Abstract
The cannabinoids are found to have particular application as neuroprotectants for mental and motor dysfuction in neurodegenerative diseases. The neuroprotective properties of cannabinoids suggest their therapeutic use for limiting neurological damage. The cannabinoids treatments should not only aim to alleviate specific symptoms but also attempt to delay/arrest disease progression and to repair the damaged structures. The author conducted a review of studies published between 1974 and 2011. The search was performed using the following PubMed search terms: “Cannabinoids” and “Neurodegenerative Diseases” and 287 papers were detected. The articles were examined and the overlapping or insufficiently clear works were excluded. Finally we chose 117 articles regarding the latest international guidelines, the pathophysiology of neurodegenerative diseases and the various therapeutic choices. The studies reported in the present review support the view that the cannabinoid signalling system is a key modulatory element in the activity of the basal ganglia. This idea is supported by different anatomical, electrophysiological, pharmacological and biochemical data. Furthermore, these studies indicate that the cannabinoid system is impaired in different neurological disorders that directly or indirectly affect the basal ganglia, which supports the idea of developing novel pharmacotherapies with compounds that selectively target specific elements of the cannabinoid system.
Key words
cannabinoids; mental and motor dysfuction, neuroprotection
Introduction
The recreational use of Cannabis Sativa preparations is known to most people [1]. However, the medicinal use of Cannabis also has a millenarian history that has been re-examined only very recently [2]. As early as 2600 BC, the Chinese emperor Huang Ti advised taking Cannabis for the relief of cramps and rheumatic and menstrual pain [3]. This long history of Cannabis medical use has resulted in the development of pharmaceutical drugs, such as Dronabinol and Cesamet. These preparations is based on ?9-tetrahydrocannabinol (THC, fig.1), which in 1964 was identified by Mechoulam and coworkers as the major psychoactive component of cannabis. They are prescribed in the United States as anti-emetic and appetite-stimulants to patients with cancer and AIDS. To date, some 60 plant terpenophenols more or less related to THC have been isolated and defined cannabinoids [4]. ?9-tetrahydrocannabinol, for its potency and abundance in cannabis, is the most important.
Cannabinoid receptors
Thus far, two cannabinoid-specific receptors have been cloned and characterized from mammalian tissues, the seven transmembrane G protein-coupled cannabinoid receptors type 1 (CB1 receptor), [5] and type 2 (CB2 receptor) [6]. Whereas the CB1 receptor expression is abundant in the central nervous system, the CB2 receptor is almost exclusively expressed in the immune system. The CB1 receptor is also expressed in peripheral nerve terminals and various extraneuronal sites such as the testis, uterus, eye, vascular endothelial, spleen and adipocytes [7-10]. Pharmacological evidence exists for the presence of other cannabinoid receptors, which, however, have not yet been cloned [11].
CB1 and CB2 receptors share only 44% overall identity and 68% within the transmembrane domains. Both cannabinoid receptors are coupled to G proteins, mostly of the Gi/o type, through whose α subunit they inhibit the activity of adenylate cyclases and stimulate mitogen-activated protein kinases. However, additional studies established that cannabinoid receptors were also coupled to ion channels, resultant in the inhibition of Ca2+ influx through N type calcium channels [12]. CB1 receptors are also implicated in activation of both phospholipase C (via the βγ subunits of the G protein) and PI-3-kinase. CB2 receptors, on the other hand, trigger a sustained activation of ceramide biosynthesis [13].
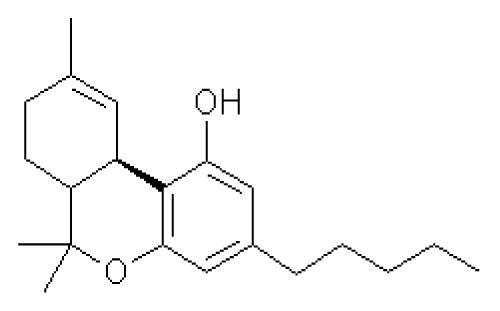
Figure 1. Chemical structure of ?9-tetrahydrocannabinol
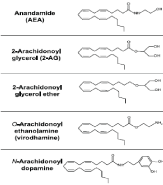
Figure 2. Chemical structure of endogenous cannabinoid
The endocannabinoid system
Several endogenous fatty-acid ligands, known as endocannabinoids, have been identified as having activity at the cannabinoid receptor. The first to discovered, in 1992, was arachidonoylethanolamide (anandamide, AEA) followed by 2-arachidonoylglycerol (2-AG). Both these compounds are derivates of arachidonic acid conjugated with ethanolamine or glycerol and are able to bind to CB1 and CB2 receptors, although with differences in affinities and activation efficacies [8]. During the last few years, several other bioactive lipid mediators have described; they appear to be active, through CB1 and/or CB2 receptors and confer specific pharmacological effects in vivo. Specifically, the compounds are 2-arachidonoyl-glyceryl-ether (noladin ether), o-arachidonoyl-ethanolamine (virodhamine), N- arachidonoyl-dopamine, and possibly oleamide [14-16,10] (fig. 2).
Cannabinoid receptors, endocannabinoids and the whole apparatus appointed of their synthesis and degradation represent the elements of a novel endogenous signalling system (the endocannabinoid system) which is implicated in a overabundance of physiological functions [17,18]. During the last few years a notable quantity of data has been reported to understand the biological roles of this system in more detail.
In general, endocannabinoid system serves several functions under physiological conditions. In the CNS, endocannabinoids intervene in the regulation of cognitive functions and emotions in neuronal circuits of the cortex, hippocampus and amygdale and to the reinforcement of substances of abuse in the mesolimbic system [19].
Endocannabinoids also modulate the control of movement and posture [20], the regulation of pain perception [21] and cardiovascular [22], gastrointestinal [23,24], respiratory and reproductive functions. CB2 receptors, instead, are involved in cellular and particularly humoral immune response, with possible implications for (neuro)inflammation and chronic pain [25].
Apart from the possible physiological functions of the endocannabinoid system briefly described above, endocannabinoid signalling undergoes dramatic tissue and blood changes under pathological conditions. Higher endocannabinoid levels are found in the case of experimental models of neurodegenerative disease, like Parkinson’s and Alzheimer’s disease and amyotropic lateral sclerosis [26].
Cannabinoids for mental and motor dysfuctions in neurodegenerative diseases
Cannabinoids for Alzheimer disease
AD is the most common neurodegenerative disease in Western Europe, and an important public health problem as the number of cases is increasing with aging of the population. It manifests with progressive decline in memory and intellectual abilities, impoverishment of language, disorientation and behavioral skills. The characteristic neuropathological aspects of AD are senile plaques (SP), neurofibrillary tangles (NFT), and amyloid angiopathy. Brain lesions associated with AD such as NFT and SP, are characterized by the presence of a broad spectrum of inflammatory mediators produced by cells residing in the brain, including neurons. Although of secondary importance compared to the fundamental cause that determines the presence of tangles and plaques, there is strong evidence that inflammation exacerbates the neuronal cell loss. Consequently, AD risk is substantially influenced by several polymorphisms in the promoter region of genes and other non-coding regions for inflammatory mediators. Alleles that support the increased expression of inflammatory mediators or alleles that favour the reduced expression of anti-inflammatory mediators are more frequent in patients with AD compared to controls. The polymorphisms are fairly common in the general population, so there is a strong probability that everyone will inherit one or more high risk alleles [27,28].
AD also is characterized by enhanced beta-amyloid peptide (betaA) deposition along with glial activation in senile plaques, selective neuronal loss, and cognitive deficits.
The role of cannabinoid receptors in AD and their possible protective effects after betaA treatment was studied by Ramirez et al. [29]. This study showed that senile plaques in AD patients express CB1 and CB2 cannabinoid receptors as well as markers of microglial activation. Furthermore, while high level of CB1-positive neurons are present in control cases, they are greatly reduced in areas of microglial activation [29]. Also, G-protein coupling and CB1 receptor protein expression are markedly decreased in AD brains wheres protein nitration is increased [29]. Cannabinoids (HU-210, WIN55,212-2, and JWH-133) prevent both betaA-induced microglial activation, cognitive impairment, and loss of neuronal markers and abrogate microglia-mediated neurotoxicity after betaA addition to rat cortical cocultures [29]. These results indicate that cannabinoid receptors are involved in the pathology of AD and that they may control the neurodegenerative process occurring in the disease.
The neuroprotective actions of CBD in AD was also studied by Iuvone et al. [30] by evaluating the effect of cannabidiol, a major non-psychoactive component of the marijuana plant (Cannabis sativa) on β-amyloid peptide-induced toxicity in cultured rat pheocromocytoma PC12 cells. β-amyloid peptide induced a strong reduction of cell survival as well as an increased reactive oxygen species (ROS) production, lipid peroxidation, caspase 3 (a key enzyme in the apoptosis cell-signalling cascade) appearance, DNA fragmentation and intracellular calcium was osbserved. Treatment of the cells with cannabidiol, prior to β-amyloid peptide exposure, significantly elevated cell survival while it decreased ROS production, lipid peroxidation, caspase 3 levels, DNA fragmentation and intracellular calcium. These results indicate that cannabidiol exerts a neuroprotective effects against β-amyloid peptide toxicity suggesting the a possible involvement of cannabidiol in the signalling pathway for this neuroprotection. This hypothesis was supported by a further study where CBD inhibited both nitrite production and nitric oxide synthase (iNOS) protein expression induced by beta-A [31]. These results of in vitro studies were confirmed in vivo with a mouse model of AD-related neuroinflammation. Mice were inoculated with human beta-A into the right dorsal hippocampus, and treated daily with vehicle or CBD (2.5 or 10 mg kg, i.p.) for 7 days. In contrast to vehicle, CBD dose-dependent significantly inhibited mRNA for glial fibrillary acidic protein and the protein expression in beta-A injected animals. Moreover, under the same experimental conditions, CBD impaired iNOS and IL-1beta protein expression, and the related NO and IL-1beta release [32]. The possibility of CBD inhibiting beta-A-induced neurodegeneration is very promising to AD prevention.
Cannabinoids for Parkinson disease
While AD is the most prevalent neurological disorder in the aged population, PD is the second most common neurodegenerative disease. Several studies indicate the presence of inflammatory mediators (including TNF-a, IL-1β, IL-6, and interferon-g (IFNγ)) in the cerebrospinal fluid (CSF) of patients with PD as well as in the post-mortem substantia nigra pars compacta in PD patient brains [28,33].
There is an increase in the levels of proinflammatory cytokines in the CSF and nigrostriatal regions of PD brains. Furthermore, large numbers of reactive microglia are found in the substantia nigra of PD patients. These may chronically produce ROS, resulting in depletion of antioxidant stores that may jeopardize mitochondrial activity. Since aerobic respiration in mitochondria is responsible for most of the ROS produced in cells, abnormalities in these organelles may exacerbate oxidative stress [28,33]
A recent study [34] showed that CBD may induce neuroprotection in animal models of Parkinson's disease (PD). Daily administration of CBD for 2 weeks reduced significantly the toxicity induced by injection of 6-hydroxydopamine into the medial forebrain bundle [34]. In this model of PD, CBD led to an up-regulation of mRNA levels of Cu/Zn-superoxide dismutase, a key enzyme in endogenous defense against oxidative stress. The conclusion was that CBD can provide neuroprotection against the progressive degeneration of nigrostriatal dopaminergic neurons that occur in PD [35]. This study was confirmed by another study showing that CBD reduced the striatal atrophy caused by 3-nitropropionic acid, in vivo, through mechanisms independent of the activation of cannabinoid, vanilloid TRPV1 and adenosine A2A receptors [36]. Also, the neuroprotective action of CBD in the human basal ganglia was supported by the strong relationship between N-acetylaspartate/total creatine ratio and CBD in the putamen/globus pallidum found in recreational cannabis users. This could reflect an enhancement of neuronal and axonal integrity in these regions by CBD [37].
Given the above preclinical evidences, for the first time, a clinical study evaluated the efficacy, tolerability and safety of CBD in PD patients [38]. In an open-label pilot study, six consecutive outpatients with the PD diagnosis received a flexible-dose regimen of CBD administration (starting with an oral dose of 150 mg/day) for four weeks, in addition to their usual therapy. The PD psychotic symptoms significantly were reduced along the CBD treatment, and the scale used to follow up the PD course exhibited a significant decrease of the total score. These preliminary data suggest that CBD may have a beneficial action in PD [39].
Cannabinoids for Amyotrophic Lateral Sclerosis
ALS is a progressive neurodegenerative disease in which motor neurons in the brain and spinal cord are selectively destroyed. Usually, the disease manifests itself during the mid-50s, although there are rare cases of early-onset ALS. The symptoms of the disease are muscle wasting and atrophy leading to eventual paralysis and death [28,40]. ALS is typically fatal within 5 years of diagnosis due to a progressive, generalized paralysis that eventually affects the muscles of respiration, causing respiratory failure [28,40]. Areas where degenerating motor neurons are present in both ALS patients and mouse models are marked by the presence of cytokines and immune cells, including T cells, activated microglia, and astrocytes [28,40].
Although a generalized neuroinflammatory response may be driving progressive loss of motor neurons, not all inflammatory mediators have been strongly implicated in ALS. For instance, IL-1β may not be critical to ALS pathogenesis as genetic deletion of IL-1β does not change the lifespan or rate of motor neurodegeneration in mutant SOD-1 mice [28,41]. Currently, no effective pharmacological agents exist for the treatment of this devastating disease and neuroinflammation may accelerate the progression of ALS. However, cannabinoids have been reported to be able to reduce the progression of ALS neuroinflammatory diseases through both CB1 and CB2 receptors [42]. The involvement of CB receptors in the development of ALS neuroinflammation was demonstrated by using an ALS animal model: G93A-SOD1 mutant mice cannabinoids [42]. The symptomatic mice showed elevated endogenous cannabinoids and the treatment with non-selective cannabinoid partial agonists prior to, or upon, symptom appearance minimally delays disease onset and prolongs survival through undefined mechanisms [42]. Furthermore, in this ALS animal model, CB2 receptors, which normally exist primarily in the periphery, are dramatically up-regulated in inflamed neural tissues associated with ALS disorders [42]. The efficacy of the selective CB2 agonist AM-1241, to increase the survival interval by 56% suggest that CB2 agonists may slow motor neuron degeneration and preserve motor function, and represent a novel therapeutic modality for treatment of ALS [42].
Cannabinoids for Multiple Sclerosis
MS is a chronic condition in which the immune system attacks the axonal myelin sheaths. The site of inflammatory damage is scarred, thus the disease name is derived from sclerosis meaning “scar” in Latin [28,40]. Since these loci of injury can occur anywhere in the brain and spinal cord, the symptoms of the disease are usually diverse in different patients. These include fatigue, numbness, vision abnormalities, incontinence, muscle weakness, and paralysis.
MS is an autoimmune condition where foci of chronic inflammation lead to compromise of oligodendrocytes and destruction of the myelin sheath. This is followed by axonal damage and consequent neuronal degeneration. Inflammation and neurodegeneration do not occur simultaneously and axonal damage and brain atrophy may follow months after an acute innate immune response [28,40]. At today, numerous drugs target the immune system to reduce the progression of MS but they are only moderately effective, and the treatment of MS remains mostly symptomatic and far from satisfactory [43].
Cannabis had been used in ancient Greece, Rome, China, and India for relieving muscle cramps, spasm, and pain [44-46] and its therapeutic application in MS is a topic of recent lively debate [47-56]. The effects of THC in MS was studied by using an experimental autoimmune encephalomyelitis (EAE) in rats [57]. In this MS experimental model, THC treatment was able both to reduce neuroinflammation and to improve neurological outcome as well as survival compared with placebo. Also, Δ8-THC, a less psychotropic and more stable analog of THC, and the nonpsychotropic dexanabinol were able to reduce the severity and incidence of neurological deficits in rats with EAE [58-60]. The efficacy of THC was also confirmed with a mouse model of chronic relapsing EAE [61] indicating a tonic control of muscle tone by the endocannabinoid system in EAE [61,62]. The use of CB1-deficient mice, which tolerated inflammatory and excitotoxic insults poorly and developed substantial neurodegeneration after the induction of EAE further confirmed the above studies [63-67].
Theiler's murine encephalomyelitis virus-induced demyelinating disease is another murine model of MS. In this MS model, the treatment with cannabinoids reduced the progression of symptoms, down-regulated delayed-type hypersensitivity reactions and interferon-γ production, and inhibited the expression of proinflammatory cytokines in the CNS [68-71].
Given the above the animal data, cannabinoids have shown promise in the treatment of MS in humans. A possible underlying mechanism is suggested by a recent study in which the endocannabinoid system was found to be highly activated during CNS inflammation in MS patients and to protect neurons from inflammatory damage by activating a negative feedback loop in microglial cells via CB1/2-mediated epigenetic regulation of mitogen-activated protein kinase phosphatase 1 expression [72].
Anecdotal reports showing the efficacy of marijuana smoking in relieving symptoms of MS [73,74], were supported by the results of early open or single-blind observations with orally given THC or smoked marijuana, involving small numbers of patients [75-81]. The results of this study showed a significative improvement in spasticity, mobility, tremor, nystagmus, mood, and bladder control. In a double-blind crossover study of a single MS patient, nabilone treatment improved muscle spasms, nocturia, and general well-being [82]. Although these encouraging reports have triggered numerous larger, population-based clinical trials of cannabis-based medicines in MS, mixed results have been reported [83-86].
A large multicenter study involving 33 clinical centers and 660 MS patients in the United Kingdom and United States and supported by the UK Medical Council aimed to explore the effects of cannabis extract (Cannador) or synthetic THC (Marinol) versus placebo on spasticity, pain, tremor, bladder function, and cognitive function [Cannabinoids in Multiple Sclerosis (CAMS) study [87,88]. After 15 weeks of treatment with Marinol or Cannador there was significant improvements in patient-reported spasticity, pain, and sleep quality as well as a reduction in hospital admissions for relapse in the two active treatment groups [87,88]. However, no benefits were observed in Ashworth score, tremor, irritability, depression, or tiredness and dverse side effects were generally minor and similar to those with placebo [87-89]. The improvements in spasm frequency and mobility by THC plus CBD in MS patients were confirmed in further studies of similar design [90-96].
In conclusion, controlled clinical trials with cannabinoids have demonstrated their efficacy in eliciting symptomatic improvements in MS patients. These results suggest that there is place for the use of cannabis in the treatment of MS, which should be confirmed in further larger-scale clinical trials.
Phytocannabinoids for Huntington disease
While neuroinflammation has been targeted in many neurodegenerative diseases ranging from AD to ALS to PD, it has not received much attention from the HD community. However, several published trials, while not having neuroinflammation per se in mind, might have also targeted this process [34].
Several studies indicate that inflammation appears in the CNS during the progression of HD and HD-like pathology. Several brain regions from HD patients and controls revealed increased gliosis and expression of inflammation-related genes, including glial fibrillary acidic protein and complement proteins. Increases were most pronounced in the caudate putamen where brain pathology is most severe in HD patients [34]. Also, increased levels of pro-inflammatory cytokines involved in the innate immune response, such as IL-6, where detected in HD patients as well as an altered immune profile before onset of clinical HD symptoms, suggesting that striatal and cortical neurodegeneration could be exacerbated by inflammation [34]. Thus, it is clear that also HD is related ti neuroinflammation process such as disruption of normal microglial functions and neuronal distress. This neuroinflammation may contribute to the death of additional neurons and once better understood, targeted interference with neuroinflammatory processes, active or reactive, could be a valuable tool for developing new therapeutic approaches [34].
Several studies have reported changes in CB1 receptor levels and CB1 receptor activity in HD and the findings from such studies suggest a role for the endocannabinoid system in the pathophysiology of these and other neurological diseases [97-99].
Animal models of HD as well as post-mortem studies carried out in HD patients suggest that CB1 receptors of basal ganglia, are downregulated and/or desensitized as a result of the expression of the mutant Huntingtin protein, and that this occurs early in the course of the disease and prior to the appearance of overt clinical symptoms [98-109]. A recent in vivo PET study of HD patients supports these findings, indicating a significative reduction in CB1 receptor availability throughout the gray matter of the cerebrum, cerebellum, and brainstem of HD patients even in early stages of the disease [110]. Also, pre-clinical and post-mortem studies in HD patients indicate an inverse relationship between the decrease in CB1 receptor levels and the increase in CB2 receptor levels in glial elements, astrocytes, and in reactive microglial cells [111]. Further clinical and pre-clinical evidence evidence strongly support that changes in the endocannabinoid system are tightly linked to the pathophysiology of HD [106-116].
The strong relationship between cannabinoids and HD was further confirmed by a study using an HD animal model [117]. The results of this study confirmed the CB1 receptors reduction in the basal ganglia leding to increases in Huntington’s-like symptom and the lower levels of BDNF in mice with a lack of CB1 receptors may futher contribute to HD development. Indeed, BDNF replacement is believed to be a possible therapeutic for HD and has been shown to decrease excitotoxicity and attenuate motor dysfunction and cell loss in animal models of HD [117]
Conclusions
In conclusion, the present paper showed a strong link between cannabinoids and neurodegenerative diseases. Neuroprotective activities of endocannabinoids appear to be mainly CB1-mediated thus promising avenues for the therapeutic targeting of different aspects of neurodegenerative diseases, by stimulating a self-protective endogenous system of the brain and by counteracting oxidative stress.
Increased research on cannabinoid medicine and modulation of the endocannabinoid system in relation to neurodegeneration has the potential to lead to novel therapies which may help to prevent progression, and potentially initiation, of these diseases.
References
- Adams IB, Martin BR (1996) Cannabis: pharmacology and toxicology in animals and humans. Addiction 91: 1585-1614. [Crossref]
- Williamson EM1, Evans FJ (2000) Cannabinoids in clinical practice. Drugs 60: 1303-1314. [Crossref]
- Mechoulam R (1986) Cannabis as Therapeutic Agent (ed. Mechoulam, R.) CRC Press Roca Ranton :1-19.
- Mechoulam R, Gaoni Y (1967) Recent advances in the chemistry of hashish. Fortschr Chem Org Naturst 25: 175-213. [Crossref]
- Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI (1990) Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 346: 561-564. [Crossref]
- Munro S, Thomas KL, Abu-Shaar M (1993) Molecular characterization of a peripheral receptor for cannabinoids. Nature 365: 61-65. [Crossref]
- Howlett AC1 (2005) Cannabinoid receptor signaling. Handb Exp Pharmacol : 53-79. [Crossref]
- Howlett AC (2002) The cannabinoid receptors. Prostaglandins Other Lipid Mediat 69: 619-631. [Crossref]
- Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR, et al. (1991) Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J Neurosci 11: 563-583. [Crossref]
- Porter AC, Felder CC (2001) The endocannabinoid nervous system. Unique opportunities for therapeutic intervention. Pharmacol Ther 90: 45-60. [Crossref]
- Begg M, Pacher P, Batkai S, Osei-Hyiaman D, Offertáler L, et al. (2005) Evidence for novel cannabinoid receptors. Pharmacol Ther 106: 133-145. [Crossref]
- Rodriguez De Fonseca F, Del Arco I, Bermudez-Silva FJ, Bilbao A, Cippitelli A, et al. (2005) The endocannabinoid system:physiology and pharmacology. Alcohol Alcohol 40: 2-14. [Crossref]
- Howlett AC, Breivogel CS, Childers SR, Deadwyler SA, Hampson RE, et al. (2004) Cannabinoid physiology and pharmacology: 30 years of progress. Neuropharmacology 47: 345-358. [Crossref]
- Hanus L, Abu-Lafi S, Fride E, Breuer A, Vogel Z, et al. (2001) 2-arachidonyl glyceryl ether, an endogenous agonist of the cannabinoid CB1 receptor. Proc Natl Acad Sci U S A 98: 3662-3665. [Crossref]
- Huang SM, Bisogno T, Trevisani M, Al-Hayani A, De Petrocellis L, et al. (2002) An endogenous capsaicin-like substance with high potency at recombinant and native vanilloid VR1 receptors. Proc Natl Acad Sci U S A 99: 8400-8405. [Crossref]
- Leggett JD, Aspley S, Beckett SR, D’Antona AM, Kendall DA (2004) Oleamide is a selective endogenous agonist of rat and human CB1 cannabinoid receptors. Br J Pharmacol 141: 253-262. [Crossref]
- Piomelli D (2003) The molecular logic of endocannabinoid signalling. Nat Rev Neurosci 4: 873-884. [Crossref]
- De Petrocellis L1, Cascio MG, Di Marzo V (2004) The endocannabinoid system: a general view and latest additions. Br J Pharmacol 141: 765-774. [Crossref]
- Gerdeman GL, Partridge J, Lupica CR, Lovinger DM. (2003) It could be habit forming: drugs of abuse and striatal synaptic plasticity. Trends Neurosci 26: 184-192. [Crossref]
- Van der Stelt M, Di Marzo V (2003) The endocannabinoid system in the basal ganglia and in the mesolimbic reward system: implications for neurological and psychiatric disorders. Eur J Pharmacol 480: 133-150. [Crossref]
- Iversen L, Chapman V (2002) Cannabinoids: a real prospect for pain relief? Curr Opin Pharmacol 2: 50-55. [Crossref]
- Randall MD, Harris D, Kendall DA, Ralevic V (2002) Cardiovascular effects of cannabinoids. Pharmacol Ther 95: 191-202. [Crossref]
- Di Carlo G, Izzo AA (2003) Cannabinoids for gastrointestinal diseases: potential therapeutic applications. Expert Opin Investig Drugs 12: 39-49. [Crossref]
- Maccarrone M, Wenger T (2005) Effects of cannabinoids on hypothalamic and reproductive function. Handb Exp Pharmacol : 555-571. [Crossref]
- Klein TW (2005) Cannabinoid-based drugs as anti-inflammatory therapeutics. Nat Rev Immunol 5: 400-411. [Crossref]
- Di Marzo V, Petrosino S (2007) Endocannabinoids and the regulation of their levels in health and disease. Curr Opin Lipidol 18: 129-140. [Crossref]
- Licastro F, Chiappelli M (2003) Brain immune responses cognitive decline and dementia: relationship with phenotype expression and genetic background. Mech Ageing Dev 124: 525-528. [Crossref]
- Pizza V, Agresta A, D'Acunto CW, Festa M, Capasso A (2011) Neuroinflamm-aging and neurodegenerative diseases: an overview. CNS Neurol Disord Drug Targets 10: 621-634. [Crossref]
- Ramírez BG, Blázquez C, Gómez del Pulgar T, Guzmán M, de Ceballos ML (2005) Prevention of Alzheimer's disease pathology by cannabinoids: neuroprotection mediated by blockade of microglial activation. J Neurosci 25:1904-13. [Crossref]
- Esposito G, De Filippis D, Carnuccio R, Izzo AA, Iuvone T (2006) The marijuana component cannabidiol inhibits beta-amyloid-induced tau protein hyperphosphorylation through Wnt/beta-catenin pathway rescue in PC12 cells. J Mol Med 84: 253-8. [Crossref]
- Esposito G, De Filippis D, Maiuri MC, De Stefano D, Carnuccio R, et,al. (2006) Cannabidiol inhibits inducible nitric oxide synthase protein expression and nitric oxide production in beta-amyloid stimulated PC12 neurons through p38 MAP kinase and NF-kappaB involvement. Neurosci Lett 399: 91-5. [Crossref]
- Esposito G, Scuderi C, Savani C, Steardo L Jr, De Filippis D, et,al. (2007) Cannabidiol in vivo blunts beta-amyloid induced neuroinflammation by suppressing IL-1beta and iNOS expression. Br J Pharmacol 151: 1272-9. [Crossref]
- Banati RB, Daniel SE, Blunt SB (1998) Glial pathology but absence of apoptotic nigral neurons in long-standing Parkinson's disease. Mov Disord 13: 221-7. [Crossref]
- Möller T (2010) Neuroinflammation in Huntington's disease. J Neural Transm (Vienna) 117: 1001-1008. [Crossref]
- Garcia-Arencibia M, Gonzalez S, de Lago E, Ramos JA, Mechoulam R, et,al. (2007) Evaluation of the neuroprotective effect of cannabinoids in a rat model of Parkinson's disease: importance of antioxidant and cannabinoid receptor-independent properties. Brain Res 1134: 162-70. [Crossref]
- Sagredo O, Ramos JA, Decio A, Mechoulam R, Fernández-Ruiz J (2007) Cannabidiol reduced the striatal atrophy caused 3-nitropropionic acid in vivo by mechanisms independent of the activation of cannabinoid, vanilloid TRPV1 and adenosine A2A receptors. Eur J Neurosci 26 :843-51. [Crossref]
- Hermann D, Sartorius A, Welzel H, Walter S, Skopp G (2007) Dorsolateral prefrontal cortex N-acetylaspartate/total creatine (NAA/tCr) loss in male recreational cannabis users. Biol Psychiatry 61 :1281-1289. [Crossref]
- Frank-Cannon TC, Alto LT, McAlpine FE, Tansey MG (2009) Does neuroinflammation fan the flame in neurodegenerative diseases?. Molecular Neurodegeneration 4: 47. [Crossref]
- Iuvone T, Esposito G, Esposito R, Santamaria R, Di Rosa M, et,al. (2004) Neuroprotective effect of cannabidiol, a non-psychoactive component from Cannabis sativa, on beta-amyloid-induced toxicity in PC12 cells. J Neurochem 89: 134-41. [Crossref]
- Nguyen MD, Julien JP, Rivest S (2001) Induction of proinflammatory molecules in mice with amyotrophic lateral sclerosis: no requirement for proapoptotic interleukin-1beta in neurodegeneration. Ann Neurol 50: 630-639. [Crossref]
- Hampson AJ, Grimaldi M, Axelrod J, Wink D (1998) Cannabidiol and (-)Delta9-tetrahydrocannabinol are neuroprotective antioxidants. Proc Natl Acad Sci U S A 95: 8268-8273. [Crossref]
- Shoemaker JL, Seely KA, Reed RL, Crow JP, Prather PL (2007) The CB2 cannabinoid agonist AM-1241 prolongs survival in a transgenic mouse model of amyotrophic lateral sclerosis when initiated at symptom onset. J Neurochem 101: 87-98. [Crossref]
- Killestein J, Uitdehaag BM, Polman CH (2004) Cannabinoids in multiple sclerosis: do they have a therapeutic role? Drugs 64: 1-11. [Crossref]
- Mechoulam R (1986) The pharmacohistory of cannabis sativa, in Cannabis as Therapeutic Agent (Mechoulam R ed) pp 1-19, CRC Press, Boca Raton, FL.
- Mechoulam R, Fride E, Di Marzo V (1998) Endocannabinoids. Eur J Pharmacol 359: 1-18. [Crossref]
- Mechoulam R, Hanus L (2000) A historical overview of chemical research on cannabinoids. Chem Phys Lipids 108: 1-13. [Crossref]
- Grundy RI (2002) The therapeutic potential of the cannabinoids in neuroprotection. Expert Opin Investig Drugs 11: 1365-1374. [Crossref]
- Pertwee RG (2002) Cannabinoids and multiple sclerosis. Pharmacol Ther 95: 165-174. [Crossref]
- Baker D, Pryce G (2003) The therapeutic potential of cannabis in multiple sclerosis. Expert Opin Investig Drugs 12: 561-567. [Crossref]
- Croxford JL (2003) Therapeutic potential of cannabinoids in CNS disease. CNS Drugs 17: 179-202. [Crossref]
- Killestein J, Uitdehaag BM, and Polman CH (2004) Cannabinoids in multiple sclerosis: do they have a therapeutic role? Drugs 64: 1-11. [Crossref]
- Sirven JI, Berg AT (2004) Marijuana as a treatment for epilepsy and multiple sclerosis? A "grass roots" movement. Neurology 62: 1924-1925. [Crossref]
- Jackson SJ1, Diemel LT, Pryce G, Baker D (2005) Cannabinoids and neuroprotection in CNS inflammatory disease. J Neurol Sci 233: 21-25. [Crossref]
- Pryce G and Baker D (2005) Emerging properties of cannabinoid medicines in management of multiple sclerosis. Trends Neurosci 28: 272-276. [Crossref]
- Robson P (2005) Human studies of cannabinoids and medicinal cannabis, in Cannabinoids (Pertwee R ed) pp 719-757, Springer, New York.
- Smith PF (2005) The safety of cannabinoids for the treatment of multiple sclerosis. Expert Opin Drug Saf 4: 443-456. [Crossref]
- Lyman WD, Sonett JR, Brosnan CF, Elkin R, and Bornstein MB (1989) Delta 9-Tetrahydrocannabinol: a novel treatment for experimental autoimmune encephalomyelitis. J Neuroimmunol 23: 73-81. [Crossref]
- Wirguin I, Mechoulam R, Breuer A, Schezen E, Weidenfeld J, et,al. (1994) Suppression of experimental autoimmune encephalomyelitis by cannabinoids. Immunopharmacology 28: 209-214. [Crossref]
- Achiron A, Miron S, Lavie V, Margalit R, and Biegon A (2000) Dexanabinol (HU-211) effect on experimental autoimmune encephalomyelitis: implications for the treatment of acute relapses of multiple sclerosis. J Neuroimmunol 102: 26-31. [Crossref]
- Berrendero F, Sanchez A, Cabranes A, Puerta C, Ramos JA, et al. (2001) Changes in cannabinoid CB1 receptors in striatal and cortical regions of rats with experimental allergic encephalomyelitis, an animal model of multiple sclerosis. Synapse 41: 195-202.
- Baker D, Pryce G, Croxford JL, Brown P, Pertwee RG, et al. (2000) Cannabinoids control spasticity and tremor in a multiple sclerosis model. Nature 404: 84-87. [Crossref]
- Ligresti A, Cascio MG, Pryce G, Kulasegram S, Beletskaya I, et,al. (2006a) New potent and selective inhibitors of anandamide reuptake with antispastic activity in a mouse model of multiple sclerosis. Br J Pharmacol 147: 83-91. [Crossref]
- Pryce G, Ahmed Z, Hankey DJ, Jackson SJ, Croxford JL, et al. (2003) Cannabinoids inhibit neurodegeneration in models of multiple sclerosis. Brain 126: 2191-2202. [Crossref]
- Jackson SJ, Diemel LT, Pryce G, and Baker D (2005a) Cannabinoids and neuroprotection in CNS inflammatory disease. J Neurol Sci 233: 21-25. [Crossref]
- Jackson SJ, Pryce G, Diemel LT, Cuzner ML, and Baker D (2005b) Cannabinoid-receptor 1 null mice are susceptible to neurofilament damage and caspase 3 activation. Neuroscience 134: 261-266. [Crossref]
- Ni X, Geller EB, Eppihimer MJ, Eisenstein TK, Adler MW, et al. (2004) Win 55212-2, a cannabinoid receptor agonist, attenuates leukocyte/endothelial interactions in an experimental autoimmune encephalomyelitis model. Mult Scler 10: 158-164. [Crossref]
- Witting A, Chen L, Cudaback E, Straiker A, Walter L, et al. (2006) Experimental autoimmune encephalomyelitis disrupts endocannabinoid-mediated neuroprotection. Proc Natl Acad Sci USA 103: 6362-6367. [Crossref]
- Croxford JL, Miller SD (2003) Immunoregulation of a viral model of multiple sclerosis using the synthetic cannabinoid R+WIN55,212. J Clin Invest 111: 1231-1240. [Crossref]
- Arevalo-Martin A, Vela JM, Molina-Holgado E, Borrell J, and Guaza C (2003) Therapeutic action of cannabinoids in a murine model of multiple sclerosis. J Neurosci 23: 2511-2516. [Crossref]
- Mestre L, Correa F, Arevalo-Martin A, Molina-Holgado E, Valenti M, et al. (2005) Pharmacological modulation of the endocannabinoid system in a viral model of multiple sclerosis. J Neurochem 92: 1327-1339. [Crossref]
- Ortega-Gutierrez S, Molina-Holgado E, Arevalo-Martin A, Correa F, Viso A, Lopez-Rodriguez ML, et al. (2005) Activation of the endocannabinoid system as a therapeutic approach in a murine model of multiple sclerosis. FASEB J 19: 1338-1343. [Crossref]
- Eljaschewitsch E, Witting A, Mawrin C, Lee T, Schmidt PM, et al. (2006) The endocannabinoid anandamide protects neurons during CNS inflammation by induction of MKP-1 in microglial cells. Neuron 49: 67-79. [Crossref]
- Grinspoon L and Bakalar JB (1993) The history of cannabis, in Marihuana: The Forbidden Medicine, pp 1-23, Yale University Press, New Haven, CT.
- Grinspoon L and Bakalar JB (1998) The use of cannabis as a mood stabilizer in bipolar disorder: anecdotal evidence and the need for clinical research. J Psychoact Drugs 30: 171-177. [Crossref]
- Dunn M, Davis R (1974) The perceived effects of marijuana on spinal cord injured males. Paraplegia 12: 175. [Crossref]
- Petro DJ (1980) Marihuana as a therapeutic agent for muscle spasm or spasticity. Psychosomatics 21: 81-85. [Crossref]
- Petro DJ, Ellenberger C Jr (1981) Treatment of human spasticity with delta 9-tetrahydrocannabinol. J Clin Pharmacol 21: 413S-416S. [Crossref]
- Clifford DB (1983) Tetrahydrocannabinol for tremor in multiple sclerosis. Ann Neurol 13: 669-671. [Crossref]
- Meinck HM, Schönle PW, Conrad B (1989) Effect of cannabinoids on spasticity and ataxia in multiple sclerosis. J Neurol 236: 120-122. [Crossref]
- Brenneisen R, Egli A, Elsohly MA, Henn V, and Spiess Y (1996) The effect of orally and rectally administered Delta 9-tetrahydrocannabinol on spasticity: a pilot study with 2 patients. Int J Clin Pharmacol Ther 34: 446-452. [Crossref]
- Schon F, Hart PE, Hodgson TL, Pambakian AL, Ruprah M, et al. (1999) Suppression of pendular nystagmus by smoking cannabis in a patient with multiple sclerosis. Neurology 53: 2209-2210. [Crossref]
- Martyn CN, Illis LS, Thom J (1995) Nabilone in the treatment of multiple sclerosis. Lancet 345: 579. [Crossref]
- Greenberg HS, Werness SA, Pugh JE, Andrus RO, Anderson DJ, et al. (1994) Short-term effects of smoking marijuana on balance in patients with multiple sclerosis and normal volunteers. Clin Pharmacol Ther 55: 324-328. [Crossref]
- Consroe P, Musty R, Rein J, Tillery W, and Pertwee R (1997) The perceived effects of smoked cannabis on patients with multiple sclerosis. Eur Neurol 38: 44-48. [Crossref]
- Killestein J, Hoogervorst EL, Reif M, Kalkers NF, Van Loenen AC, et al. (2002) Safety, tolerability, and efficacy of orally administered cannabinoids in MS. Neurology 58: 1404-1407. [Crossref]
- Thompson AJ, Baker D (2002) Cannabinoids in MS: potentially useful but not just yet! Neurology 58: 1323-1324. [Crossref]
- Zajicek J, Fox P, Sanders H, Wright D, Vickery J, et al. (2004) The cannabinoids in MS study—final results from 12 months follow-up. Mult Scler 109: S115.
- Zajicek J, Fox P, Sanders H, Wright D, Vickery J, et al. (2003) Cannabinoids for treatment of spasticity and other symptoms related to multiple sclerosis (CAMS study): multicentre randomised placebo-controlled trial. Lancet 362: 1517-1526. [Crossref]
- Fox P, Bain PG, Glickman S, Carroll C, Zajicek J (2004) The effect of cannabis on tremor in patients with multiple sclerosis. Neurology 62: 1105-1109. [Crossref]
- Vaney C, Heinzel-Gutenbrunner M, Jobin P, Tschopp F, Gattlen B, et al. (2004) Efficacy, safety and tolerability of an orally administered cannabis extract in the treatment of spasticity in patients with multiple sclerosis: a randomized, double-blind, placebo-controlled, crossover study. Mult Scler 10: 417-424. [Crossref]
- Vaney C, Heinzel-Gutenbrunner M, Jobin P, Tschopp F, Gattlen B, et al. (2004) Efficacy, safety and tolerability of an orally administered cannabis extract in the treatment of spasticity in patients with multiple sclerosis: a randomized, double-blind, placebo-controlled, crossover study. Mult Scler 10: 417-424. [Crossref]
- Wade DT, Robson P, House H, Makela P, and Aram J (2003) A preliminary controlled study to determine whether whole-plant cannabis extracts can improve intractable neurogenic symptoms. Clin Rehabil 17: 21-29. [Crossref]
- Smith PF (2004) GW-1000. GW Pharmaceuticals. Curr Opin Investig Drugs 5: 748-754. [Crossref]
- Wade DT, Makela P, Robson P, House H, and Bateman C (2004) Do cannabis-based medicinal extracts have general or specific effects on symptoms in multiple sclerosis? A double-blind, randomized, placebo-controlled study on 160 patients. Mult Scler 10: 434-441. [Crossref]
- Russo E, Guy GW (2006) A tale of two cannabinoids: the therapeutic rationale for combining tetrahydrocannabinol and cannabidiol. Med Hypotheses 66: 234-246. [Crossref]
- Russo EB (2004) Clinical endocannabinoid deficiency (CECD): can this concept explain therapeutic benefits of cannabis in migraine, fibromyalgia, irritable bowel syndrome and other treatment-resistant conditions? Neuro Endocrinol Lett 25: 31-39. [Crossref]
- Fernández-Ruiz J (2009) The endocannabinoid system as a target for the treatment of motor dysfunction. Br J Pharmacol 156: 1029-1040. [Crossref]
- Glass M, Dragunow M, and Faull RL (2000). The pattern of neurodegeneration in Huntington's disease: a comparative study of cannabinoid, dopamine, adenosine and GABA(A) receptor alterations in the human basal ganglia in Huntington's disease. Neuroscience. 97: 505-519. [Crossref]
- Lastres-Becker I, Fezza F, Cebeira M, Bisogno T, Ramos JA et al. (2001) Changes in endocannabinoid transmission in the basal ganglia in a rat model of Huntington's disease. Neuroreport. 12: 2125-2129. [Crossref]
- Denovan-Wright EM and Robertson HA (2000). Cannabinoid receptor messenger RNA levels decrease in a subset of neurons of the lateral striatum, cortex and hippocampus of transgenic Huntington's disease mice. Neuroscience 98: 705-713. [Crossref]
- Lastres-Becker I, Gomez M, de Miguel R, Ramos JA and Fernández-Ruiz J (2002) Loss of cannabinoid CB(1) receptors in the basal ganglia in the late akinetic phase of rats with experimental Huntington's disease. Neurotox Res 4: 601-608. [Crossref]
- Naver B, Stub C, Moller M, Fenger K, Hansen AK, et al. (2003) Molecular and behavioral analysis of the R6/1 Huntington's disease transgenic mouse. Neuroscience 122: 1049-1057. [Crossref]
- McCaw EA, Hu H, Gomez GT, Hebb AL, Kelly ME, et al. (2004). Structure, expression and regulation of the cannabinoid receptor gene (CB1) in Huntington's disease transgenic mice. Eur J Biochem 271: 4909-4920. [Crossref]
- Centonze D, Rossi S, Prosperetti C, Tscherter A, Bernardi G, et al. (2005) Abnormal sensitivity to cannabinoid receptor stimulation might contribute to altered gamma-aminobutyric acid transmission in the striatum of R6/2 Huntington's disease mice. Biol Psychiatry 57: 1583-1589. [Crossref]
- Pazos MR1, Sagredo O, Fernández-Ruiz J (2008) The endocannabinoid system in Huntington's disease. Curr Pharm Des 14: 2317-2325. [Crossref]
- Dowie MJ, Bradshaw HB, Howard ML, Nicholson LF, Faull RL, et al. (2009) Altered CB1 receptor and endocannabinoid levels precede motor symptom onset in a transgenic mouse model of Huntington's disease. Neuroscience 163: 456-465. [Crossref]
- Blázquez C, Chiarlone A, Sagredo O, Aguado T, Pazos MR, et al. (2011) Loss of striatal type 1 cannabinoid receptors is a key pathogenic factor in Huntington's disease. Brain 134: 119-136. [Crossref]
- Casteels C, Vandeputte C, Rangarajan JR, Dresselaers T, Riess O, et al. (2011) Metabolic and type 1 cannabinoid receptor imaging of a transgenic rat model in the early phase of Huntington disease. Exp Neurol 229: 440-449. [Crossref]
- Mievis S, Blum D, Ledent C (2011) Worsening of Huntington disease phenotype in CB1 receptor knockout mice. Neurobiol Dis 42: 524-529. [Crossref]
- Van Laere K, Casteels C, Dhollander I, Goffin K, Grachev I, et al. (2010) Widespread decrease of type 1 cannabinoid receptor availability in Huntington disease in vivo. J Nucl Med 51: 1413-1417. [Crossref]
- Palazuelos J, Aguado T, Pazos MR, Julien B, Carrasco C, et al. (2009) Microglial CB2 cannabinoid receptors are neuroprotective in Huntington's disease excitotoxicity. Brain. 132: 3152-3164. [Crossref]
- Dowie MJ, Howard ML, Nicholson LF, Faull RL, Hannan AJ, et al. (2010) Behavioural and molecular consequences of chronic cannabinoid treatment in Huntington's disease transgenic mice. Neuroscience 170: 324-336. [Crossref]
- Consroe P, Laguna J, Allender J, Snider S, Stern L, et al. (1991) Controlled clinical trial of cannabidiol in Huntington's disease. Pharmacol Biochem Behav 40: 701-708. [Crossref]
- Curtis A, Mitchell I, Patel S, Ives N, Rickards H (2009) A pilot study using nabilone for symptomatic treatment in Huntington's disease. Mov Disord 24: 2254-2259. [Crossref]
- Müller-Vahl KR, Schneider U, Emrich HM (1999) Nabilone increases choreatic movements in Huntington's disease. Mov Disord 14: 1038-1040. [Crossref]
- Curtis A and Rickards H (2006) Nabilone could treat chorea and irritability in Huntington's disease. J Neuropsychiatry Clin Neurosci 18: 553-554. [Crossref]
- Blázquez C, Chiarlone A, Sagredo O, Aguado T, Pazos MR, et al. (2011) Loss of striatal type 1 cannabinoid receptors is a key pathogenic factor in Huntington's disease. Brain 134: 119-136. [Crossref]