Abstract
Neurofibromatosis type 1 (NF 1) is a very often emerging neurocutaneous genetic disease. It is originated from the mutation in NF 1 gene which exists in chromosome, number 17 and appears once in 2500-3000 live birth. Also, spontaneous mutation is in charge of about 50% of cases. Endemic clinical features are cafe-au-lait stains on the skin, cutaneous neurofibromas, freckling on cleavages and Lisch nodules. More serious problems which ruin quality of life are plexiform neurofibromas, central neural system tumors, endocrine disorders, orthopedic problems, behavior disorders and cognitive incompetence. In addition, a discriminating diagnosis was performed with Proteus syndrome which the asymmetric and progressive extremity hypertrophy could be seen with.
In this study, we found appropriate to evaluate a 25 year old male patient with no family history, whose quality of life was seriously spoiled, who had hydrocephaly due to aquaduct stenosis, which created both aesthetic and functional problems in his left hand, who had an asymmetric extremity hypertrophy because of cutaneous neurofibromas and painful hip dysplasias and due to arthropathic neuropathy developed, additionally who was an inpatient with cognitive incompetence in the lights of literature data.
Key words
Cutaneous neurofibromas, hypertrophy, aquaduct stenosis, hydrocephaly, skeleton anomalies, neurofibromatosis type 1, proteus syndrome
Introduction
Neurofibromatosis type 1(NF 1) is also known as von Recklinghausen disorder and the most common reported neuro cutaneous genetically disease in single gene disorders. It occurs once in 2500-3000 live births without race, gender and ethnic feature indications [1,2]. The disorder which shows autosomal dominant transition results from a heterozygote mutation in NF 1 gene which is located in chromosome, number 17 q11. 2. The gene, NF 1 encodes bigger cytoplasmic protein called as neurofibroma and this protein is the negative regulator of ‘Ras’ protooncogene. Neurofibroma which is the protein product of the gene, NF 1 plays a crucial role in transmission of cell signal related with learning, memory and synaptic plasticity [2-6]. The diagnosis in NF 1 is based on the clinical criteria. But, if it is not possible to diagnose with the criteria or needed a prenatal diagnosis, then, today, gene mutation analysis can be performed and they are sensible at the rate of 95% [5]. In half of the cases, this disorder emerges sporadically by spontaneous mutations which cause to grow abnormally in nerve cells and fibrous tissues. The gene, NF 1 is distinguished by a series of consecutive clinical symptoms and can almost affect systems of all organs and cause to multiple disorders. If two or more of the symptoms below are substantial, clinical diagnosis is then expressed:in a diameter larger than 5mm in the phase before puberte, in a diameter larger than 15mm in the phase after puberte, 6 or more cafe au lait macules, 2 or more neurofibromas of any type or a plexiform neurofibroma; axils or inguinal freckling; optic glioma;2 or more Lisch nodules in iris;it is that he got diagnosis of NF – 1 having criteria existing in one of his first-degree relatives or a significant osseous lesion like pseudoarthrosis or sfenoid wing dysplasia [2,3]. Clinical features of neurofibromatosis type 1 (NF-1)patients cannot be distinguished before, they are quite variable and progressive. Diagnosis becomes precise when 2 of the 7 criteria of diagnosis identified by international health institute are noticed (NF 1988). But, a discriminating diagnosis is required according to many other syndroms distinguishing themselves by asymmetric neoplasm [1-3]. In those syndroms, especially Proteus has an importance [4-9]. The most common clinical features in NF-1 are cafe-au-lait stains on the skin, cutaneous neurofibromas, freckling in skinfolds and Lisch nodules (iris hamartoma). There are also lesions featuring cafe-au-lait stains indicating a mosaic diffusion and asymmetric, unproportional excessive extremity growing in Proteus syndrom. But, as against neurofibromas observed in NF -1, nodules which are like brain substance and noticed as asymmetric especially on foot soles occur in Proteus syndrom [10-13]. It is important that radiological inspecting as well as clinical features are leading the way in the discriminating diagnosis of these syndroms [14]. The other problems increasing morbidity and mortality, disrupting quality of life in neurofibromatosis type 1 are deformities due to plexiform neurofibromas, central neural system tumors (optic glioma), various organ malignities, mental retardation, problems of language, learning disabilities, lack of attention, hyperactivity disorder, autism, depression and anxiety [15-20].
Case presentation
A 25 year old male patient with complaints of acute pain in his right hip and being unable to walk was brought to neurology clinic. He was conscious, cooperative and oriented. He had a look of acute pain on his face. His retardation of psychosocial and psychomotor was distinctive. He had macrocephaly and many prevalent subcutaneous neurofibromas. He had a limitation of movement in both hips and a progression of flexion contractures in his knees. On the left in his upper extremity, there was a view of dysmorphic and hypertrophy on the hand and forearm and was unable to use his hand. Besides, he had many cafe au lait stains of 15 mm or larger size (Figures 1-3).
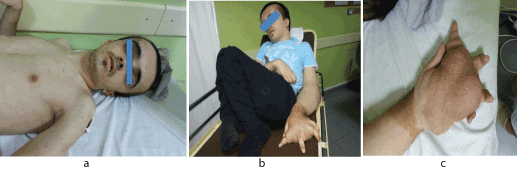
Figure 1. a. Thorax front Walland cafe au lait stainsinextremities, b. position ofthecase and contractures inextremities, c. congenital anatomic anomalyonthelefthand
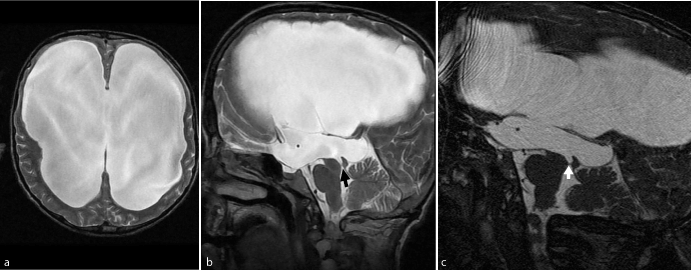
Figure 2. a. T2A Axial MR, b. T2 ASagittalMR c. FIESTA
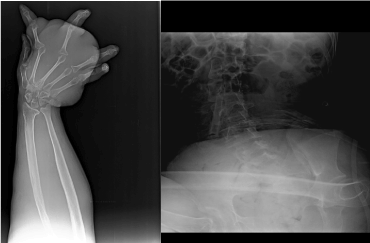
Figure 3. a. graph of left forearm and hand, b. Lumbar vertebra and graph of hip
With the anamnesis taken from his parents;it was found out that he was the fourth male child of the family which was not an intermarriage and born by normal vaginal canal at a hospital with a birth-weight of 3800 gr. and no progression of birth asphyxia. It was stated that he had a convulsion on postnatal 20th day and he was hospitalized for 2 weeks, examined and treated, macrocephaly started to progress in the 3rd month, offered an operation as diagnosed hydrocephaly, but he could not be operated and his macrocephaly had a progression, his neuromotor history was undeveloped and he started to walk at the age of 2.5-3. It was found out that he started to use his left arm after the age of 3-4 because of his skeleton anomaly and dysmorphic hand by birth, got educated till 3rd year class at primary school and learnt to write and read, but he was bad at maths . It was stated that he had difficulty in walking after the age of 8 and could walk skittering, had an operation on the right hip 5 years ago, had pain on the hipsdue to arthropathic neuropathy and he was confined to bed for 5 years .
There was an advanced degree of hydrocephaly in his studies of cranial MR and therefore a very obvious parenchymal atrophy developed at frontal levels of periventricular White substance . A light membrane limiting the canal at the level of aquaduct was taking the attention and it was understood that hydrocephaly was linked to aquaduct stenosis. Corpus callosum was highly atrophic (Figure 2).
Electroencephalographic examination was performed and epileptiform activity was not observed . With the battery of neuropsychological test, logical memory-instant reminiscence, test of face recognition, abstract thinking, tests of clock drawing and Boston naming, test of Luria Alternan drawings, copying shape and standard mini mental test were applied. He got 17 points in mini mental test after those tests. He was effective in the test of Luria Alternan drawings, but he failed the test of copying shape. In the test of Boston naming, the number of parameters he could never name was 6. He could do nothing in the tests of abstract thinking and number range . He failed instant reminiscence. According to Shulman evaluation, he got 3 points in the test of clock drawing . His instant memory was not enough, his attention and ability of keeping attention was very low and he got a middle level of cognitive incompetence. Straight skeleton graphs were taken and it was established that there was a sight of osteoporotic on all his skeleton bones, deformations and luxations in his left humerus distal epiphyses and humeroradial and humeroulnar joints. It was determined that there was deformation on the At the level of elbow joint on the left there was determined deformation, decreasing at the top of the radius and osteophytic changes in its sides. It was being observed that there was remodelling and from place to place focal thinning in cortical bones at the level of ulna diaphysis and radius on the forearm on the left. The symptoms were linked to neurofibromas in perosseous regions or periosteal bone resorption in this place. Similarly, there was also clear thickening in soft tissues at the level of forearm and hand . It was noticed that slight deformation occured in metacarpophalangeal joints on the left hand. It was defined that there was a rotation and a clear scoliotic view in lumbar vertebrae. There was deformation at the top of right femur in hip films and both acetabular angles significantly increased on the right. There was a bilateral progressive hip dysplasia (Figure 3). As for Lisch nodules, he had a detailed eye test and it was found normal.
In order to publish the pictures of the case, a written permission document was taken from his father.
Dıscussıon and result
Neurofibromatosis type 1 (NF 1) is a common neurocutaneous genetic disorder, it is evident with both focal and generalized skeleton anomalies. But, Proteus syndrom whic has been known as ‘elephant man’disorder for many years has also been defined as NF type 1 [5]. But Proteus syndrom (PS) is a very rare noticed congenital disorder, however it is observed with excessive assymetric and progressive growing . There is also excessive assymetric and progressive growing in our case. Facial hemihypertrophy which plenty of soft tissue nodules accompany in the mouth at Proteus syndrom is a crucial symptom. Proteus syndrom has a highly variable clinic spectrum. Patients are characteristically normal at birth, but indications and symptoms begin to grow progressively during childhood time. This feature can be a overlapping when it is compared with the symptoms of NF-1. But, many of those soft tissue nodules noticed at Proteus syndrome are in the type of connective tissue nodules alike brain with their external appearances and they immediately hold the skin. Focal atrophies can also appear in lipoma or fatty tissue besides those assymetric and unproportional growing nodules . Those brain -like nodules observed at Proteus syndrome generally occur increasingly on hands and foot soles. But, the nodules in NF-1 are in the type of subcutaneous and /or plexiform neurofibromas [5,7,9,10]. Although NF 1 is seen equally in both types, Proteus syndrome occurs once in 2 male patients instead of 1 female . There is not a defined gene in Proteus syndrome and how it occurs is sporadic, in addition it is defined postzygotic somatic mutation [4-6]. NF-1 gene which is in chromosome, number 17 q11. 2 is held accountable in NF 1 and it shows autosomal dominant progression. NF-1 gene also encodes the large cytoplasmic protein called as neurofibroma and this protein is the negative regulator of ‘Ras’ protooncogene [2,3]. Diffusion of cafe au lait stains in Proteus syndrome shows a mosaic structure and comes into sight sporadically. Especially capillary, parts from NF type 1 with venous and lymphatic malformations. Before the second decate, specific tumors (two sided ovarian cystadenoma or monomorphic adenoma in parotis) can be noticed. Proteus syndrome affects the skeleton very often . Its characteristic features are macrodactylia, clinodactyly, excessive assymetric growing in extremities, scoliosis, hyperostosis, focal calvarial thickening, macrocephalus due to hyperostosis, enlargement of ribs. A discriminating diagnosis can also be made with accompanying face features. Characteristic features like a sight of a long face, dolichocephaly, ptosis, a flattened nose, anteversion of nostrils are considerable face symptoms out of NF-1 [5-14].
In NF-1, tibial dysplasia, acute angled scoliosis and sphenoid wing dysplasia are frequently observed. Also, pathologic hip dislocation is slightly reported in patients of NF-1 . In cases of dislocations, intra articular growing neurofibromas are kept responsible. Similarly, neurofibromas cause feeling of inadequacy at hip joint and leading neuropathic arthropathy, they make progression of dislocation easy [1-3]. Another common feature in NF 1 is macrocephaly occured in nearly 30% of cases and it is distinguished by abnormal growth of both white and grey substance. But, it is stated that the rate of macrocephaly due to aquaduct stenosis is 1% [1-7]. Furthermore, common NF 1 complications are neurocognitive inadequacies. In order to plan the training, neuropsychological evaluation should be performed in an earlier phase as possible as it could be [5,6]. Features noticed at a high rate in cognitive phenotype of NF-1 are lack of attention, hyperactivity disorder (ADHD), autistic spectrum disorders, behavioural disorders and psychosocial retardation [5,6,16,20].
The cause of macrocephaly was aquaduct stenosis in our case. It could be displayed by a special sequence studied out of membrane conventional MR at the level of Aquaduct Silvi. Progressive hip dysplasia was bilateral and although right hip was operated the case worsened and occured a matter of progressive neuropathic pain. Besides, hypertrophy in forearm and hand together with upper left extremity dysplasia led to dysmorphic position, the case was unable to use the left hand. Also, confinement to bed affected the quality of life [15,22].
When the literature was inspected, the matter of macrocephaly which had an abnormal structural link as aquaduct stenosis in our case, two sided display of the rare hip dislocation and occurence of neuropathic osteoarthropathy, dysmorphic and hypertrophic structure of left hand were found suitable to be presented together with progressive clinic and neurocognitive incompetence, furthermore, in an NF-1 case, importance of discriminating diagnosis with Proteus syndrome was explained.
As a result, observation of more than one anomaly in only one case, initiation of two sided congenital hip dysplasia and in spite of an operation on the right hip, occurence of arthropathy neuropathic due to intensive subcutaneous neurofibromas and confinement to bed suggested a reevaluation of the case. Besides, to be able to demonstrate membrane causing aquaduct stenosis with special techniques will make possible to learn the definite reason of hydrocephaly and identify treatment methods accordingly.
Acknowledgement
Thanks to Mr. Hakan Uzun who translated this case presentation into English.
Disclosure
This case was presented as an E-poster at the 49th national neurology congress.
References
- Williams VC, Lucas J, Babcock MA, Gutmann DH, Korf B, et al. (2009) Neurofibromatosis type 1 Minor disease features in neurofibromatosis type 1 (NF1) and their possible value in diagnosis of NF1 in children ≤ 6 years and clinically suspected of having NF1. Neurofibromatosis team of Sophia Children's Hospital. Pediatrics 123: 124-133.
- Jouhilahti EM, Peltonen S, Heape AM, Peltonen J (2011) The pathoetiology of Neurofibromatosis 1. Mini-Review. Am J Pathol 178: 1932-1939 [Crossref]
- Stumpf D, Alksne J, Annegers J, Brown S, Conneally P, et al., (1988) Neurofibromatosis. Conference Statement; National Institutes of Health Consensus Development Conference. Arch Neurol 45: 575-578
- Thiele EA, Korf BR 2021 Copyright OAT. All rights reservions in : Pediatric Neurology Principles & Practice. 771-796
- Boyd KP, Korf BR, Theos A (2009) Neurofibromatosis type 1. J Am ACAD Dermatol 61
- Hachon C, Iannuzzi S, Chaix Y (2011) Behavioural and cognitive phenotypes in children with neurofibromatosis type 1(NF-1):The link with the neurobiological level. Brain&Development 33: 52-61
- Lacerda LS, Alves UD, Zanier JFC, Machado DC, Camilo GB et al. (2014) Differential diagnoses of overgrowth syndromes: The most ımportant clinical and radiological disease manifestations. Radiology Research and Practice
- Legendre CM, Charpentier-Cote C, Drouin R, Bouffard C (2011) Neurofibromatosis Type 1: Persisting Misidentificationof the “Elephant Man” Disease. JABFM 6: e16409. [Crossref]
- Caballero PEJ, Espuela FL, Cuenca JCP, Sevilla RMR, Marrero JAF, et al. (2013) Clinical and neuroradiological signs in adults with type 1 neurofibromatosis. Neurologia 28: 361-365. [Crossref]
- Beachkofsky TM, Sapp JC, Biesecker LG and Darling TN. Progressive overgrowth of the cerebriform connective tissue nevus in patients with Proteus syndrome. J Am Acad Dermatol 63: 799-804. [Crossref]
- Dietrich RB, Glidden DE, Roth GM, Martin RA, Demo DS (1998) The Proteus Syndrome: CNS Manifestations. AJNR Am J Neuradiol 19: 987-990. [Crossref]
- Biesecker L (2006) The challenges of proteus syndrome: diagnosis and management . European Journal of Human Genetics 14: 1151-1157. [Crossref]
- Linton JA, Seo BK,Oh CS (2002) Proteus syndrome: A Natural Clinical Course of Proteus Syndrome . Yonsei Medical Journal 43: 259-266
- Kaduthodil MJ, Prasad DS, Lowe AS, Punekar AS, Yeung S, et al. Imaging manifestations in Proteus Syndrome:an unusual multisystem developmental disorder. The British Journal of Radiology 85: 793-799. [Crossref]
- Steen RG,Taylor JS, Langston JW, Glass JO, Brewer VR, et al. (2001) Prospective Evaluation of the Brain in Asymptomatic Children with Neurofibromatosis Type 1: Relationship of Macrocephaly to T1 Relaxation Changes and Structural Brain Abnormalities. Am J Neuroradiol 22: 810-817. [Crossref]
- Costa DS, Paula JJ, Rezende NA, Rodrigues LOC, Malloy-Diniz LF, et al. (2014) Neuropsychological impairments in elderly Neurofibromatosis type 1 patients. European Journal of Medical Genetics. 57: 216-219. [Crossref]
- Johnson BA, MacWilliams BA, Carey JC, Viskochil DH, D'Astous JL, et al. (2010) Motor proficiency in children with neurofibromatosis type 1. Pediatr Phys Ther 22: 344-348. [Crossref]
- Cnossen MH, Moons KG, Garssen MP, Pasmans NM, de Goede-Bolder A, et al. (1998) Minor disease features in neurofibromatosis type 1 (NF1) and their possible value in diagnosis of NF1 in children ≤ 6 years and clinically suspected of having NF1. Neurofibromatosis team of Sophia Children's Hospital. J Med Genet 35: 624-627.
- Clementi M, Milani S, Mammi I, Boni S, Monciotti C, et al. (1999) Neurofibromatosis type 1 growth charts. Am J Med Genet 87: 317-323. [Crossref]
- Saltık S, Başgül ŞS (2013) Quality of Life in Children with Neurofibromatosis Type 1, Based on Their Mothers’ Reports. Turk Psikiyatri Derg 24: 25-34. [Crossref]
- Galbraith JG, Butler JS, Harty JA (2011). Recurrent spontaneous hip dislocation in a patient with neurofibromatosis type 1: a case report. J Med Case Rep 5: 106
- Jones EA, Manaster BJ, May DA, Disler DG (2000) Neuropathic osteoarthropathy: diagnostic dilemmas and differential diagnosis. Radiographics 20: 279-293. [Crossref]