back pocket of the MET kinase domain [72]. Therefore, switching from type I to type II MET inhibitors may overcome MET Y1230 and D1228 resistance mutations. In a case report a patient with advanced lung adenocarcinoma harboring EGFR exon 19 deletion mutation with high level MET amplification as mechanism of acquired resistance, was treated with a combination of osimertinib and savolitinib. At disease progression, analysis with next-generation sequencing (NGS) of the biopsy obtained following the development of resistance, detected MET D1228V. Subsequently patient was successfully treated with erlotinib combined with type II MET inhibitor cabozantinib [73]. Further studies are necessary to analyse the efficacy of switching from type I to type II MET inhibitors. MET-addicted tumor cells, which are characterized by ligand-independent, constitutive MET-activation, upon pharmacological MET inhibition, become dependent for survival on HGF produced by mesenchymal cells of microenvironment [74]. Vertical inhibition of a tyrosine-kinase receptor with antibodies against MET or against HGF can overcome HGF-mediated resistance. Ficlatuzumab is a monoclonal antibody (mAb) which binds to the soluble ligand HGF preventing the interaction of HGF to its receptor c-Met and the activation of the HGF/c-Met signaling pathway [75]. In preclinical models ficlatuzumab sensitizes MET-addicted tumors to MET-targeted agents [72]. In a phase II trial, conducted in asian patients with previously untreated lung adenocarcinoma, ficlatuzumab has been evaluated in combination with gefitinib or alone versus gefitinib [75]. FOCAL study is a phase II study evaluating efficacy of ficlatuzumab versus placebo when administered with erlotinib in subjects with previously untreated metastatic EGFR-mutated NSCLC [76]. Emibetuzumab (LY2875358) is a humanized, bivalent, anti-MET IgG4 which blocks HGF from activating MET both in vitro and in vivo and induces MET receptor internalization and degradation. In preclinical studies emibetuzumab inhibits proliferation of tumor cells with MET amplification and demonstrated antitumor activity in xenograft models of NSCLC [77]. Limited single-agent activity of emibetuzumab was observed in a phase I study in patients with MET-positive solid tumors including NSCLC [78]. Another strategy involves the use of drugs that deliver cytotoxic agents directly to tumor cells with targeted antibodies. ABBV-399 is an antibody-drug conjugate (ADC) comprised of the anti-c-Met antibody, ABT-700, and monomethyl auristatin, an antimicrotubule agent. In a phase I trial in advanced solid tumors with an expansion cohort of patients with MET-positive NSCLC, ABBV-399 demonstrated promising anti-tumor activity [79].
Immunotherapy in MET-deregulated NSCLC
Tumor mutational burden (TMB) can be defined as the number of somatic base substitution or indel alterations per megabase (MB). To analyse how mutational landscape of NSCLCs may influence response to anti–PD-1 therapy, exome sequencing was conducted on two independent cohorts of patients with NSCLC treated with pembrolizumab highlighting that higher somatic mutational burden was associated with higher clinical efficacy of pembrolizumab [80]. The average TMB in METex14+ NSCLC patients was 6.9 mutations/Mb, slightly lower than the overall average of 10.7 mutations per MB for all lung cancers [15], but higher than the average TMB for EGFR-mutated (mean, 4.5) and ALK-positive NSCLC (mean, 2.8). The median number of mutations per MB was 4.4 for non-MET amplificated cases and 6.8 for MET amplificated [15]. Lower efficacy was observed in EGFR-mutant/ALK-positive patients treated with PD-1/PD-L1 inhibitors compared to a cohort of EGFR- wild type and ALK negative/unknown patients [81,82]. A recent review of clinicopathologic, molecular features, and an analysis of response to immune checkpoint inhibition on patients with MET exon 14-deregulated NSCLC confirmed that the median TMB is lower than in unselected NSCLCs and that the overall clinical efficacy to PD-1 blockade is modest in this subgroup of patients [83-87].
Conclusion
MET is an emerging molecular target for non–small cell lung carcinoma. Several studies have contributed to define clinicopathologic characteristics of tumors with MET alteration although further researches are necessary to investigate some significant features as the incidence and management of brain metastases. Encouraging data are emerging from clinical trials investigating agents targeting MET. A deeper understanding of mechanisms of acquired resistance are necessary to develop strategies to overcome the inevitable arise of acquired resistance to targeted therapies. A broad, molecular profiling, assessing potential genetic alterations and emerging biomarkers, is recommended to offer patients the best treatment option even recruiting in clinical trial.
References
- NCCN guidelines (2018) Version 5. Last accessed June 27, 2018.
- MET gene-Genetics Home Reference–NIH.
- Organ SL, Tsao MS (2011) An overview of the c-MET signaling pathway. Ther Adv Med Oncol 3: S7-S19. [Crossref]
- Comoglio P.M. and Trusolino L. (2002) Invasive growth: from development to metastasis. J Clin Invest 109: 857-862. [Crossref]
- Kawakami H, Okamoto I, Okamoto W, Tanizaki J, Nakagawa K, et al. (2014) Targeting MET amplification as a new oncogenic driver. Cancers (Basel) 6: 1540-1552. [Crossref]
- JR Sierra, M-S Tsao (2011) c-MET as a potential therapeutic target and biomarker in cancer. Ther Adv Med Oncol 3: S21-35.[Crossref]
- Drilon A, Cappuzzo F, Ou SI, Camidge DR (2017) Targeting MET in Lung Cancer: Will Expectations Finally Be MET?. J Thorac Oncol 12: 15-26. [Crossref]
- Tong JH, Yeung SF, Chan AW, Chung LY, Chau SL, et al. (2016) MET amplification and exon 14 splice site mutation define unique molecular subgroups of non-small cell lung carcinoma with poor prognosis. Clin Cancer Res 22: 3048-3056. [Crossref]
- Bean J, Brennan C, Shih JY, Riely G, Viale A, et al. (2007) MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A 104: 20932-20937. [Crossref]
- Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, et al. (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316: 1039-1043. [Crossref]
- Noonan SA, Berry L, Lu X, Gao D, Baron AE, et al. (2016) Identifying the appropriate FISH criteria for defining MET copy number driven lung adenocarcinoma through oncogene overlap analysis. J Thorac Oncol 11: 1293-1304.
- Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, et al. (2013) Analysis of tumor specimens at the time of acquired resistance to EGFR TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res 19: 2240-2247. [Crossref]
- Onozato R, Kosaka T, Kuwano H, Sekido Y, Yatabe Y, et al. (2009) Activation of MET by gene amplification or by splice mutations deleting the juxtamembrane domain in primary resected lung cancers. J Thorac Oncol 4: 5-11.
- Kong-Beltran M, Seshagiri S, Zha J, Zhu W, Bhawe K, et al. (2006) Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res 66: 283-289.
- Schrock AB, Frampton GM, Suh J, Chalmers ZR, Rosenzweig M, et al. (2016) Characterization of 298 lung cancer patients harboring MET exon 14 skipping (METex14) alterations. J Thorac Oncol 11: 1493-1502. [Crossref]
- Awad MM, Oxnard GR, Jackman DM, Savukoski DO, Hall D, et al. (2016) MET Exon 14Mutations in Non–Small-Cell Lung Cancer Are Associated With Advanced Age and Stage-Dependent MET Genomic Amplification and c-Met Overexpression. J Clin Oncol 34: 721-730. [Crossref]
- Liu X, Jia Y, Stoopler MB, Shen Y, Cheng H, et al. (2015) Next-Generation Sequencing of Pulmonary Sarcomatoid Carcinoma Reveals High Frequency of Actionable MET Gene Mutations. J Clin Oncol 34: 794-802.
- Cappuzzo F, Marchetti A, Skokan M, Rossi E, Gajapathy S, et al. (2009) Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J Clin Oncol 27: 1667-1674. [Crossref]
- Albertson DG (2006) Gene amplification in cancer. Trends Genet 22: 447-455. [Crossref]
- Hellman A, Zlotorynski E, Scherer SW, Cheung J, Vincent JB, et al. (2002) A role for common fragile site induction in amplification of human oncogenes. Cancer Cell 1: 89-97.
- Tong JH, Yeung SF, Chan AW, Chung LY, Chau SL, et al. (2016) MET amplification and exon 14 splice site mutation define unique molecular subgroups of non-small cell lung carcinoma with poor prognosis. Clin Cancer Res 22: 3048-3056. [Crossref]
- Tanaka A, Sueoka-Aragane N, Nakamura T, Takeda Y, Mitsuoka M, et al. (2012) Co-existence of positive MET FISH status with EGFR mutations signifies poor prognosis in lung adenocarcinoma patients. Lung Cancer 75: 89-94.
- Jurmeister P, Lenze D, Berg E, Mende S, Schaper F, et al. (2015) Parallel screening for ALK, MET and ROS1 alterations in non-small cell lung cancer with implications for daily routine testing. Lung Cancer 87: 122-129.
- Cheng TL, Chang MY, Huang SY, Chyun SC, Long E, et al. (2005) Overexpression of circulating c-met messenger RNA is significantly correlated with nodal stage and early recurrence in non-small cell lung cancer. Chest 128: 1453-1460.
- Beau-Faller M, Ruppert AM, Voegeli AC, Neuville A, Meyer N, et al. (2008) MET gene copy number in non-small cell lung cancer: Molecular analysis in a targeted tyrosine kinase inhibitor naïve cohort. J Thorac Oncol 4: 331-339.
- Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G (2012) Targeting MET in cancer: rationale and progress. Nat Rev Cancer 12: 89-103. [Crossref]
- Backes A, Zech B, Felber B, Klebl B, Muller G, et al. (2008) Small-molecule inhibitors binding to protein kinase: part II: the novel pharmacophore approach of type II and type III inhibition. Expert Opin Drug Discov 3: 1427-1449.
- Reungwetwattana T, Liang Y, Zhu V, Ou SI (2017) The race to target MET exon 14 skipping alterations in non-small cell lung cancer: The Why, the How, the Who, the Unknown, and the Inevitable. Lung Cancer 103: 27-37. [Crossref]
- Camidge DR, Bang YJ, Kwak EL, Iafrate AJ, Varella-Garcia M, et al. (2012) Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol 13: 1011-1019. [Crossref]
- Kim DW, Ahn MU, Shi Y, De Pas T, Yang PC, et al. (2012) Results of a global phase II study with crizotinib in advanced ALK positive non-small cell lung cancer. J Clin Oncol 30: 7533.
- Shaw AT, Kim DW, Nakagawa K, Seto T, Crinó L, et al. (2013) Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med 368: 2385-2394. [Crossref]
- Mok T, Kim DW, Wu YL, Solomon BJ, Nakagawa K, et al. (2014) First-line crizotinib versus pemetrexed-cisplatin or pemetrexed-carboplatin in patients (pts) with advanced ALK-positive non-squamous non-small cell lung cancer (NSCLC): results of a phase III study (PROFILE 1014). J Clin Oncol 32: 8002.
- Malik SM, Maher VE, Bijwaard KE, Becker RL, Zhang L, et al. (2014) U.S. Food and drug administration approval: crizotinib for treatment of advanced or metastatic non-small cell lung cancer that is anaplastic lymphoma kinase positive. Clin Cancer Res 20: 2029-2034.
- Kazandjian D, Blumenthal GM, Luo L, He K, Fran I, et al. (2016) Benefit-Risk summary of crizotinib for the treatment of patients with ROS1 alteration-positive,metastatic non-small cell lung cancer. Oncologist 21: 974-980.
- Shaw AT, Ou SH, Bang YJ, Camidge DR, Solomon BJ, et al. (2014) Crizotinib in ROS1-Rearranged Non–Small-Cell Lung Cancer. N Engl J Med 371: 1963-1971. [Crossref]
- Available from: https://clinicaltrials.gov/.ClinicalTrials.gov Identifier: NCT00585195.
- Camidge DR, Ou SI, Shapiro G, Otterson GA, Villaruz LC, et al. (2014) Efficacy and safety of crizotinib in patients with advanced c-MET-amplified non-small cell lung cancer(NSCLC). J Clin Oncol 32: 8001.
- Camidge DR, Otterson GA, Clark JW (2018) Crizotinib in patients (pts) with MET-amplified non-small cell lung cancer (NSCLC): Updated safety and efficacy findings from a phase 1 trial. J Clin Oncol 36: 9062-9062.
- Drilon AE, Camidge DR, Ou SI (2016) Efficacy and safety of crizotinib inpatients (pts) with advanced MET exon 14-altered non-small cell lungcancer (NSCLC). J Clin Oncol 34: 108.
- Available from: https://clinicaltrials.gov/.ClinicalTrials.gov Identifier: NCT02465060.
- Available from: https://clinicaltrials.gov/.ClinicalTrials.gov Identifier: NCT02664935.
- Landi L, Chiari R, Dazzi C, Tiseo M, Chella A, et al. (2017) Crizotinib in ROS1 rearranged or MET deregulated non-small-cell lung cancer (NSCLC): final results of the METROS trial. Ann Oncol 28: 6.
- Liu X, Wang Q, Yang G, Marando C, Koblish HK, et al. (2011) A novel kinase inhibitor, INCB28060, blocks c-MET-dependent signaling, neoplastic activities, and cross-talk with EGFR and HER-3. Clin Cancer Res 17: 7127-7138.
- Smit EF, Kopp H, Kim D (2016) GEOMETRY duo-1: A phase (Ph) Ib/II, multicenter trial of oral cMET inhibitor capmatinib (INC280)± erlotinib vs platinum + pemetrexed in adult patients (pts) with epidermal growth factor receptor (EGFR)-mutated, cMET-amplified, locally advanced/metastatic non-small cell lung cancer (NSCLC) with acquired resistance to prior EGFR tyrosine kinase inhibitor (TKI) therapy. J Clin Oncol 34: TPS9109.
- Bladt F, Faden B, Friese-Hamim M, Knuehi C, Wilm C, et al. (2013) EMD 1214063 and EMD 1204831 constitute a new class of potent and highly selective c-Met inhibitors. Clin Cancer Res 19: 2941-2951.
- Falchook GS, Kurzrock R, Amin HM (2015) Efficacy, safety, biomarkers, and phase II dose modeling in a phase I trial of the oral selective c-Met inhibitor tepotinib (MSC2156119J). J Clin Oncol 33: 2591.
- Available from: https://clinicaltrials.gov/.ClinicalTrials.gov Identifier: NCT02864992.
- Felip E, Horn L, Patel JD (2016) Tepotinib in patients with advanced non-small cell lung cancer (NSCLC) harboring MET exon 14-skipping mutations: Phase II trial. https://clinicaltrials.gov/ct2/show/NCT02864992.
- Cruz DC, Frigault M, Adam A, Ren Y, Barry E, et al. (2014) Targeting MET in preclinical models tosupport the clinical development of Volitinib in NSCLC. Cancer Res 74: 3114.
- Gan HK, Lickliter J, Millward M (2014) First-in-human phase I study of aselective c-Met inhibitor volitinib (HMP504/AZD6094) in patients with advanced solid tumors. J Clin Oncol 32: 11111.
- Available from: https://clinicaltrials.gov/.ClinicalTrials.gov Identifier: NCT02897479.
- Adding the MET inhibitor savolitinib to osimertinib was beneficial for certain pre-treated lung cancer patients in TATTON trail. Available online: https://www.aacr.org/Newsroom/Pages/News-Release-Detail.aspx?ItemID=1294.
- Kwak E, LoRusso P, Hamid O, Janku F, Kittaneh M, et al. (2015) Clinical activity of AMG337, an oral MET kinase inhibitor, in adult patients (pts) with MET-amplified gastroesophageal junction (GEJ), gastric (G), or esophageal(E) cancer. J Clin Oncol 33: 1.
- gov. Available from: https://clinicaltrials.gov/.
- Besterman JM, Fournel M, Dupont I (2010) Potent preclinical antitumoractivity of MGCD265, an oral Met/VEGFR kinase inhibitor in phase II clinical development, in combination with taxanes or erlotinib. J Clin Oncol 28: e13595.
- Kollmannsberger CK, Sharma S, Shapiro G (2015) Phase I study of receptor tyrosine kinase (RTK) inhibitor, MGCD265, in patients (pts) with advanced solid tumors. J Clin Oncol 33: 2589.
- Available from: https://clinicaltrials.gov/.ClinicalTrials.gov Identifier: NCT02544633.
- Yan SB, Peek VL, Ajamie R, Buchanan SG, Graff JR, et al. (2013) LY2801653 is an orally bioavailable multi-kinase inhibitor with potent activity against MET, MST1R, and other oncoproteins, and displays anti-tumor activities in mouse xenograft models. Invest New Drugs 31: 833-844. [Crossref]
- Available from: https://clinicaltrials.gov/.ClinicalTrials.gov Identifier: NCT02920996.
- Choueiri TK, Escudier B, Powles T, Mainwaring P, Rini BI, et al. (2015) Cabozantinib versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med 373: 1814-1823. [Crossref]
- Choueiri TK, Escudier B, Powles T, Tannir NM, Mainwaring PN, et al. (2016) Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR): final results from a randomised, open-label, phase 3 trial. Lancet Oncol 17: 917-927. [Crossref]
- Choueiri TK, Halabi S, Sanford BL, Hahn O, Michaelson MD, et al. (2017) Cabozantinib Versus Sunitinib As Initial Targeted Therapy for Patients With Metastatic Renal Cell Carcinoma of Poor or Intermediate Risk: The Alliance A031203 CABOSUN Trial. J Clin Oncol 35: 591-597. [Crossref]
- Hellerstedt BA, Edelman G, Vogelzang NJ (2012) Activity of cabozantinib (XL184) in metastatic NSCLC: Results from a phase II randomized discontinuation trial (RDT). J Clin Oncol 30: 7514.
- Hellerstedt BA, Edelman G, Vogelzang NJ (2012) Activity of cabozantinib (XL184) in metastatic NSCLC: Results from a phase II randomized discontinuation trial (RDT). J Clin Oncol 30: 7514. [62] Neal JW, Dahlberg SE, Wakelee HA, Aisner SC, Bowden M, et al. (2016) Erlotinib, cabozantinib, or erlotinib plus cabozantinib as second- or third-line treatment of patients with EGFR wild-type advanced non-small cell lung cancer (ECOG-ACRIN 1512): a phase 2 randomised controlled trial. Lancet Oncol 17: 1661-1671. [Crossref]
- Wakelee HA, Gettinger SN, Engelman J, Janne PA, West H, et al. (2010) A phase Ib/II study of XL184 (BMS 907351) with and without erlotinib in patients (pts) with non-small cell lung cancer (NSCLC). J Clin Oncol 28: 3017.
- Reckamp KL, Mack PC, Ruel N (2015) Biomarker analysis of a phase II trial of cabozantinib and erlotinib in patients (pts) with EGFR-mutant NSCLC with epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor(TKI) resistance: a California Cancer consortium phase II trial (NCI 9303). J Clin Oncol 33 15: 8087.
- Available from: https://clinicaltrials.gov/.ClinicalTrials.gov Identifier: NCT01639508.
- Klempner SJ, Borghei A, Hakimian B, Ali SM, Ou S-HI, et al. (2017) Intracranial activity of cabozantinib in MET exon 14 positive NSCLC with brain metastases. J Thorac Oncol 12: 152-156.
- Lovly CM, Shaw AT (2014) Molecular pathways: resistance to kinase inhibitors and implications for therapeutic strategies. Clin Cancer Res 20: 2249-2256. [Crossref]
- Tiedt R, Degenkolbe E, Furet P, Appleton BA, Wagner S, et al. (2011) A drug resistance screen using a selective MET inhibitor reveals a spectrum of mutations that partially overlap with activating mutations found in cancer patients. Cancer Res 71: 5255-5264.
- Heist RS, Sequist LV, Borger D, Gainor JF, Arellano RS, et al. (2016) Acquired resistance to crizotinib in non-small cell lung cancer with MET exon 14 skipping. J Thorac Oncol 11: 1242-245.
- Ignatius SH, Ou L, Young AB, Schrock A, Johnson A, et al. (2016) Emergence of pre-existing MET Y1230C mutation as a resistance mechanism to crizotinib in NSCLCwith MET exon 14 skipping. J Thorac Oncol 12: 119.
- Bahcall M, Sim T, Paweletz CP, Patel JD, Alden RS et al. (2016) Acquired MET D1228V mutation and resistance to MET inhibition in lung cancer. Cancer Discov 6: 1334-1341. [Crossref]
- Pennacchietti S, Cazzanti M, Bertotti A, Rideout III WM, Ham M, et al. (2014) Microenvironment-derived HGF overcomes genetically determined sensitivity to anti-MET drugs. Cancer Res 74: 6598-6609.
- Mok TSK, Park K, Geater SL (2012) A randomized phase (PH) 2 study with exploratory biomarker analysis of ficlatuzumab (F) a humanized hepatocyte growth factor (HGF) inhibitory mAb in combination with gefitinib (G) versus G in asian patients (PTS) with lung adenocarcionma (LA), Ann Oncol 23: 1198P.
- Available from: https://clinicaltrials.gov/.ClinicalTrials.gov Identifier: NCT02920996NCT02318368.
- Liu L, Zeng W, Wortinger MA, Yan SB, Cornwell P, et al. (2014) LY2875358, a neutralizing and internalizing anti-MET bivalent antibody, inhibits HGF-dependent and HGF-independent MET activation and tumor growth. Clin Cancer Res 20: 6059-6070.
- Banck MS, Chugh R, Natale RB (2015) Phase 1 results of emibetuzumab (LY2875358), a bivalent MET antibody, in patients with advanced castration-resistant prostate cancer, and MET positive renal cell carcinoma, non-small cell lung cancer, and hepatocellular carcinoma. Mol Cancer Ther 14: A55.
- Strickler JH, Nemunaitis JJ, Weekes CD (2016) Phase 1, open-label,dose-escalation and expansion study of ABBV-399, an antibody drug conjugate (ADC) targeting c-Met, in patients (pts) with advanced solid tumors. J Clin Oncol 34: 2510.
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, et al. (2015) Cancer immunology: mutational landscape determines sensitivity to PD-1 blockade in non-small cell lungcancer. Science 348: 124-128. [Crossref
- Gainor JF, Shaw AT, Sequist LV, Fu X, Azzoli CG, et al. (2016) EGFR mutations and ALK rearrangements are associated with low response rates to PD-1 pathway blockade in non-Small cell lung cancer (NSCLC): a retrospective analysis. Clin Cancer Res 22: 4585-4593. [Crossref]
- Sabari JK, Leonardi GC, Shu CA, Umeton R, Montecalvo J, et al. (2018) PD-L1 expression, tumor mutational burden, and response to immunotherapy in patients with MET exon 14 altered lung cancers. Ann Oncol 29: 2085-2091.
- K. Paik, A. Drilon, P.D. Fan, Yu H, Rekhtman N, et al. (2015) Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon14 skipping. Cancer Discov 5: 842-849.
- Wolf J, Seto T, Han J, Reguart N, Garon EB, et al. (2018) Results of the GEOMETRY mono-1 phase II study for evaluation of the MET inhibitor capmatinib (INC280) in patients (pts) with METΔex14 mutated advanced non-small cell lung cancer (NSCLC). Ann Oncol 8: 45-47.
- Wen PY, McLaren Black P, Loeffler JS (1999) Metastatic brain cancer. In De Vita VT Jr, Hellman S, Rosenberg SA (eds): Cancer: Principles and Practice of Oncology, 6th edition. Philadelphia, PA: Lippincott Williams and Wilkins pp: 2655-2670.
- Costa DB, Shaw AT, Ou SH, Solomon BJ, Riely GJ, et al. (2015) Clinical experience with Crizotinib in patients with Advanced ALK-rearranged non- small-cell lung cancer and brain metastases. J Clin Oncol 33: 1881-1888. [Crossref]
- Solomon B, Felip E, Blackhall F, Mok TSK, Kim D, et al. (2014) Overall and incranial (IC) efficacy results and time to symptom deterioration in PROFILE 1014: 1st line crizotinib vs. premetrexed-platinum chemotherapy (PPC) in patients with advanced ALK-positive non-squamous non-smeall cell lung cancer (NSCLC). Ann Oncol 25: iv426–iv470.