Hypophosphatasia (HPP) is a rare genetic disorder characterized by the abnormal development of bones and teeth. These abnormalities occur due to defective bone and tooth mineralization associated with low serum and bone alkaline phosphatase activity. Which caused by a number of loss-of-function mutations in the ALPL gene leading to diminished activity of the enzyme in bone, liver, and kidney. The clinical presentation of this disease is extremely variable. Ranging from extreme life-threatening forms revealed at birth in young infants presenting with severely impaired bone mineralization, seizures, and hypercalcemia, to young adults with premature exfoliation of their teeth without any other symptom. Herein, we report a case of HPP who presented with pyridoxine-responsive seizures in the early neonatal period and was found to have hypercalcemia, low alkaline phosphate and skeletal demineralization
alkaline phosphatase, infantile hypophosphatasia, ALPL gene, hypercalcemia
Hypophosphatasia (HPP) [Online Mendelian Inheritance in Man (OMIM) 146300, 241500 and 241510] is a rare metabolic disease defined by a deficiency of serum tissue nonspecific alkaline Phosphatase (TNSALP). It was first described in 1948 (Rathbun 1948) and has a variable clinical presentation. Seven forms have been reported based primarily on the age at which skeletal lesions are discovered [1].
Perinatal HHP: associated with profound inactivity of alkaline phosphatase and markedly impaired mineralization with clinically apparent skeletal deformities and pathognomonic radiographic changes with rapidly progressive clinical course. Some pregnancies end in stillbirth. In other cases, affected newborns pass away from respiratory failure in several days.
Prenatal benign HHP: is associated with bowed limbs at birth. The skeletal malformations associated with this form improve after birth. Infantile HHP: has symptoms similar to, but typically less severe, than perinatal form and recognized before 6 months of age. Childhood HHP: is highly variable, from severe to mild forms, but are less severe than the infantile form. Affected children may sometimes have craniosynostosis and exhibit signs of intracranial hypertension.
Adult HPP: osteopenia, recurrent fractures, and pseudofractures with early loss of adult dentition common. Odontohypophosphatasia: is characterized by the premature loss of deciduous teeth in childhood, or loss of teeth in adulthood. Pseudohypophosphatasia: is similar to infantile HPP but with unremarkable ALP levels.
The genes of the other, tissue-specific, ALP isoenzymes are located on the long arm of chromosome 2 (2q34-37) [2-4]. The main symptoms are diagnosis before age 6 months because of pyridoxine-responsive seizures, failure to thrive, muscular hypotonia, mainly in perinatal and infantile forms of hypophosphatasia and the common clinical signs are rickets, osteomalacia, fractures, teeth loss, nephrocalcinosis, craniosynostosis, fractures and respiratory failure. The severely affected babies often die at or soon after birth from respiratory insufficiency due to pulmonary hypoplasia, a consequence of poorly mineralized bones of the chest [5,6].
The main diagnostic laboratory abnormalities are low serum ALP and TNSALP activity (hypophosphatasemia) and increased levels of ALP substrates (inorganic pyrophosphate (PPi), pyridoxal-5’-phosphate (PLP, the active metabolite of vitamin B6), and phosphoethanolamine (PEA)
Nine months -old female infant was referred to genetic metabolic physician at age of on week due to neonatal seizure since birth and hypercalcemia. She was born at the 37th week of gestation by Caesarean section with a birth weight of 2010 g and the Apgar score was 8 at 1 minute and 10 at 5 minutes after birth. Her prenatal ultrasound was normal and she the second child of consanguineous Saudi parents with was significant family history, the oldest sibling baby girl was died at age of 4 months with respiratory failure and unclear diagnosis. Metabolic acidosis and seizures were observed in the first day of life. She kept in NICU and intubation was attempted. She has frequent myoclonic seizures that were not controlled after phenobarbital and phenytoin therapy. On her physical examination at one month old, her weight was 2370 g (<3th centile), her height was 48.6cm (3th-10th centile), and her head circumference was 35.6 cm (10th-50th centile). A flattened facial appearance, broad forehead, flattened nasal bridge, bilateral low-set ears, short neck, narrow thorax, no organomealy and heart examination murmur was normal. (Figure 1)

Figure 1. At age of 4 months .Facial phenotype showing broad forehead, flattened nasal bridge, bilateral low-set ears, short neck.
Her seizures controlled after pyridoxine (vitamin B6) administration (100 mg/day initial, 50 mg/day maintenance dose, PO). Intravenous hydration with saline plus furosemide was started at the time of admission to treat hypercalcemia, this therapy had minimal effect on calcium levels, and serum calcium was 14.1 mg/dL on the 6rd hospital day. The hypercalcemia was resistant to other treatment options including prednisolone (2 mg/kg/d). Normocalcemia was achieved with a calcitonin (4 U/kg/d).
Her skull and long bones radiographies showed on dedicated shoulder radiograph widening of growth plate and proximal humeral metaphyseal flaying and fraying in keeping with history of hypophosphatemia (Figure 2). Chest x-ray was showed Rachitic rosary appearance of the ribs. Left humeral head ossification center is not aligned with metaphysis Left perihilar consolidation with blunting of both costophrenic angles. Craniosynostosis was not observed. Renal ultrasonography showed both kidneys are normal in size, showing mild increased echogenicity texture. No hydronephrosis seen. EEG was done and showed voltage suppression but no epileptiform. Her echocardiography was normal. Her ophthalmologic examination, cranial magnetic resonance imaging, and electroencephalography were normal.
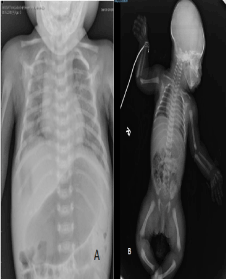
Figure 2. ( A,B,) Radiographic imaging of the patient before Asfotase alfa treatment showing distorted trabeculation, reduced mineralization in the epiphyses and metaphyses, demineralizationin the skull.
The results of blood serum tests were as follows: calcium 13.6 mg/dL ,phosphorus 7.3 mg/dL , Alkaline phosphate 6 U/L .magnesium 1.5 mg/dL ,parathyroid hormone (PTH) 5.5 pg/mL and 25-hydroxy vitamin D3 52.5 ng/mL (the cut-off for vitamin D3 deficiency is <20). Thyroid funcrions teste was normal . Urinary calcium excretion was high with a calcium/creatinine ratio of 0.64. Her complete blood count and routine biochemistry tests for renal, liver, immunoglobulins, tandem mass spectrometry, urinary and plasma amino acid analysis, urinary organic acid analysis were normal (Figure 3).
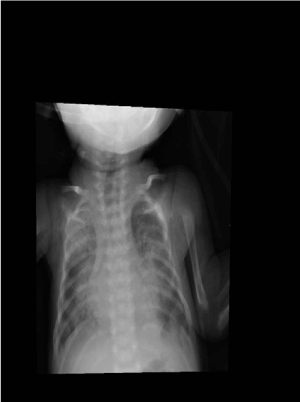
Figuer 3. After 20 weeks of ERT substantial mineralization is apparent.
At age of 4 months Asfotase alfa was given (2 mg/kg three times weekly subcutaneously). After 20 weeks of asfotase alfa administered SC, the patient showed great improvement in rickets, and pulmonary function. Even level of alkaline phosphatase improved significantly gradually, on discharged the ALP 102 UL .She weaned from ventilator support. This better respiratory outcome accompanied radiographic improvements in skeletal mineralization with in 4 weeks of treatment. She was doing well and discharged home in stable condition with no more seizure and normalized alkaline phosphates and calcium. But unfortunately she died with sepsis after one month in private clinic.
Exon 2 to 12 of ALPL gene (NM_000478) were amplefied by PCR. The amplified produces were then sequenced using ABI 3730 sequencers and analyzed for Sequence variations. The significanceof the variations wasdetermined by comparison with wild type Sequence,previously reported mutations, and correlation with the structure of the alkaline phosphatase enzyme (liver/bone/kideny type) Although DNA sequencing is a highly sensitive methodology, mutation detection may not 100% DNA based testing is confirmatory A negative test result reduces but dose not eliminate agenetic cause for this individuals phenotype.
Molecular analysis performed by saudi diagnostic laboratory SDL , in saudia confirmed the diagnosis of HPP. A homozygous c.173T>C transition in exon 3 pf the ALP gene. This change converts a codon for leucine (CTG) to 3 codon for proline (CCG). This change has not been previuosly reported as either amutation or a polymorphism. Additionally, this change is not listed in the dbSNPor ESP database . comparison of ALPL amino acid sequences derivedfrom numerous species indicates that Leu58 is highly conserved (37/37 species). These finding suggest that this change is pathogenic.
an Hypophosphatasia is highly variable in its clinical symptoms and signs d is thought to reflect the severity of the mutation in the ALPL gene and and depending on the clinical form. which can range from stillbirth with no mineralized bone to early loss of teeth in late adulthood, but with no bone symptoms [8,9]. Patients with severe forms suffer from respiratory failure, which is the main cause of death. clinical characteristics of infantile type of HPP are respiratory complications, premature craniosynostosis, demineralization,rachitic changes in the metaphyses, hypercalcemia, shortstature, and elevated serum concentrations of calcium and phosphorus, and low serum ALP enzyme activity helps differentiate HPP from other conditions. Differential diagnosis of infantile HPP includes Osteogenesis imperfecta (OI) with deformation (typically type III in infancy or type IV later on) may resemble hypophosphatasia clinically.Pseudohypophosphatasia is characterized by clinical, biochemical, and radiographic findings reminiscent of infantile hypophosphatasia, with the exception that clinical laboratory assays of serum alkaline phosphatase activity are in the normal range , thanatophoric dysplasia and Radiological findings may be similar to rickets but It can be distinguished from rickets by low ALP levels . Hypophosphatasia is highly variable in its clinical presentation, which can range from stillbirth with no mineralized bone to early loss of teeth in late adulthood, but with no bone symptoms [7,8].
The transmission of disease is autosomal recessive mainly sever type, while milder forms may be transmitted as dominant or recessive autosomal traits.
The disease is due to mutations in the alkaline phosphatase liver-type (ALPL) gene, also named the TNSALP gene. Hypophosphatasia is a monogenic disease that is mainly caused by various mutations in the TNSALP gene. Currently, at least 267 distinct mutations and 16 polymorphisms are known to correlate with hypophosphatasia. The most prevailing mutation type is the missense mutation which accounts for 74.5% of all mutations. our patient muttion has not been reported befor , A homozygous c.173T>C transition in exon 3 pf the ALP gene , The molecular gene diagnosis also done for the parents and both heterozygous for same gene ALPL.
An established treatment for HPP was not available until the recent development of ERT with a modified human TNAP (asfotase alfa) it was shown to prevent their infantile hypophosphatasia including its associated seizures and dental abnormalities [10-12]. Asfotase Although enzyme replacement therapy may provide a therapeutic option, but still there is no current therapy for HPP till now.
- Whyte MP (2012) Hypophosphatasia In: Thakker RV, Whyte MP, Eisman J, Igarashi T (eds) Genetics of bone biology and skeletal disease. Academic Press, London. pp: 337-360
- FRASER D (1957) Hypophosphatasia. Am J Med 22: 730-746. [Crossref]
- Mornet E (2007) Hypophosphatasia. Orphanet J Rare Dis 2: 40. [crossref]
- Griffin CA, Smith M, Henthorn PS, Harris H, WeissMJ, et al. (1987) Human placental and intestinal alkaline phosphatase genes map to 2q34eq37. Am J Hum Genet 41: 1025-1034 [Crossref]
- Whyte MP (2008) Hypophosphatasia: nature’s window on alkaline phosphatase function in humans. In: Bilezikian JP, Raisz LG, Martin TJ (eds) Principles of Bone Biology, 3rd edn. Academic Press, San Diego. Pp: 1573-1598
- Mornet E (2000) Hypophosphatasia: the mutations in the tissue-nonspecific alkaline phosphatase gene. Hum Mutat 15: 309-315. [Crossref]
2021 Copyright OAT. All rights reserv
- Harris H (1990) The human alkaline phosphatases: what we know and what we don't know. Clin Chim Acta 186: 133-150. [Crossref]
- Mornet E (2007) Hypophosphatasia. Orphanet J Rare Dis 2:40.
- Zurutuza L, Muller F, Gibrat J, Taillandier A, Simon-Bouy B, et al. (1999) Correlations of genotype and phenotype in hypophosphatasia. Hum Mol Genet 8: 1039-1046. [Crossref]
- Millan JL, Whyte MP (2015) Alkaline phosphatase and hypophosphatasia. Calcif. Tissue Int
- McKee M (2011) Enzyme replacement therapy prevents dental defects in a model of hypophosphatasia. J Dent Res 90: 470-476. [Crossref]
- Millán JL, Narisawa S, Lemire I, Loisel TP, Boileau G, et al. (2008) Enzyme replacement therapy for murine hypophosphatasia. J Bone Miner Res 23: 777-787. [Crossref]