Introduction
Nail Patella Syndrome (NPS) is a mutational, hereditary disease resulting in a loss of functionality in transcription factor LMX1B. It is transmitted with an autosomal dominant inheritance pattern with complete penetrance. The pleiotropic gene LMX1B, a member of the “homeogene” family involved in development, participates in the normal configuration of the dorsal-ventral axis and the glomerular basement membrane during embryonic development [1,2].
It is estimated that it occurs in one out of every 50,000 births and is transmitted through autosomal dominant inheritance [1]. Little, in 1897 first described the disease, and Hawkins and Smith established its association with kidney disease in 1950. The gene responsible for this syndrome was described by Dreyer in 1998 [1] and has several names, such as: Nail-Patella Syndrome or Nail and Patella, hereditary onycho-osteodysplasia, hereditary osteo-onychodysplasia (HOOD), arthro-osteoonychodysplasia, osteo-onycho-dysostosis, onycho-meso-dysplasia, and fong disease. It is described as a disease that affects nails, skeletal system, kidneys and eyes [1,3,4]. We present a case diagnosed at the Service of Pediatric Nephrology, Hypertension, Dialysis and Transplantation at Roosevelt Hospital in Guatemala City. Early diagnosis and follow up is important for successful treatment. Because of the infrequency of this syndrome presentation, its signs and symptoms are often overlook.
Case report
An 8-year-old Mayan girl, who lives in a small village in a rural area of Guatemala, was referred by a pediatrician to the emergency room of Roosevelt Hospital in Guatemala City due to elevated creatinine and blood urea nitrogen (BUN) serum levels. The patient had a difficult birth because the mother has a hip dislocation. She is the second of three girls; her birth weight was 6 pounds (2.72kg). The family history is remarkable for limited elbow extension, hypoplasia of thumb nails, interphalangeal abnormalities, and hip dislocation in the mother; all consistent with NPS. Additionally, the patient’s younger sister demonstrated also positive signs for NPS: limited elbow extension and nail hypoplasia (Figure 1).
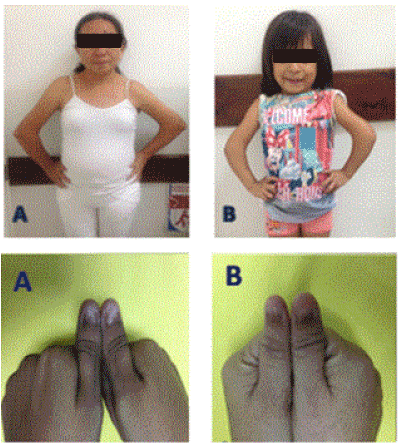
Figure 1. Mother (A) and younger sister (B) with arthrogryposis and hypoplasia of the nail thumbs.
Developmental milestones: The patient reportedly crawled at the age of 9 months and walked without assistance when she was two years old. At eight years of age, the mother noticed a decrease in her daughter’s growth and consulted several physicians during two years wherein several studies were performed. Thyroid tests were within normal limits, and hand radiography calculated a bone age of 5 years old. Blood chemistry showed evidence of hyperkalemia and elevated serum creatinine (4.18 mg/dl) and a BUN (42 mg/dl) and was referred to a tertiary level hospital (Hospital Roosevelt) in Guatemala City.
Upon physical exam, blood pressure was 90/60 mm/hg (90th percentile for gender and height is 112/73 mm/hg) with a height of 105 cm, corresponding to minus two standard deviations below the mean on the z score of the World Health Organization Child Growth Standards. The patient had a broad forehead, low-set hair and broad nasal philtrum as shown in Figure No. 2. Motion limitations were evident in the upper limbs with reduced extension in the elbows. The limitation was also demonstrated in flexion, pronation and supination of the hands. Hypoplasia of the thumbs was evident, thin nails with longitudinal grooves and absence of the crescent-shaped pale area at the base of the nail (moon nail) is malformed and/or triangular (Figure 2). Palpable iliac spines were prominent in the hips. There were no palpated kneecaps. Additional studies showed normocytic normochromic anemia, elevated potassium and phosphorus, decrease serum calcium levels. Creatinine was 4.18 mg/dl and estimated glomerular filtration rate (Schwartz formula) was 13 ml/min/1.73m². Random urine protein to creatinine ratio was 3.5 and venous blood gases were positive for metabolic acidosis (pH: 7.30 y HCO3 14).
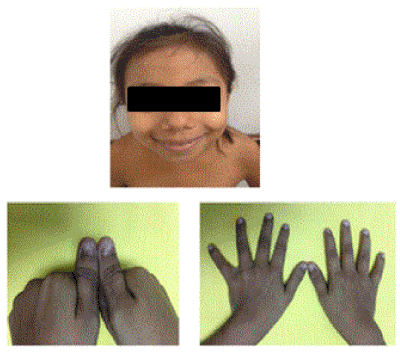
Figure 2. (A) Head: Broad forehead, low-set hair and broad nasal philtrum. (B) Thumbs: Thin nail hypoplasia with longitudinal grooves.
Discussion
Nail Patella Syndrome is an autosomal dominant disease evidenced by hypoplasia or absence of patella, nail dysplasia, limited elbow motion, and presence of iliac horns. About 40% of patients will show abnormalities in urinalysis, consisting in proteinuria and hematuria [1,5]. Of those with kidney damage, 5-10% develop nephrotic range proteinuria in childhood or adolescence and can progress to End Stage Renal Disease (ESRD) [6]. In 2015, the population in Guatemala was 16,000 million [7]. The estimated incidence of NPS is 22 per million inhabitants [8]. Base on this statistics, the existence of 352 cases can be predicted for Guatemala, however to date no cases have been reported.
Our 8-year-old patient demonstrated the following phenotypic characteristics of the NPS:
- Nail abnormalities: This is the most common sign of NPS. 98-100% of cases show nail hypoplasia of thumbs, brittle nail, longitudinal grooves and absence of moon nail [3,8,9]. (Figure No. 2)
- Elbow dysplasia: This sign occurs in 70% - 84% of patients with NPS. It is evident upon physical examination through motion limitation, accompanied by reduced extension, flexion, pronation and supination [2,3,8,9]. (Figure No.3)
- Knee abnormalities: These occur in 74-88% of patients suffering from NPS. They may present with absence of kneecaps. Lateral and bilateral radiography of the patella demonstrates hypoplasia of the medial femoral condyle [3,8,9]. (Figure No. 4).
- Bilateral iliac horns: Iliac bone horns consist of asymptomatic symmetrical bony outgrowths derived from the superior iliac crest (simulating flared elephant ears). Most commonly observed in the anteroposterior pelvic radiography. Although iliac horns are pathognomonic of NPS, they occur in only 70% of individuals with NPS [1,2,3,8]. (Figure No.5).
- Kidney disease: Renal involvement is present in approximately 40% of patients with NPS; it affects men and women equally. The degree of renal involvement varies between families and within members of the same family [1,3,8]. The glomerular basement membrane is the main target of this pathology. The LMX1B gene is expressed in podocytes and is responsible for the transcription of four genes, COL4A3, COL4A4, CD2AP and NPHS2 [10]. Together these genes produce an alteration in the permeability of the glomerular basement membrane and podocyte stability. Elimination of pericellular processes of podocytes as well as scattered deposits of collagen fibrils which are increased in the mesangial matrix have been observed through electron microscopy [1,8]. The first sign of renal disease as a result of NPS is proteinuria. Proteinuria in NPS can occur at any age from birth and it varies greatly among people.
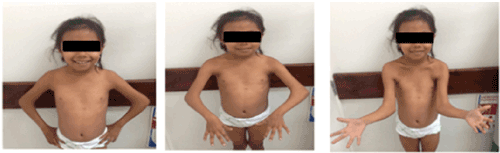
Figure 3. Arthrogryposis, elbows with reduced extension, flexion, pronation and supination.
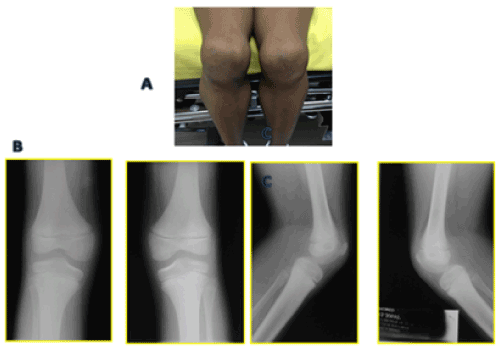
Figure 4. (A) No kneecaps observed. (B) Radiography shows absence of kneecaps. (C) Lateral patella Radiography show hypoplasia above the medial femoral condyle.
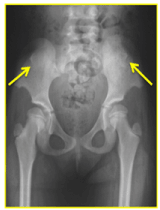
Figure 5. Horns Iliac bones, ridges that flared ears elephant.
In the case presented here, the patient reported evidence of proteinuria in the nephrotic range, with a protein/creatinine ratio of 3.5 based on a random urine sample. Estimated glomerular filtration rate was 13 ml/min/1.73m2 corresponding to stage V of chronic kidney disease (CKD) according to National Kidney Foundation Disease Outcomes Quality Initiative (KDOQI) classification.
Due to the patient's advanced kidney disease, a biopsy was not performed. Normochromic normocytic anemia, metabolic acidosis, hyperkalemia and hyperphosphatemia were all clear signs of ESRD due to NPS in our patient. Renal ultrasound showed changes in renal echogenicity with loss of cortico-medullary definition, decreased size in both kidneys and changes consistent with CKD (Figure No. 6).
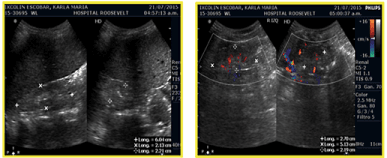
Figure 6. Both kidneys decreased in size, changes consistent with Chronic Kidney Disease.
In voiding phase cystourethrogram with fluoroscopy, there was no presence of vesicoureteral reflux. Dysgraphia was seen in L5 and S1 (Figure No.7) which is a rare finding related to NPS [6].
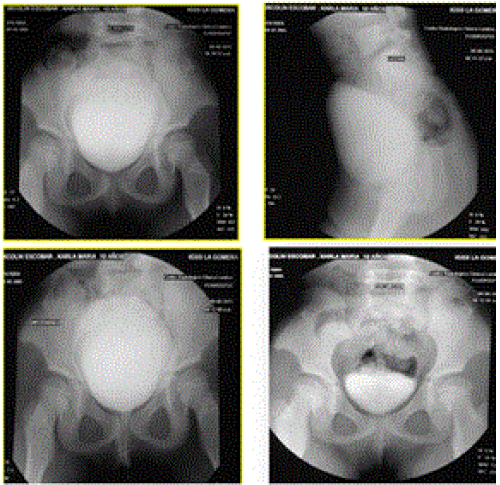
Figure 7. Voiding cystourethrogram with fluoroscopy.
Kidney failure may appear to happen rapidly or after many years of asymptomatic proteinuria. For patients with stage V of CKD as a result of NPS, renal transplantation has resulted favorably. No ultrastructural abnormalities are reported and no cases of disease recurrence have been documented [1,3,7].
Genetic analysis were not performed in the patient and relatives. Phenotypic manifestations of NPS were present in the mother and sister, such as arthrogryposis and hypoplasia of the nail thumbs, however, findings of renal disease were not present. Despite autosomal dominant hereditary disease, this syndrome exhibits variable penetrance and expression [11,12,13]. Therefore, one of the relatives might have mild or no symptoms while others might have severe symptoms of NPS (although they have the same genotype).
NPS is also associated with ophthalmologic abnormalities such as the Lester sign, a hyperpigmentation of the iris and glaucoma. Our patient did not present any ophthalmologic abnormalities.
Recommendations for pediatric patient care in NPS
Patients with a history or family history of NPS should be examined for kidney disease annually by the primary care. This includes blood pressure monitoring and urinalysis. An ophthalmologic evaluation may also be considered in cases of NPS including optic disc, visual field and intraocular pressure evaluation in order to detect glaucoma. If abnormalities are detected, the patient should be referred to a nephrologist and ophthalmologist for evaluation and follow up. Genetic counseling, when available, should be offered to all patients with NPS.
References
2021 Copyright OAT. All rights reserv
- Lemley K (2009) Kidney disease in nail–patella syndrome. Pediatr Nephrol 24: 2345–2354. [Crossref]
- Álvarez-Martín N, Gamundi M, Hernan I, Carbello M, Luis-Yanes MI, et al. (2013) Síndrome uña-rótula. Un caso con una mutación de novo en el gen LMX1B no descrita previamente. Revista Nefrología 33: 585-586.
- Ghoumid J, Petit F, Holder-Espinasse M, Anne-Sophie J, Guerra J, et al. (2016) Nail–Patella Syndrome: clinical and molecular data in 55 families raising the hypothesis of a genetic heterogeneity. Eur J Hum Genet 24: 44–50. [Crossref]
- Letts M (1991) Hereditary onycho-osteodysplasia (nail-patella syndrome). A three-generation familial study. Orthop Rev 20: 267-272. [Crossref]
- Bongers E, Gubler MC, Knoers N (2002) Nail-patella syndrome Overview on clinical and molecular findings. Pediatr Nephrol 17: 703–712. [Crossref]
- Goshen E, Schwartz A, Zilka LR, Zwas ST (2000) Bilateral accessory iliac horns: pathognomonic findings in Nail Patella Syndrome. Scintigraphic evidence on bone scan. Clin Nucl Med 25:476-477.
- Instituto Nacional de Estadística Guatemala (2015) Disponible en.
- Sweeney E, Fryer A, Mountford R, Green A, McIntosh I, et al. (2003) Nail patella syndrome: a review of the phenotype aided by developmental biology. J Med Genet 40: 153–162. [Crossref]
- Arenas-Planelles A, Arenas Miquélez A, Pombo Manero V, Ortega Arruti JA, Repáraz Padrós J, et al. (2005) The nail-patella Syndrome Report of 5 cases Revista Española de Cirugía Osteoarticular. 40: Nº 222.
- Morello R, Zhou G, Dreyer SD, Harvey SJ, Ninomiya Y, et al. (2001) Regulation of glomerular basement membrane collagen expression by LMX1B contributes to renal disease in nail patella syndrome. Nat Genet 27: 205–208. [Crossref]
- Grody W. Molecular diagnosis of genetic diseases. Henry's Clinical Diagnosis and Management by Laboratory Methods. McPherson-Pincus. El Sevier. 23th edition. Chapter 71, 1377-1395e2.
- Millá E, Hernan I, Gamundi MJ, Martínez-Gimeno M, Carballo M, et al. (2007) Novel LMX1B mutation in familial nail-patella syndrome with variable expression of open angle glaucoma. Mol Vis 13: 639-48. [Crossref]
- Bongers E, Huysmans F, Levtchenko E, de Rooy JW, Blickman JG, et al. (2005) Genotype–phenotype studies in nail-patella syndrome show that LMX1B mutation location is involved in the risk of developing nephropathy. Eur J Hum Genet 13: 935–946. [Crossref]