Introduction: Autoimmune myasthenia is a neuromuscular junction disorder that is poorly described in sub-Saharan Africa because of its rarity and difficult diagnosis.
Objectives, patients and methodology: We present the socio-demographic, clinical, paraclinical and evolutionary characteristics of a series of 14 patients prospectively collected from March 2012 to January 2017, in Tingandogo University Hospital, Ouagadougou, Burkina Faso.
Results: There was a predominance of females (8 females / 6 males), the age at onset of symptoms was between 20 and 40 years for the majority of women and between 40 and 60 years for males. Diagnostic delay was noted with an average delay of 24.73 months. In the initial clinical examination, according to the classification of Myasthenia Gravis Foundation of America, 9 patients (64.3%) were in class IV; Classes II and III were each represented by 2 patients (14.3%), whereas there was only one patient (7.1%) in Class I. Anti-acetylcholine receptor antibodies and Anti-muscle specific kinase antibodies were positive in 6 patients (50%) and 3 patients (25%), respectively. Thymic CT showed a thymic abnormality in 10 patients, including 4 cases of thymoma (28.6%) and 6 cases of thymic hyperplasia (42.8%). Thymectomy was performed in 7 patients (50%), followed by clinical remission in 4 patients (54.1%) and a death in one patient (7.1%).
Conclusion: Our results are consistent with those of the literature, except for a low prevalence of anti RACh antibodies and a diagnostic delay at the generalized severe stage.
myasthenia,anti RACh antibodies,thymome,thymectomy
Myasthenia Gravis (MG) is a rare autoimmune disease due to specific autoantibodies directed against neuromuscular junction proteins: acetylcholine nicotinic receptor (AChR), muscle specific kinase (MuSK), low-density lipoprotein receptor-related protein 4 (LRP4) [1,2], which induce a dysfunction of the neuromuscular transmission, the consequence of which is an excessive fatigability of the musculature striated to the effort. Its prevalence of 100 to 200 per million is similar in the majority of populations [2], constantly increasing, specifically in the elderly [3-5]. The MG at its beginnings is influenced by age and gender in a bimodal way: predominantly female before age 40 (F/H = 3), gender balance between 40 and 50 years of age, male preponderance from of the 6th decade [3,6,7]. MG was categorized into subtypes according to the age of onset (early or late), clinical (ocular or generalized), serological (seropositive or seronegative) and presence or absence of thymoma [8].
While MG is abundantly published in the different regions of the world, in sub-Saharan black Africa, the publications devoted to it seem rather rare: illness little known to the health professionals, under notification of the cases, diagnostic difficulty or little of interest for the disease? [9]. Does MG have the same socio-demographic, diagnostic and evolutionary profile in sub-Saharan Black Africa in general and Burkina Faso in particular, as in other regions of the world or are there any differences? According to a multiracial study conducted in Cape Town, South Africa [9], demographic data, excluding the age of onset, did not vary significantly among white or black patients with similar proportions of seropositive, seronegative and of thymoma. However, black patients had an increased proportion of myasthenic ophthalmoplegia complete with ptosis whereas whites were more likely to develop a severe generalized myasthenia gravis that responded poorly to treatment and had repeated seizures. Phenotypic differences in MG have already been observed between white and Chinese subjects [10]; eastern subjects were more likely to develop juvenile ocular MG (50%), had lower AChR titres and were less likely to develop a few generalized severe forms [1,10,11]. To answer this question and to contribute to a better understanding of MG in black Africa, we describe for the first time in Ouagadougou (Burkina Faso) a hospital series of 14 patients diagnosed and followed for myasthenia autoimmune.
It was a prospective, observational, descriptive, multicenter collaborative study, involving all patients diagnosed or treated for myasthenia autoimmune in the various public or private health structures of the city of Ouagadougou, over a period 4 years and 10 months from March 2012 to January 2017. In order to include the maximum number of patients, we included in the study all neurologists and ophthalmologists as well as some general practitioners and gastroenterologists from the city of Ouagadougou. We have also benefited from the support of the Burkina Faso Association of patients with myasthenia gravis. As soon as the diagnosis of myasthenia autoimmune was established, all patients were referred to the neurology department of the Tingandogo University Hospital in Ouagadougou, where they had at least regular quarterly neurological follow-up. In case of operative indication, thymectomy was performed in the thoracic surgery unit of the visceral surgery department of the Tingandogo University Hospital. In patients with a myasthenic outbreak or crisis, or for any intercurrent episode with hospitalization, the patients were either hospitalized in a neurological unit or in intensive care at the Tingandogo University Hospital.
Diagnostic criteria were based on clinical symptoms suggestive of MG, associated with at least one of the following criteria: high anti-AchR antibodies (> 0.4 nmol / ml), a decrement of ≥10% of the amplitude of the overall motor driving potential in an electroneuromyography (ENMG) with repetitive stimulation at 3 Hz. For each patient, socio-demographic data, age at onset of disease, age at diagnosis, diagnosis time, initial clinical picture and course of disease [clinical signs, motor muscle score (MMS), Myasthenia Gravis Foundation of America (MGFA) classification], ENMG and chest CT scan results. The anti-AChR autoantibody assay was performed using the radioimmunoprecipitation technique. Anti-MuSK antibodies were assayed by immunofluorescence on transfected cells. Immunological status, particularly for AChR and MuSK antibodies, has not been known for some patients because of the very high cost; none of our patients were able to carry out AChR antibodies assay with low affinity, LRP4 antibodies, titin antibodies, ryanodine antibodies ... There was no immunological monitoring report.
All patients received drug therapy (anticholinesterase +/- corticotherapy +/- immunosuppressant +/- motor kinesitherapy) and some patients underwent thymectomy followed by anatomopathological examination of the surgical specimens. No patients were treated with corticosteroids or immunosuppressants prior to myasthenia gravis diagnosis.
In the course of our study, the anonymity of the patients was guaranteed. The oral consent of all patients was collected. The data were entered and analyzed on a microcomputer using the Epi info software in version 7.1.1.14.
Socio-demographic characteristics of patients
During the study period, 14 patients were consecutively collected, including 8 female (57.1%) and 6 male (42.9%). The average age was 35 years (range 04-56 years). The age group of 20 to 40 years, with 08 patients (57.1%), was the most representative. The mean age of onset of symptoms was 28.8 years ± 14.9 (range of 04 - 55 years). There were 2 cases (14.3%) of forms with a juvenile onset (starting age <15 years), 10 cases (71.4%) of young adult forms (starting age between 15 and 50 years), 2 cases (14.3%) of late-onset forms (starting age> 50 years). The age groups of symptom onset were predominantly between 20 and 40 years for women (50%) and between 40 and 60 years for men (50%).
Clinical characteristics of patients
The mean time between the first symptoms and the first neurological visit was 24.7 months (range 0.3 - 108 months); the mean time between first symptoms and diagnosis was 24.9 months (range 0.3 - 108 months). The average time between symptom onset and surgical treatment was 36 months (3 years) (extremes 7 and 120 months), and the interval between diagnosis and thymectomy was 17.50 months (extremes of 3 and 60 months).
The clinical presentation was multi-symptomatic in 78.6% or 11 patients, whereas it was mono-symptomatic in 21.4% of cases (3 patients).
In the initial clinical examination, 13 patients (92.8%) had MG with generalized form and 1 patient (7.2%) had ocular form. A proximal motor deficiency of the scapular and pelvic belts in 8 patients (57.1%), limb muscle amyotrophy in 5 patients (35.7%), a decrease in thoracic ampliation in 3 patients (21.4 %), lingual amyotrophy in 2 patients (14.3%) were the main abnormalities observed during the initial clinical examination. The initial motor muscle score (MMS) averaged 50.8 out of 100 (extremes 22-90); 50% of patients (7 patients) had an initial MMS of between 25 and 50 out of 100. According to the clinical classification of MGFA, 9 patients (64.3%) were in class IV (severe muscle deficit other than ocular muscles), including 5 patients (35.7%) in the class Iva and 4 patients (28, 6%) in the class ІVb. Classes II (discrete deficit of muscles other than ocular muscles) and III (moderate deficit of muscles other than ocular muscles) were each represented by 2 patients (14.3%), one patient (7.1%) were in class I (ocular muscle deficit).
Paraclinical examinations
The ENMG of repetitive stimulation was performed in our 14 patients (100%) and showed a significant decrement (> 10%) in 13 patients (92.8%). AchR antibodies and MuSK antibodies responses were performed in 12 patients (85.7%); AChR antibodies returned positive in 6 patients (50%), with an average of 13.7 nmol / L (extremes 0.2 and 48 nmol / L); the MuSK antibodies were found in 3 patients (25%), or 50% of the 6 patients without AchR antibodies; 3 patients (25%) had returned negative. Thoracic CT scan was performed in all our patients and showed a thymic abnormality in 10 patients (71.4%): 4 cases of thymoma (28.6%) and 6 cases of thymic hyperplasia (42.8 %), the thymus was normal in 4 cases (28.6%). Thyroid hormone levels (TSH, T3,T4) and autoimmunity were determined in eight patients (57.1%) and two patients (14.3%), respectively, with normal income in all cases. The anatomopathological examination of the 6 surgical specimens confirmed 3 cases of type A thymoma already diagnosed with thoracic CT scan and 3 cases of thymic hyperplasia of follicular lymphoid type.
Evolution-complications
Nine (9) patients (64.3%) had an average of 3.7 relapses (range 1 to 6 relapses) during the course of MG with an average delay of 8.8 months month for the first outbreak, caused by the following triggers: warm periods of the year 5 cases, infection of upper airways 3 cases, menstruation 3 cases, taking contraindicated drugs (quinine, chloroquine) 2 cases, overwork or stress 2 cases, therapeutic rupture 2 cases, surgical intervention 1 case. Only one patient out of the 14 (7.1%) experienced a myasthenic crisis, favored by a ruptured treatment and bronchopulmonary superinfection with resistant germ, requiring intubation in intensive care and causing death, with a delay of 60-month (5 years).
Seven (7) patients (50%) had thymectomy, including 4 patients (28.6%) with MG + thymoma and 3 patients (21.4%) with generalized MG associated with thymic hyperplasia and low response to medical treatment.
The evolution of MMS in non-operated patients showed an average progression index of 8 points per semester under drug therapy, whereas in operated patients, this progression index was on average 4 points per semester before thymectomy, before moving to 5 points per semester after thymectomy. This index was on average 1.8 points per semester in patients with thymic abnormalities whereas it was 7.4 points per semester in patients with normal thymus. The evolution of the MMS in the operated and non-operated patients and the differences of evolution according thymic abnormalities are illustrated in FIGS. 1, 2 and 3 (confers annexes).
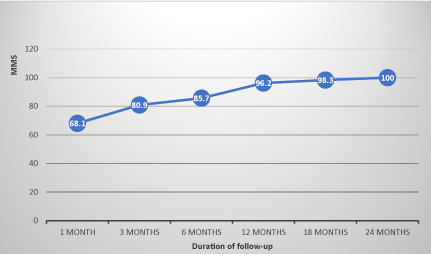
Figure 1. Evolution of the MMS of non operated patients.
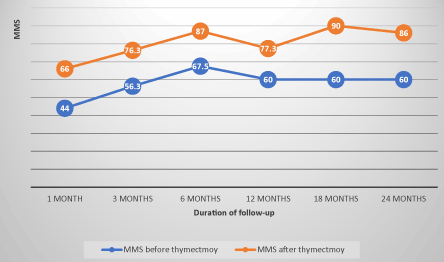
Figure 2. Evolution of the MMS of patients before and after thymectomy correlation coefficient=0.85.
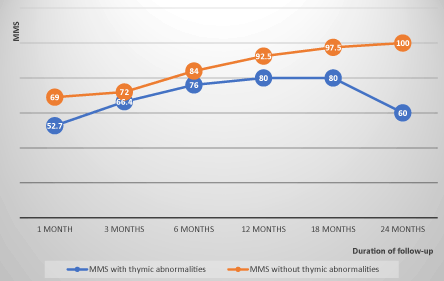
Figure 3. Evolution of the MMS of patients with and without thymic abnormalities correlation coefficient=0.5.
At the end of follow-up of the 7 patients operated, 4 patients (57.1%) were in remission, 2 in pharmacological remission and 2 in complete stable remission; 2 patients (28.6%) remained stationary; a patient (14.3%) with AChR antibodies and thymic hyperplasia died in a context of myasthenic crisis after rupture of drug therapy despite thymectomy. Patients in remission had all AchR antibodies, had either a thymoma (50%) or thymic hyperplasia (50%). The two patients with unfavorable clinical evolution (stationary clinical state) had no AChR antibody, one had MuSK antibodies and the other was seronegative, with one thymoma each.
MG with AChR antibodies accounted for 50% of the patients, with an average age of 26.7 years, a female predominance, exclusively consisting of severe generalized forms during diagnosis, consisting of 66.7% and 33.3% thymic hyperplasia (4/6) and thymoma (2/6); a clinical improvement was observed in 83.3% (5/6) and one death in 16.7% of the patients (1/6).
MG with MuSK antibodies accounted for 25% of the patients, with an average age of 26.5 years, a female predominance and severe generalized forms (2/3) during diagnosis, a normal thymus in 1/3 of the patients, and thymic hyperplasia or thymoma in 1/3 of each; the evolution was stationary in 2/3 of the cases, slightly improved in 1/3 of the cases.
Seronegative MG accounted for 25% of cases with an average age of onset at age 30, female predominance, a majority of ocular or generalized forms at diagnosis (2/3), a balanced distribution between thymic hyperplasia, thymoma and normal thymus, respectively 1/3 of the cases; the evolution was stationary in 2/3 of the cases.
Clinical features, chest CT characteristics and evolutionary characteristics of patients with MG, according to immunoassay results are summarized in Table 1.
Table 1. Clinical, CT scan and evolution characteristics of myasthenic patients, based on immunoassay results
Caractéristiques |
MG with AChR
antibodies
n=6 (50%) |
MG with MuSK
antibodies
n=3 (25%) |
seronegatives MG
(AChR/MuSK antibodies-)
n=3 (25%) |
MG with undefined
antibodies n=2 |
Clinical features |
Average age of onset |
26.7 years (18-55) |
26.5 years (25-28) |
30 years (16-52) |
22.3 years (4-43) |
Ratio F/H |
2/1 |
2/1 |
2/1 |
0/3 |
Initial MMS (n/100) |
43.9 (30-48) |
37.3 (22-55) |
66.7 (30-83) |
66.7 (50-90) |
MGFA classification at diagnosis |
IV |
III : 1
IV : 2 |
I : 2
IV : 1 |
I : 1
II : 1
IV : 1 |
Results of thoracic CT |
Thymic hyperplasia (n=6) (50%) |
4 |
1 |
1 |
1 |
Thymoma (n=4) (28,6%) |
2 |
1 |
1 |
0 |
Normal thymus (n=3) (21,4%) |
0 |
1 |
1 |
1 |
Treatment |
Medical treatment |
6 |
3 |
3 |
3 |
Thymectomy |
5 |
1 |
1 |
0 |
Evolution |
Stable or pharmacological clinical remission |
5 |
0 |
1 |
3 |
Clinical improvement |
0 |
1 |
0 |
0 |
Stationary or aggravated |
0 |
2 |
2 |
0 |
Death |
1 |
0 |
0 |
0 |
MG associated with a thymoma accounted for 28.6% of patients, with an average age of 27 years, female exclusivity, ¾ at the generalized stage of diagnosis, half had AchR antibodies. In terms of progression, half of the patients were either in clinical remission or in a steady clinical state. MG associated with thymic hyperplasia accounted for 42.8% of the patients, with an average age of 26 years, a female predominance (F / H ration = 2/1), 60% of patients had AchR antibodies, the majority of the patients had a generalized form during the diagnosis (5/6 of the patients), of serious evolution with a patient deceased (16.7%) following a myasthenic crisis.
The clinical, serological and evolutionary characteristics of MG according to the results of thoracic CT scan are summarized in Table 2.
Table 2. Clinical, serological and evolutionary characteristics of myasthenia gravis according to the results of thoracic CT
Clinical, serological and
evolutionary characteristics |
Results of thoracic CT scan |
Thymoma
n=4 (28.6%) |
Thymic hyperplasia
n=6 (42.8%) |
Normal thymus
n=4 (28.6%) |
Initial clinical features |
Mean age of onset |
27 years
(16-46 years) |
26 years
(4-55 years) |
35 years
(20-52 years) |
Ratio F/M |
4/0 |
2/1 |
0/4 |
Initial MMS |
46.7 (22-83) |
|
|
MGFA classification at diagnosis |
II : 1
IV : 3 |
I : 1
IV : 5 |
II : 2
III : 1
IV : 1 |
Serological status |
MG with AChR antibodies |
2 |
3/5 (60%) |
1/3 |
MG with MuSK antibodies |
1 |
1/5 (20%) |
1/3 |
Seronegative MG |
1 |
1/5 (20%) |
1/3 |
- |
Thymectomy |
4 (100%) |
3 (50%) |
0 |
Clinical evolution
|
Clinical remission |
2 |
4 (66.6%) |
2 |
Clinical improvement |
0 |
1 (16.7%) |
0 |
Stationary |
2 |
0 |
2 |
Death |
0 |
1 (16.7%) |
0 |
Autoimmune myasthenia gravis is a rare disease with an estimated prevalence of between 50 and 200 per million people worldwide and appears to be increasing in recent decades [12,13]. In our study, we identified 14 cases in 4 years and 10 months between March 2012 and January 2017. We found a female predominance with a ratio F/M 1.3, a predominance in young adults (<50 years), and only two cases each of late MG and juvenile MG (<15 years) were recorded, which is in line with data from the literature [12-20]. Sex hormones appear to play a central role and some studies involve estrogens as the main mediator of the disease [21,22]. At the beginning, MG is influenced by sex and age in a bimodal way: in women under 40, women predominate, but in the fifth decade cases are equally divided between men and women, and after 50 years, new cases are slightly more frequent in men [7,16-18,22,23]. This characteristic has also been observed in our patients.
The mean time between the first symptoms and the first neurological visit was 24.7 months and that between the first symptoms and the diagnosis of 24.9 months for our patients, similar to those of El Mahdi in Morocco (24 months) [15], but significantly higher than those of Barbaud et al. in Montpellier [24], Oöpik et al. in Estonia [20] and Boughammoura-Bouatay et al. in Tunisia [25], respectively of 17 months, 6.1 months and 5 months.
This diagnostic delay results in our context from the sub - medicalization and the frequent diagnostic wandering, because myasthenia, especially for the general practitioner, is not well known due to its relative rarity, the frequently intermittent nature of the clinical manifestations and, absence of objective signs at the time of the consultation; the risk is great to neglect symptoms such as fatigue, too early to account for an early stress or depression [26].
In our series, 5 patients (35.7%) had amyotrophy of the root of the limbs during the initial examination, which could be explained by the long duration of the disease evolution before the diagnosis and malnutrition due to disorders of swallowing and mastication. Majority of our patients had severe and generalized forms on initial clinical examination, due to long diagnostic delays. In comparison, 2/3 of the patients of El Mahdi were in class II MGFA during the diagnosis [15].
AChR antibodies were found in the serum of 6 of the 12 patients who were able to perform this assay, ie 50%. On the other hand, Fall [17], El Mahdi [15], and Nouvel [19] had superior results with 80%, 89% and 70% of myasthenia with AchR antibodies. In the literature, the prevalence of AchR antibodies varies from 75% to 90% for general MG, and from 50% to 60% for ocular MG [27,28]. In most patients with MG, the disease is caused by an immune humoral response directed against the ACh nicotinic muscle receptor [29]. Several reasons may explain the low prevalence of AchR antibodies in our patients. First, in the early forms, the levels of antibodies may be zero, before they subsequently appear, which justifies in view of a recent seronegative myasthenia, to repeat the dosage after a few months [30].
Secondly, in our series, 2 patients (14.3%) could not carry out the assay of the AChR antibodies, for want of financial means. Third, the prevalence of seronegative MG in our series is high, 25% versus 10-15% in the literature. Some (20%) have anti-LRP4 antibodies, mainly IgG1s able of binding complement, interfering in the binding of agrin to its receptor and modifying aggregation of AChR on muscle cells [1,31,32].
Others (60%) have AChR antibodies that are not detected by the available classical tests because they recognize only the AChR in a native cellular configuration, whereas in these myasthenic forms the AChRs are aggregated to the cell surface [33-35]. MuSK antibodies were found in 3 patients (25%), half of the 6 patients without AchR antibodies. MG with MuSK antibodies is of recent discovery and much less frequent than that with AchR antibodies. MuSK and AchR antibodies can coexist in the same patient. In one series, this association was found in 10.5% of MG with AchR antibodies [36]. Thymic abnormalities (thymic hyperplasia or thymoma) were detected in 71.4% of our patients, thymic hyperplasia in 42.8% and thymoma in 28.6 %, whereas data from the literature indicate the prevalence of thymic abnormalities in MG at about 75% [15,17,24], ie 10-15% for thymoma and 65% for thymic hyperplasia [37,38]. Thus, the prevalence of 28.6% of thymoma in our serie is much higher than in the literature and of Nouvel et al results (16.7%) in France, near Barbaud et al. (28%) in France, but lower than El Mahdi et al in Morocco (33%) and Fall et al in Senegal (40%). This observation leads us to hypothesize a possible higher prevalence of thymoma in myasthenic patients in Africa [15,17,19,24]. However, a study in South Africa found no significant racial differences in the prevalence of MG, seropositive, seronegative or associated with a thymoma [9].
In our study, only patients with thymic abnormalities (4 cases of thymoma and 3 cases of thymic hyperplasia) benefited from thymectomy. Evolution showed an average MMS progression index of 7.8 points per semester in non-operated patients, whereas it was only 4 points per semester before thymectomy, before moving to 5 points per semester after thymectomy. This index was on average 1.8 points per semester in patients with thymic abnormalities whereas it was 7.4 points per semester in patients with normal thymus, probably indicating a pejorative prognosis of MG with thymic abnormalities, with or without thymectomy. Subsequent studies with a large sample of patients are needed to answer this question. This post-thymectomy evolutionary status quo could also be explained by the long delays between the onset of the disease and thymectomy (3 years). Many studies suggest that the earlier the thymectomy is performed in the course of the disease, the better the results of the treatment, especially during the first two years of the disease. The therapeutic effect of thymectomy usually occurs after about one year [39]. The interest and indications of thymectomy in MG are still debated and are still not the subject of a consensus. Very broad for some, its indications are selective for others, reserved for thymoma, myasthenia generalized of the young subject or the patient who did not respond favorably to the immusuppressors and the corticoids; this explains the variable proportion of thymectomy from one serie to another. In our series, there was only one patient belonging to the myasthenia subgroup with pure ocular form, whereas El Mahdi (Morocco) [15], Maharaj (Trinidad) [18], Matuja (Tanzania) [40] and Ojini (Nigeria) [41] found 17%, 40%, 47% and 50% ocular form respectively in their series, while Zhang in China had 58% ocular form, whose 75% were children [1]. The proportion of ocular MG varies according to the authors and the populations, it is of 10-15% in the Caucasian populations and goes to more than 50% in Asians, with a clear predilection for the children [42]. The generalized form of MG is heterogeneous in terms of serological status, thymic abnormalities and age of onset. It is usually subdivided into early-onset (<50 years), late onset (> 50 years) and very late onset (> 60 years) [43]. In our series, 71.4% and 14.3%of our patients had an early onset and a late onset respectively; none of our patients had a very late onset (> 60 years), probably because of the low sample size, still low life expectancy, and diagnostic difficulties in our context. The increased incidence of this very late form of the disease in the developed countries is due to increased longevity in the population combined with a tendency to increase autoimmune diseases with age and the accumulation of predisposing environmental factors [43-45]. Approximately 40% of MG patients without AChR antibodies have MuSK antibodies. They are mostly young women [46], with severe clinical symptoms, with facial, bulbar and respiratory muscles symptoms, but rarely with ocular symptoms. Muscular atrophy is common in these patients [47]. The chances of achieving clinical remission of the disease are low [36,48]. In general, no thymic pathology was observed in this subgroup of patients [47,49]. In our series, we found the same characteristics described above, except that this subgroup represented 25% of all the MG with known serology and 50% of the generalized MG without AChR and a thymic anomaly (thymoma) was present in 1/3 of patients. In our series, 25% of patients were seronegative (AChR and MuSK antibodies absent). Seronegative patients are heterogeneous and may have a pure ocular form or a generalized form of MG [34]. They are near of classical myasthenia with AchR antibodies: young fringe, feminine preponderance, involvement in generalized moderate and ocular forms, involutive or hyperplasic thymus. These characteristics have been observed in our patients. Up to 60% of these patients appear to have low-affinity for AChR antibodies that are not detectable by conventional immunoprecipitation examinations because their auto antibodies recognize AChR only in its native configuration. Approximately 20% of the seronegative generalized myasthenia (2% of the total of the MG), have LRP4 antibodies. This recently described form [1,31], mostly composed of young women with moderate clinical form, but little is known about the associated thymic abnormalities [43].
We were unable to perform this type of serological examination. Even after LRP4 antibodies identification, some myasthenic patients are still triple-negative (2-5% of patients) [50].
Fifteen to 30% of myasthenic patients and 10-20% of generalized MG, according to the series, present a thymoma; these patients are usually over 40 years of age, both sexes are affected equally, the presence of AchR antibodies is almost constant; the elevation of the muscle striated and titine acids antibodies is in favor of a thymoma, but can also be seen in the elderly without thymoma. Overall, MG associated with a thymoma is more severe, almost always generalized with bulbar disorders, most often requiring treatment with corticosteroids and immunosuppressants, with a probability of complete remission without treatment unless in MG without thymoma. Nevertheless, with a well-conducted treatment, which should in most cases be continued, about 2/3 of the patients are very much improved, of which more than 20% are in remission without treatment (recent Italian series of 192 patients); mortality remains low, even if the thymoma is invasive [43,51,52]. Identical data have been found in our series. Thymic hyperplasia associated with MG is characterized by early onset (<40 years), clinically generalized and severe and high levels of AchR antibodies [53]. In our series 6 patients had thymic hyperplasia with CT, 3 of which had a generalized form, with 1 year, 38 years and 50 years at the onset of the disease; three patients underwent thymectomy.
Limitations of our study: Our study population was limited to 14 patients and the immunological assessment, which was only possible for 12 patients (85.7%), could have biased our results.
We recorded 14 cases of MG in 4 years and 10 months, indicating a greater rarity or under diagnosis of autoimmune myasthenia in Ouagadougou. The socio-demographic, clinical, immunological and CT scan profile seems comparable to the literature. However, late-onset forms were not observed in our study. In addition, a low prevalence of AchR antibodies (50%) was observed. Post-thymectomy clinical remission involved only 51.7% of patients. Additional studies are needed to better characterize the profile of MG in Ouagadougou (Burkina Faso).
2021 Copyright OAT. All rights reserv
- Zhang B, Tzartos JS, Belimezi M, Ragheb S, Bealmear B, et al. (2012) Autoantibodies to lipoprotein-related protein 4 in patients with double-seronegative myasthenia gravis. Arch Neurol 69: 445-451. [Crossref]
- Gilhus NE (2009) Autoimmune myasthenia gravis. Expert Rev Neurother 9: 351-358. [Crossref]
- Jacob S, Viegas S, Lashley D, Hilton-Jones D (2009) Myasthenia gravis and other neuromuscular junction disorders. Pract Neurol 9: 364-371. [Crossref]
- Alkhawajah NM, Oger J (2013) Late-onset myasthenia gravis: a review when incidence in older adults keeps increasing. Muscle Nerve 48: 705-710. [Crossref]
- Aarli JA (2008) Myasthenia gravis in the elderly: Is it different? Ann N Y Acad Sci 1132: 238-243. [Crossref]
- Keesey JC (2004) Clinical evaluation and management of myasthenia gravis. Muscle Nerve 29: 484-505. [Crossref]
- Grob D, Brunner N, Namba T, Pagala M (2008) Lifetime course of myasthenia gravis. Muscle Nerve 37: 141-149. [Crossref]
- Phillips LH 2nd (2003) The epidemiology of myasthenia gravis. Ann N Y Acad Sci 998: 407-412. [Crossref]
- Heckmann JM, Owen EP, Little F (2007) Myasthenia gravis in South Africans: Racial differences in clinical manifestations. Neuromuscul Disord 17: 929-934. [Crossref]
- Chui HC, Vincent J, Newsom-Davis J, et al. (1987) Myasthenia gravis: population differences in disease expression and acetylcholine receptor antibody titers between Chinese and Caucasians. Neurology 37: 1854-1857. [Crossref]
- Kawaguchi N, Kuwabara S, Nemoto Y (2004) Treatment and outcome of myasthenia gravis: retrospective multi-center analysis of 470 Japanese patients, 1999-2000. J Neurol Sci 224: 43-47. [Crossref]
- Zhang X, Yang M, Xu J. (2007) Clinical and serological study of myasthenia gravis in HuBei province, China. J Neurol Neurosurg Psychiatry 78: 386-390. [Crossref]
- Carr AS, Cardwell CR, McCarron PO, McConville J (2010) A systematic review of population based epidemiological studies in myasthenia gravis. BMC Neurol 10: 46. [Crossref]
- Somnier FE (2005) Increasing incidence of late-onset anti-AChR antibody- seropositive myasthenia gravis. Neurology 65: 928-930. [Crossref]
- El Mahdi B (2016) Myasthenia gravis: Experience of the neurology department of the Moulay Ismaël military hospital in Meknes. [thesis: Med]. Sidi Mohammed Ben Abdellah University Faculty of Medicine and Pharmacy Fes: 141.
- Eymard B (2009) Antibodies in myasthenia gravis. Rev Neurol (Paris) 165: 137-143. [Crossref]
- Fall M, Awbeck Fall A, Léye A, Ndiaye M, Moreira Diop T (2015) La myasthénie auto-immune de l’adulte lors d’une consultation décentralisée de neurologie au centre hospitalier national de Pikine dans la banlieue de Dakar-Sénégal. Rev Neurol 171: A153.
- Maharaj J, Bahadursingh S, Ramcharan K (2013) Myasthenia gravis in South Trinidad. West Indian Med J 62: 510-514. [Crossref]
- Nouvel L, Karkoutly C, Santerre N, Racoussot F, Vermersch P, et al. (2008) 656 La myasthénie?: étude descriptive de 12 cas. Journal Français d’Ophtalmologie 31: 199.
- Oöpik M, Kaasik AE, Jakobsen J (2003) A Population Based Epidemiological Study on Myasthenia Gravis in Estonia. J Neurol Neurosurg Psychiatry 74: 1638-1643. [Crossref]
- Nancy P, Berrih-Aknin S (2005) Differential estrogen receptor expression in autoimmune myasthenia gravis. Endocrinology 146: 2345-2353. [Crossref]
- Eymard B (2014) Myasthenia, from the internist's point of view. Rev Med Interne 35: 421-429. [Crossref]
- Onyekwulu FA, Onwuekwe IO (2010) Critical care of myasthenia gravis in a resource poor setting: a study of South East Nigeria. Neurologist 16: 368-370. [Crossref]
- Barbaud A, Carlander B, Pagès M. (2006) Late forms of myasthenia gravis. Comparative study with myasthenia gravis. Rev Neurol 162: 990-996.
- Boughammoura-Bouatay A, Chebel S, Younes-Mhenni S, Frih-Ayed M (2008) Myasthenia gravis of late revelation: about a Tunisian population. NPG Neurologie-Psychiatrie-Gériatrie 47: 21-24.
- Eymard B, Stojkovic T, Sternberg D, Richard P, Nicole S, et al. (2013) Congenital myasthenic syndromes: difficulties in the diagnosis, course and prognosis, and therapy--The French National Congenital Myasthenic Syndrome Network experience. Rev Neurol 169: S45-S55. [Crossref]
- Vincent A, Newsom-Davis J (1985) Acetylcholine receptor antibody as a diagnostic test for myasthenia gravis: results in 153 validated cases and 2967 diagnostic assays. J Neurol Neurosurg Psychiatry 48: 1246-1252. [Crossref]
- Vincent A (2002) Unravelling the pathogenesis of myasthenia gravis. Nat Rev Immunol 2: 797-804. [Crossref]
- Meriggioli MN, Sanders DB (2012) Muscle autoantibodies in myasthenia gravis: beyond diagnosis? Expert Rev Clin Immunol 8: 427-438. [Crossref]
- Eymard B (2009) Antibodies in myasthenia gravis. Rev Neurol (Paris) 165: 137-143. [Crossref]
- Higuchi O, Hamuro J, Motomura M, Yamanashi Y (2011) Autoantibodies to low-density lipoprotein receptor-related protein 4 in myasthenia gravis. Ann Neurol 69: 418-422. [Crossref]
- Kim N, Stiegler AL, Cameron TO, Hallock PT, Gomez AM, et al. (2008) Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell 135: 334-342. [Crossref]
- Cossins J, Belaya K, Zoltowska K, Koneczny I, Maxwell S, et al. (2012) The search for new antigenic targets in myasthenia gravis. Ann N Y Acad Sci 1275: 123-128. [Crossref]
- Jacob S, Viegas S, Leite MI, Webster R, Cossins J, et al. (2012) Presence and pathogenic relevance of antibodies to clustered acetylcholine receptor in ocular and generalized myasthenia gravis. Arch Neurol 69: 994-1001. [Crossref]
- Leite MI, Jacob S, Viegas S, Cossins J, Clover L, et al. (2008) IgG1 antibodies to acetylcholine receptors in “seronegative” myasthenia gravis. Brain 131: 1940-1952. [Crossref]
- Ohta K, Shigemoto K, Fujinami A, Maruyama N, Konishi T, et al. (2007) Clinical and experimental features of MuSK antibody positive MG in Japan. Eur J Neurol 14: 1029-1034. [Crossref]
- Maggi G, Casadio C, Cavallo A, Cianci R, Molinatti M, et al. (1989) Thymectomy in myasthenia gravis. Results of 662 cases operated upon in 15 years. Eur J Cardiothorac Surg 3: 504-509. [Crossref]
- Maggi L, Andreetta F, Antozzi C, Baggi F, Bernasconi P, et al. (2008) Thymoma-associated myasthenia gravis: outcome, clinical and pathological correlations in 197 patients on a 20-year experience. J Neuroimmunol 15: 237-244. [Crossref]
- Kim JY, Park KD, Richman DP (2011) Treatment of myasthenia gravis based on its immunopathogenesis. J Clin Neurol 7: 173-183. [Crossref]
- Matuja WB, Aris EA, Gabone J, Mgaya EM (2001) Incidence and characteristics of Myasthenia gravis in Dar Es Salaam, Tanzania. East Afr Med J 78: 473-476. [Crossref]
- Ojini FI, Danesi MA, Ogun SA (2004) Clinical Manifestations of Myasthenia Gravis - Review of Cases Seen at the Lagos University Teaching Hospital. Niger Postgrad Med J 11: 193-197. [Crossref]
- Zielinski M, Hauer L, Hauer J, Pankowski J, Nabialek T, et al. (2010) Comparison of complete remission rates after 5 year follow-up of three different techniques of thymectomy for myasthenia gravis. Eur J Cardiothorac Surg 37:1137-1143. [Crossref]
- Berrih-Aknin S, Le Panse R (2014) Myasthenia Gravis: A comprehensive review of immune dysregulation and etiological mechanisms. J Autoimmun 52: 90-100. [Crossref]
- Aragonès JM, Bolíbar I, Bonfill X, Bufill E, Mummany A, et al. (2003) Myasthenia gravis: a higher than expected incidence in the elderly. Neurology 60: 1024-1026. [Crossref]
- Casetta I, Groppo E, De Gennaro R, Cesnik E, Piccolo L, et al. (2010) Myasthenia gravis: a changing pattern of incidence. J Neurol 257: 2015-2019. [Crossref]
- Lavrnic D, Losen M, Vujic A, De Baets M, Hajdukovic LJ, et al. (2005) The features of myasthenia gravis with autoantibodies to MuSK. J Neurol Neurosurg Psychiatry 76: 1099-1102. [Crossref]
- Evoli A, Tonali PA, Padua L, Monaco ML, Scuderi F, et al. (2003) Clinical correlates with anti-MuSK antibodies in generalized seronegative myasthenia gravis. Brain 126: 2304-2311. [Crossref]
- Zhou L, McConville J, Chaudhry V, Adams RN, Skolasky RL, et al. (2004) Clinical comparison of muscle-specific tyrosine kinase (MuSK) antibody-positive and -negative myasthenic patients. Muscle Nerve 30: 55-60.
- Leite M, Strobel P, Jones M, Micklem K, Moritz R, et al. (2005) Fewer thymic changes in MuSK antibody-positive than in MuSK antibody-negative MG. Ann Neurol 57: 444-448. [Crossref]
- Sieb JP1 (2014) Myasthenia gravis: an update for the clinician. Clin Exp Immunol 175: 408-418. [Crossref]
- Suster S, Moran CA (2006) Thymoma classification: current status and future trends. Am J Clin Pathol 125: 542-554. [Crossref]
- Filosso PL, Galassi C, Ruffini E, Margaritora S, Bertolaccini L, et al. (2013) Thymoma and the increased risk of developing extrathymic malignancies: a multicentre study. Eur J Cardiothorac Surg 44: 219-224. [Crossref]
- Goulan-Goeau C, Gajdos P (2002) Myasthénie et syndromes myasthéniques, EMC Neurologie 17: 172.