Abstract
Shiga toxin-producing E. coli hemolytic-uremic syndrome (STEC-HUS) is characterized by acute renal failure associated with thrombocytopenia and mciroangiopathic hemolytic anemia following exposure to STEC. It may present with spectrum of multi-organ dysfunction syndrome (MODS). Pathogenesis of STEC-HUS is partially understood. Proposed is two sequential molecular pathogeneses. First path is from Shiga toxins to endotheliopathy as a result of organotropism and endothelial heterogeneity. Shiga toxins binding to Gb3-endowed ECs, leads to endotheliopathy in the kidneys and other organs. Also, Shiga toxins downregulating CD59 provoke unprotected activation of complement, in which C5b-9 causes channel formation and promotes additional endotheliopathy. Second path is from endotheliopathy to microthrombogenesis. Endotheliopathy triggers activation of two independent molecular pathways (i.e., inflammatory and microthrombotic), promoting inflammation and formation of “microthrombi” composed of platelet and unusually large von Willebrand factor multimers (ULVWF) complexes. Microthrombogenesis is initiated by the activated platelet and endothelial exocytosis of ULVWF. If ADAMTS13 is insufficient due to excessive production of ULVWF, uncleaved ULVWF are anchored to ECs. ULVWF anchored to Gb3-endowed and CD59-disendowed ECs in situ are long elongated strings and recruit the activated platelets to assemble microthrombi. Microthrombi cause disseminated intravascular microthrombosis (DIT), leading to vascular microthrombotic disease (VMTD). The result is organ phenotype syndromes of MODS such as HUS, encephalopathy, acute respiratory distress syndrome (ARDS), etc. DIT/VMTD triggers thrombotic thrombocytopenic purpura (TTP)-like syndrome. Current understanding of Gb3 and CD59 roles and experience with complement inhibition therapy support the complex molecular pathogenesis of STEC-HUS is due to heterogeneity-associated endotheliopathy, leading to impaired ADAMTS13 function.
Indexing terminology: ADAMTS13; CD59; C5b-9 [TCC: MAC]; Gb3; Complement activation;Disseminated intravascular microthrombosis; Endotheliopathy; Endothelial heterogeneityHemolytic-uremic syndrome; Microthrombogenesis; Organotropism; Shiga toxins; TTP-like syndrome; Vascular microthrombotic disease
Abbreviations: AFH; acute fulminant hepatic failure; aHUS; atypical hemolytic-uremic syndrome;ARDS, acute respiratory distress syndrome; ARF, acute renal failure; “C”, complement; DIC, disseminated intravascular coagulation; DIT, disseminated intravascular microthrombosis; ECs, endothelial cells HFRS, hemorrhagic fever with renal syndrome; HUS, hemolytic uremic-syndrome; MAHA, microangiopathic hemolytic anemia; MAC, membrane attack complex; MODS, multi-organ dysfunction syndrome; SIRS, systemic inflammatory response syndrome; SNP, single nucleotide polymorphism; STEC, Shiga toxin producing E. coli; Stx, Shiga toxin; TMA, thrombotic microangiopathy; TPE, therapeutic plasma exchange;TTP, thrombotic thrombocytopenic purpoura; ULVWF, unusually large von Willebrand factor multimers;VMTD, vascular microthrombotic disease; Vtx, verotoxin
Introduction
Shiga toxin-producing E. coli hemolytic-uremic syndrome (STEC-HUS) is a serious kidney disease associated with intravascular microthrombosis resulting in organ phenotype of acute renal failure due to endothelial heterogeneity and Shiga toxin-caused organotropism. Hematologic phenotype of thrombocytopenia and microangiopathic hemolytic anemia (MAHA) is the result of impaired ADAMTS13 activity leading to disseminated intravascular microthrombosis (DIT) and thrombotic microangiopathy (TMA). The extra-renal manifestations commonly occur as the phenotypes of MODS that are associated with DIT in selected organs.
The pathogenesis of STEC-HUS is still poorly understood and its morbidity and mortality are high. Mortality is reported to be between 3% and 5%, and death due to STEC-HUS is almost always associated with severe extra-renal diseases, including serious central nervous system involvement [1].
In this brief review, the unified concept of molecular pathogenesis of STEC-HUS, leading to two sequential paths, 1) first, from STEC to endotheliopathy and 2) second, from endotheliopathy to microthrombogenesis, are presented as illustrated in . This conceptual framework is based on “two-activation theory of the endothelium” .
Role of Shiga toxins
STEC-HUS is typically preceded by a prodrome of diarrhea, which is sometimes bloody, and is caused by Shiga toxin-producing Escherichia coli bacteria, of which E. coli O157:H7 is the most common serotype.
Two different Shiga toxin 1 (Stx1) and Shiga toxin 2 (Stx2) show the similar molecular structure. However, their role in STEC-HUS seems to be different because STEC cells could produce one or both of two major toxins, and these toxins appear to be crossing the epithelial cell barrier by different pathways [3]. Also, Stx2 is more frequently associated with severe STEC-HUS in humans for reasons as yet clearly undetermined [4-5]. Recently it was found that Shiga toxins activate complement and binds to factor H, which is a strong evidence for an active role of complement system in STEC-HUS [6].
Shiga toxins are multi-subunit protein complexes that bind to a glycosphingolipid receptor Gb3, on select eukaryotic cell types. Localization of Gb3-endowed ECs in the kidneys and other organs is predictive of the sites of action of Shiga toxins. However, the toxins are cytotoxic to some, but not to all cell types that express Gb3 [5]. Despite related primary amino acid sequences, Stx1 and Stx2 are immunologically distinct. In addition, Stx1 and Stx2 do not target exactly the same tissues and organs although both bind Gb3 and are thought to be capable of causing STEC-HUS [5]. These findings suggest the molecular pathogenesis of STEC-HUS is a complex process.
In addition, Shiga toxins are found to activate complement system [6,7]. Thus, as seen in atypical HUS (aHUS), the pathogenesis of STEC-HUS is also suspected to be complement-mediated endotheliopathy resulting in thrombotic microangiopathy [7,8]. Activation of the complement system produces terminal C5b-9 (MAC [membrane attack complex]), which insertion to unprotected ECs causes of channel (pore) formation on the membrane of ECs [9]. In order to protect themselves from the complement attack, ECs express several regulatory molecules, including the terminal complex regulator CD59 [9] that suppresses assembly of the large MACs through inhibition of polymerization of additional C9 molecules. Since Stx2 downregulates CD59 mRNA and protein levels on tubular epithelial and glomerular endothelial cells, CD-59-disendowed ECs likely contributes to the destruction of the kidneys and other organs in STEC-HUS [10,11]. Therefore, it is suspected that STEC-HUS as well as aHUS is partly caused by “unprotected” complement activation, rather than due to the traditional concept of “uncontrolled” complement activation.
Endothelial heterogeneity
Microvascular ECs within different tissues and organs are endowed with distinct but as yet unrecognized structural, phenotypic, and functional attributes [12-14]. STEC-HUS is a typical example of organ phenotypic diversity related to functional molecules of ECs. For example, the distribution of CD59-disendowed ECs and Gb3-endowed ECs also regulates endothelial heterogeneity in different target organs. Diversified endothelial heterogeneity, which could be genetically determined or altered by an acquired event [5,11-12,15-19], responds to the environmental factor (i.e., STEC) and contributes to diversified organ phenotypes of the disease. The extrarenal manifestations of STEC-HUS are caused by altered expression of these endowed molecules resulting in endothelial heterogeneity in various organs. As shown in Table 2, organ specific phenotypes occurring in STEC-HUS include gastroenteritis, acute renal failure (ARF), encephalopathy [20-27], acute respiratory distress syndrome (ARDS) [23,24], myocardial infarction [23,26], acute pancreatitis [25,24], rhabdomyolysis [25,27], skin necrosis [25,27], etc.
Table 2. Target organs in STEC-HUS associated with endothelial heterogeneity and observed clinical phenotypes.
Target organ due to endothelial heterogeneity |
Organ phenotype disease |
Hematologic phenotype |
References of known Shiga toxin targeted organ dysfunction |
Incidence of organ phenotype in STEC-HUS |
Bowels
Kidneys
Brain
Lungs
Heart
Pancreas
Muscle
Skin |
Gastroenteritis
Acute renal failure
Encephalopathy
CNS dysfunction syndrome
ARDS
Myocardial infarction
Acute pancreatitis
Rhabdomyolysis
Skin necrosis |
Thrombocytopenia
MAHA
DIT
VMTD
TTP-like syndrome |
Well documented
Well documented
[20] [21] [22] [23] [24]
[25] [26] [27]
[23] [24]
[23] [26]
[25] [24]
[25] [27]
[25] [27] |
Almost 100% [25]
100% [25]
5-69% [20] [23]
5-10% [23]
<1% [25]
20% [25]
<1-3% [25]
<1-2% [24] |
ARDS, acute respiratory distress syndrome; CNS, central nervous system; DIT, disseminated intravascular microthrombosis; MAHA, microangiopathic hemolytic anemia; STEC-HUS, Shiga toxin-induced hemolytic-uremic syndrome |
Because of organ phenotypic diversity associated with endothelial heterogeneity, in STEC-HUS, ARF could present simultaneously with encephalopathy and ARDS as a result of Shiga toxin-induced DIT. In such situation, first physician (e.g., nephrologist) would call this condition “HUS” with involvement of the brain and lungs; second physician (e.g., neurologist) might call it “encephalopathy” with the involvement of the kidneys and lungs; and third physician (e.g. pulmonologist) call it “ARDS” with involvement of the kidneys and brain. Yet, the fourth (e.g., hematologist) would call it “thrombotic thrombocytopenic purpura (TTP)-like syndrome” with MODS. Certainly, nature has created this remarkable endothelial heterogeneity and also endowed the physician with the endless perplexity and complexity of human diseases.
Molecular Pathogenesis (first path) leading to Endotheliopathy
Shiga toxins are some of the most potent toxins among various pathogens and are known to cause an injury to ECs leading to endotheliopathy. Shiga toxins bind to the cellular receptor Gb3 that is commonly endowed to ECs [5,12,17,19,28]. These toxins bound to Gb3-endowed tubular epithelium, as well as the glomerular endothelial cells, leading to cellular apoptosis and necrosis, which result in endotheliopathy and subsequently induce DIT in selected organs (e.g., ARF, ARDS, and pancreatitis due to vascular microthrombosis) seen in STEC-HUS [24,26,29]. Separately, normal ECs are also endowed with CD59 [10,11,15,16], which is the membrane inhibitor of reactive cell lysis following complement activation. It might be disendowed due to gene mutation [30] or due to acquired events such as Stx2 intrusion or immune diseases [10,11,31]. This CD59-disendowed ECs could independently contribute to additional endotheliopathy through channel formation on the endothelial membrane [15,16] when complement activation produces C5b-9 (MAC).
Shiga toxins inducing the above independent molecular alterations initiate first path of molecular pathogenesis from STEC to endotheliopathy (Table 1). Gb3-endowed or CD59-disendowed endotheliopathy could cause functional and/or anatomical changes of various organs resulting in MODS (e.g., ARF, encephalopathy, ARDS, etc.), and trigger to second path of molecular pathogenesis, which is from endotheliopathy to microthrombogenesisa, promoting molecular process that leads to TTP-like syndrome as illustrated in Figure 1.
Table 1. Unified concept of “two-sequential pathg molecular pathogenesis the pathogenesis of STEC-HUS.
Pathogenesis of STEC-HUS |
First path: from STEC
to endotheliopathy |
Second path: from endotheliopathy
to microthrombogenesis |
Pathogenic source
Gene involved
Modifying molecule
Molecular mechanism
Examples of target organs due to endothelial heterogeneity
Phenotype examples |
Shiga toxins (stx1 and stx2)
Shiga-like toxins (vtx1 and vtx2)
GLA
CD59
“C” regulator
Gb3 (overexpressed)
CD59 (underexpressed)
Shiga toxins binding to Gb3-endowed ECs leading to apoptosis & endotheliopathy
“C” activation by Shiga toxins
Shiga toxin2 causing CD59-disendowed ECs leading to “unprotected” “C” activation and channel formation & endotheliopathy
Bowels; kidneys; lungs; heart; brain; pancreas; muscle; skin
Gastroenteritis/hemorrhagic
enteritis; acute renal failure; encephalopathy; ARDS; acute pancreatitis; myocardial infarction; rhabdomyolysis; skin necrosis
|
Endotheliopathy
ADAMTS13 insufficiency
ADAMTS13
eULVWF (from endothelial exocytosis)
ADAMTS13 (insufficiency)
Activation of microthrombotic pathway
Microthrombogenesis
ECs
DIT
Consumptive thrombocytopenia
MAHA
MODS |
Contemporary clinical disorder |
Acute renal failure; STEC-HUS |
TTP-like syndrome |
Disease entity |
VMTD |
ARDS, acute respiratory distress syndrome; DIT, disseminated intravascular microthrombosis; ECs. Endothelial cells; MAHA, microangiopathic hemolytic anemia; STEC, Shiga toxin producing E. Coli; STEC-HUS; Shiga toxin-producing E. Coli hemolytic-uremic syndrome; TTP, thrombotic thrombocytopenic purpura; eULVWF, endothelial unusually large von Willebrand factor multimers; VMTD, vascular microthrombotic disease |
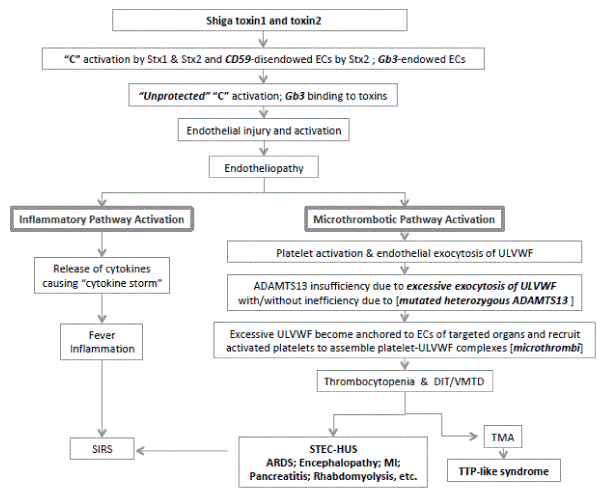
Figure 1. Molecular Pathogenesis of STEC-HUS.
Molecular pathogenesis of STEC-HUS is presented in this schematic illustration. Shiga toxins are transported to upregulated Gb3 receptors in ECs. Gb3-endowed heterogeneity of ECs is present in many different organs such as kidneys, brain, heart, pancreas, lungs, muscle, and skin, etc. The binding of Shiga toxins to Gb3 promotes endotheliopathy. Also, Shiga toxins activate complement system. Additionally, Shiga toxin2 downregulates CD59. This disendowed CD59, which is the membrane protector of ECs, contributes to “unprotected” complement activation leading to additional endotheliopathy through the channel formation in the endothelial membrane. Combined endotheliopathy activates both inflammatory and microthrombotic pathways. Excessive endothelial exocytosis of ULVWF would occur from Weibel Palade bodies. If ADAMTS13 were insufficient due to imbalance between ADAMTS13 activity and exocytosed ULVWF with/without inefficiency due to ADAMTS13 gene mutation, uncleaved ULVWF are anchored ECs as long elongated strings and recruit activated platelet to assemble platelet-ULVWF complexes, which are “microthrombi” that trigger endotheliopathy-associated DIT/VMTD. These multi-step processes cause TTP-like syndrome and MODS.
ARDS, acute respiratory distress syndrome; CNS, central nervous system; DIT, disseminated intravascular microthrombosis; ECs. Endothelial cells; HUS, hemolytic-uremic syndrome; MAHA, microangiopathic hemolytic anemia; MODS, multi-organ dysfunction syndrome; SNP, single nucleotide polymorphism; STEC, Shiga toxin producing E. Coli; TMA, thrombotic microangiopathy; TCTTP, thrombotic thrombocytopenic purpura; ULVWF, unusually large von Willebrand factor multimers
Molecular Pathogenesis (second path) leading to Microthrombogenesis
According to novel thesis of “two-activation theory of the endothelium” (Figure 1) [2,32], combined Gb3 and C5b-9-induced endotheliopathy promotes activation of two independent endothelial pathways (i.e., inflammatory and microthrombotic). In short, two important molecular events are: 1) release of inflammatory cytokines (e.g., interleukin (IL)-1, IL-6, tumor necrosis factor-a, and others) [33,34], and 2) activation of the platelet [35] and exocytosis of ULVWF [36-38]. The former triggers inflammation through “activation of inflammatory pathway”, and the latter mediates microthrombogenesis via “activation of microthrombotic pathway”. The activation of microthrombotic pathway promotes DIT, which would lead the clinical disorder “vascular microthrombotic disease” (VMTD) [2,32,39]. VMTD is characterized by intravascular microthrombosis in various organs as seen in MODS, and hematologic features of thrombocytopenia and MAHA. DIT/VMTD might be localized in the kidneys or can become systemic by involving other target organs depending upon the distribution of heterogeneity of ECs in a particular patient.
The hematologic phenotype, which is characterized by thrombocytopenia and MAHA, is the same in different etiology-associated HUS, including 1) STEC-HUS, 2) aHUS, and 3) secondary HUS associated with infection and other critical illness/condition (e.g., virus, bacteria, rickettsia, pregnancy, trauma, transplant, cancer, and drugs) [40-44]. Although some hematologists, nephrologists and clinicians would designate them as combined “TTP-HUS” syndrome, the proper diagnostic term should be “TTP-like syndrome” because it is characterized by endotheliopathy-associated DIT/VMTD [2,32,39]. The difference between TTP and TTP-like syndrome in their pathophysiological mechanisms as well as etiologies is clearly defined as shown in Tables 3 and 4 [2,32]. The tables are self-explanatory.
Table 3. Genesis of VMTD and characteristics of TTP and TTP-like syndrome.
|
Hereditary TTP: ADAMTS13 gene mutation-associated VMTD
Acquired TTP: ADAMTS13 antibody-associated VMTD |
TTP-like syndrome: Endotheliopathy-associated VMTD |
Primary event (causes)
Secondary event
Tertiary event
Final event
|
Hereditary ADAMTS13 gene mutation
Acquired ADAMTS13 antibody formation
¯
Excessive circulating mULVWF
¯
Microthrombogenesis leading to platelet-mULVWF complexes in circulation
¯
Microthrombi lodged in arteriolar capillary lumens of organs
¯
VMTD
¯
TMA (microthrombotic microangiopathy)
¯
TTP |
Pathogen (e.g., viruses; bacteria; fungi; rickettsia; parasites)
Polytrauma (e.g., chest/lung; bones; skull/brain injury)
Pregnancy (e.g., preeclampsia; abruptio placenta; amniotic fluid embolism)
Cancer (e.g., stomach; breast; lung)
Transplant (e.g., liver; kidney; bone marrow)
Drug and chemical (e.g., cyclosporine; mytomycin C; Shiga toxins; ricin)
¯
Complement activation (C5b-9 production) ® endothelial injury ® endotheliopathy
¯
Cytokine release ® Inflammation ® SIRS
Endothelial exocytosis of eULVWF & anchored to ECs as long elongated strings ® DIT
¯
Microthrombogenesis leading to platelet-eULVWF complexes anchored to ECs
¯
VMTD
¯
TMA (microthrombotic angiopathy)
¯
TTP-like syndrome |
Hematologic features
Platelet
Red blood cell
Clinical syndromes
Inflammation
Cytokine storm
SIRS
Encephalopathy
ARDS
AFH
ARF/HUS
“DIC” (see Ref. 2 & 32) |
Consumptive thrombocytopenia
MAHA
Uncommon
Absent
Absent
Very common
Probably absent
Probably absent
Very common
Doesn’t occur |
Consumptive thrombocytopenia
MAHA
Very common
Often present in sepsis and MODS
Often present in sepsis and MODS
Common, especially in HUS
Common
Common, sometimes with hepatic coagulopathy
Common
Identical to TTP-like syndrome |
Laboratory features
ADAMTS13 activity
ADAMTS13 antibody
LDH
Haptoglobin
Schistocytosis |
Markedly decreased (<5% of normal)
Positive in acquired TTP
Increased
Markedly decreased
++ to ++++ |
Mild to moderately decreased (20-70% of normal) or normal (?)
Negative
Increased
Markedly decreased
None to +++ |
Therapeutic response to
TPE
Platelet transfusion
rADAMTS13 |
Very good response
Contraindicated
Unknown at this time; expected to be effective in hereditary TTP |
Excellent and fast response if treated in early stage
Contraindicated
Unknown at this time; expected to be very effective in non-infectious conditions |
AFH, acute fulminant hepatitis; ARF/HUS; acute renal failure/ hemolytic-uremic syndrome; ARDS, acute respiratory distress syndrome; “DIC”, disseminated intravascular coagulation of McKay; ECs, endothelial cells; eULVWF/mULVWF, endothelial unusually large von Willebrand factor multimers/megakaryocytic ULVWF; LDH, lactate dehydrogenase; MAHA, microangiopathic hemolytic anemia; rADAMTS13, recombinant ADAMTS13; SIRS, systemic inflammatory response syndrome; TMA, thrombotic microangiopathy; TPE, therapeutic plasma exchange; TTP, thrombotic thrombocytopenic purpura; VMTD, vascular microthrombotic disease |
Table 4. Characteristics of two different ULVWF.
|
mULVWF |
eULVWF |
Synthesized in
Stored in
Primary distribution at release
Availability
Exposure to ADAMTS13
Interaction with platelets causing
Localization of
platelets-ULVWF complexes
Example of its attachment to
TMA manifestation
Hematopathologic manifestation
Associated inflammation
Associated clinical syndrome |
Megakaryocytes
a granules of platelets
In circulation
In blood
As platelet-adherent form
Platelet aggregation and adhesion
Arteriolar capillary lumens lodged as
microthrombi
Auto-ADAMTS13 antibody
Microthrombotic microangiopathy
Thrombocytopenia
MAHA
Encephalopathy; ARF
None to minimal (?)
TTP |
Endothelial cells
Weibel-Palade bodies of ECs
On the membrane of ECs
At activated ECs after endothelial exocytosis
As ECs-adherent form
Platelet recruitment and aggregation
Intravascular ECs-anchored as platelet-decorated long elongated ULVWF strings
ECs in sepsis-induced endotheliopathy
Microthrombotic angiopathy
Thrombocytopenia
MAHA
MODS (ARF; HUS; encephalopathy; ARDS; MI)
None to severe
TTP-like syndrome |
ARF, acute renal falure; ARDS, acute respiratory distress syndrome; ECs, endothelial cells; eULVWF/mULVWF, endothelial unusually large von Willebrand factor multimers/ megakaryocytic ULVWF; HUS, hemolytic-uremic syndrome; MAHA, microangiopathic hemolytic anemia; MI, myocardial infarction; MODS, multi-organ dysfunction syndrome; SS, stroke syndrome; TMA, thrombotic microangiopathy; TTP, thrombotic thrombocytopenic purpura |
TTP occurs due to severe ADAMTS13 deficiency (i.e., familial ADAMTS13 gene mutation or acquired antibody production against ADAMTS13), resulting in excessive accumulation of megakaryocytic ULVWF in circulation. On the other hand, TTP-like syndrome occurs in ECs in situ due to insufficiency of ADAMTS13 following excessive endothelial exocytosis of ULVWF in endotheliopathy with or without moderate inefficiency of ADAMTS13 (e.g. heterozygous gene mutation/polymorphism) [2,32,36,39]. Megakaryocytic ULVWF triggers microthrombogenesis to cause TTP in circulation, but endothelial ULVWF, anchored to ECs, promote TTP-like syndrome in situ.
Microthrombogenesis in endotheliopathy is the process, in which long elongated ULVWF strings are anchored to ECs after release from Weibel-Palade bodies and recruit activated platelets, promoting the formation of platelet-ULVWF complexes [45-47]. It occurs when ADAMTS13 is insufficient to cleave the excessively exocytosed ULVWF. Platelet-ULVWF complexes are “microthrombi”, which lead to DIT and VMTD [2,32] in CD59-disendowed and Gb3-endowed ECs of certain vital organs.
Organotropism
Organotropism of pathogens occurs due to their metabolic versatility and ability to produce diverse bioactive compounds such as hydrolytic enzymes, antibiotics, anti-tumorals and also toxins [48]. Toxins can be produced by prokaryotes such as bacteria [1], but also by eukaryotes such as fungi [48].
In first path of molecular pathogenesis of STEC-HUS, renal organotropism of Shiga toxins is expressed by their affinity to ECs in the kidneys and other organs that are associated with unique endothelial heterogeneity characterized by distribution of endowed/disendowed molecules. Shiga toxins appear to trigger combined endotheliopathy in association with Gb3 upregulation and CD59 downregulation in ECs.
The virulence of Shiga toxin might be expressed by organotropism too (e.g., brain vs. kidneys) and combined role of molecular effects of Gb3 binding and CD56-unprotected complement activation (e.g., STEC-HUS vs. a HUS). Also, second path of molecular pathogenesis associated with the expressivity of ADAMTS13 gene mutation on microthrombogenesis could play a role to the degree of virulence of Shiga toxins in producing hematologic phenotype of TTP-like syndromes.
Endothelial heterogeneity and pathogen/toxin-induced organotropism are two sides of the same coin in the molecular pathogenesis. It should be emphasized that HUS is not a specific syndrome by itself, but is only the manifestation (i.e., ARF) from the localization associated with endothelial heterogeneity.
Thus, the proper term for the variety of TTP-syndromes (i.e., thrombocytopenia, MAHA and various organ phenotypes) can be defined as follows with designation of prominent organ failure: for examples,
- TTP-like syndrome with ARF (including HUS, hepato-renal syndrome, hemorrhagic fever with renal syndrome (HFRS) due to hanta virus)
- TTP-like syndrome with encephalopathy (including HUS, hepatic encephalopathy)
- TTP-like syndrome with acute fulminant hepatic failure (AFH) (including HELLPs)
- TTP-like syndrome with ARDS (including cardio-pulmonary syndrome)
- TTP-like syndrome with acute adrenal insufficiency (including Warter house Friderichsen syndrome, septic shock)
- TTP-like syndrome with stroke syndrome (including transient ischemic attack)
- TTP-like syndrome with pancreatitis
- TTP-like syndrome with rhabdomyolysis
- TTP-like syndrome with myocardial infarction
- TTP-like syndrome with sepsis syndrome (including “TTP-mimicking syndrome”)
- TTP-like syndrome with peripheral digit ischemic syndrome
- TTP-like syndrome with “DIC” (disseminated intravascular coagulation) [2,32]
- TTP-like syndrome with MODS (multi-organ dysfunction syndrome) and so on.
VMTD in TTP and TTP-like syndrome
Succinctly speaking, TTP is either gene mutation-associated (hereditary) or antibody-associated (acquired) DIT/VMTD. TTP is well defined. In contrast, TTP-like syndrome is endotheliopathy-associated DIT/VMTD [2,32], which clinical manifestation is associated with endothelial heterogeneity. So, TTP-like syndrome is well defined too. Better understanding of distinct nature of molecular pathogenesis of TTP-like syndrome clearly explains how TTP-like syndrome is different from TTP. The same hematologic phenotype is probably due to the similar nature of “microthrombi” composed of platelet-ULVWF complexes (Table 3). Thus, it is appropriate to call both disorders as VMTD.
Hematologic phenotype of thrombocytopenia and MAHA is almost identical in both TTP and TTP-like syndrome and is the result of microthrombogenesis. Because TTP occurs in circulation and TTP-like syndrome occurs at the endothelial membrane in association with endothelial heterogeneity, the organ phenotype is dissimilar. TTP typically tends to involve in the kidneys and brain, which is due to that circulating microthrombi become easily lodged in those organs, but TTP-like syndrome develops in microthrombogenesis at ECs endowed with specific molecule(s) in situ.
In STEC-HUS, if the target organ is localized and confined only to the kidneys, the effect of activation of inflammatory pathway is muted, which would cause minimal inflammation if ever present. However, if endothelial heterogeneity is significantly expressed not only in the kidneys, but also other organs [11,21,49], moderate to severe inflammation, including systemic inflammatory response syndrome (SIRS), could develop [49-51]. According to “two-activation theory of the endothelium”, in TTP, inflammation typically is absent because activation of inflammatory pathway is incited by endothelial response.
Insight on the molecular pathogenesis from therapeutic experiences
First, because of the same hematologic phenotypic feature of HUS to TTP, therapeutic plasma exchange (TPE) has been employed and evaluated for both STEC-HUS and aHUS. There is insufficient evidence of benefit with TPE in the treatment of Shiga toxin-mediated disease [52]. For aHUS, TPE had been the mainstay of treatment prior to the use of eculizumab [53] and has been thought it might be helpful especially in patients with no identified complement mutation or in whom TTP is suspected [54]. Although TPE has reduced the high mortality rate and prevented relapses, it is far from optimal.
Second, because complement inhibitor eculizumab has been used successfully, it is presumed “uncontrolled” complement activation is associated with complement mutation. Eculizumab is now recognized as the treatment of choice for aHUS [55,56]. However, the benefit of eculizumab and/or TPE for STEC-HUS should be established with well-designed clinical trials. Some cases reports have suggested significant benefits with eculizumab [57-59]. An analysis of registry data presented at the XLIX European Renal Association-European Dialysis and Transplant Association (ERA-EDTA) Congress suggested that the combination of eculizumab with TPE for treating the accompanying STEC-HUS is no better than TPE alone [60]. In a review of the therapy for HUS, complement inhibition was effective in most patients with aHUS but was beneficial only in some patients with STEC-HUS or secondary HUS [61].
Although complement clearly contributes to the molecular pathogenesis in patients with STEC-HUS [62], only limited patients seem to benefit from early administration of eculizumab. Most of STEC-HUS patients do not respond to this drug [60]. These conflicting findings can be explained by the “combined two-leg two-path molecular pathogeneses” of STEC-HUS. In first path, combined (two-leg) Gb3-endowed ECs, which is complement independent, and CD59-disendowed ECs, which is complement dependent, Gb3-induced endotheliopathy could be the culprit for poor response. Gb3-induced endothelial damage would not benefit from complement inhibition alone. This is the critical difference between STEC-HUS and aHUS. Eculizumab would work only in the earlier stage of complement dependent aHUS. Once the hematologic phenotype of TTP-like syndrome is diagnosed, aHUS is in the second path and microthrombogenesis already must have started and is in advanced stage. In this case, TPE could be more beneficial treatment for DIT/VMTD.
Design of molecular therapy for STEC-HUS
According to proposed molecular pathogenesis of STEC-HUS, first path is caused by organotropism of Shiga toxins attracting to CD59 and Gb3-associated endothelial heterogeneity, and second path is caused by microthrombogenesis, leading to DIT, renal necrosis and end stage renal diseases in sequence. Theoretically, first path should be treated with Gb3 inhibition (e.g., anti-Gb3 antibody) and complement inhibition using anti-C5 (eculizumab) or CD59 replacement (e.g., recombinant CD59) if the disease is in very early stage. However, the better target is second path, which molecular pathogenesis should be treated with anti-microthrombotic therapy (e.g., TPE, but better yet recombinant ADAMTS13).
For the practicality, it is impossible to begin timely treatment for STEC-HUS to prevent both first path and second path. If thrombocytopenia and MAHA were recognized with advanced DIT and organ necrosis, the benefit targeting first path is long passed and second path would be in advancing stage. Thus, the chance of recovery may be not very high. Since full-blown DIT is the crux of problem leading to irreversible end stage renal disease and other vital organ involvement, antimicrothrombotic therapy using recombinant ADAMTS13 (currently only approved for clinical trial of hereditary TTP) should be more effective molecular therapy.
The best approach should be the initiation of anti-microthrombotic therapy in the earliest possible time. This can be accomplished by close monitoring of platelet count and intravascular hemolysis for the patient during the outbreak of STEC, especially when presented with gastroenteritis. Unexplained thrombocytopenia and intravascular hemolysis with gastroenteritis should confirm the diagnosis of STEC-HUS and be the indication for immediate treatment with TPE at this time. Since STEC infection is self-limited disease, the better treatment may be with prophylactic recombinant ADAMTS13 administration if it is approved.
Complement inhibition therapy and CD59 replacement trial should be contraindicated if the patient is suspected to have any kind of coexisting infection or is immunosuppressed. The discussion of this issue is not the scope of this article.
Conclusion
One environmental factor and several mutated genotypes participate in the molecular pathogenesis of STEC-HUS. The severity of STEC-HUS depends upon the endotheliopathy related to: 1) Shiga toxin organotropism, 2) endothelial heterogeneity and 3) functionality of ADAMTS13 gene. Shiga toxins are the initiators via “unprotected complement activation” and Gb3 binding, endothelial heterogeneity is the fabricator of organ phenotypes, and ULVWF are the molecular mediator of microthrombogenesis leading to TTP-like syndrome. These several molecular ingredients make up the complex nature of STEC-HUS.
STEC-HUS is the prototype of TTP-like syndrome revealing how interaction takes place between an environmental factor and mutated gene(s). Endotheliopathy-associated DIT/VMTD (i.e., TTP syndromes) should be a good place to study the nature of endothelial heterogeneity in many human diseases.
References
- Scheiring J, Andreoli SP, Zimmerhackl LB (2008) Treatment and outcome of Shiga-toxin-associated hemolytic uremic syndrome (HUS). Pediatr Nephrol 23: 1749-1760. [Crossref]
- Chang JC (2017) Thrombocytopenia in critically ill patients due to vascular microthrombotic disease: pathogenesis based on two-activation theory of the endothelium”. Vascul Dis Ther 2:1-7.
- Hurley BP, Jacewicz M, Thorpe CM, Lincicome LL, King AJ, et al. (1999) Shiga toxins 1 and 2 translocate differently across polarized intestinal epithelial cells. Infect Immun 67: 6670-6677. [Crossref]
- Lee JE, Reed J, Shields MS, Spiegel KM, Farrell LD, et al. (2007) Phylogenetic analysis of Shiga toxin 1 and Shiga toxin 2 genes associated with disease outbreaks. BMC Microbiol 7:109. [Crossref]
- Obrig TG (2010) Escherichia coli Shiga Toxin Mechanisms of Action in Renal Disease. Toxins (Basel) 2: 2769-2794.
- Karpman D, Tati R (2016) Complement contributes to the pathogenesis of Shiga toxin-associated hemolytic uremic syndrome Kidney Int 90: 726-729. [Crossref]
- Conway EM (2015) HUS and the case for complement. Blood 126: 2085-2090. [Crossref]
- Keir LS (2015) Shiga toxin associated hemolytic uremic syndrome. Hematol Oncol Clin North Am 29: 525-39. [Crossref]
- Farkas I, Baranyi L, Ishikawa Y, Okada N, Bohata C, et al. (2002) CD59 blocks not only the insertion of C9 into MAC but inhibits ion channel formation by homologous C5b-8 as well as C5b-9. J Physiol 539: 537-545. [Crossref]
- Ehrlenbach S, Rosales A, Posch W, Wilflingseder D, Hermann M, et al. (2013) Shiga toxin 2 reduces complement inhibitor CD59 expression on human renal tubular epithelial and glomerular endothelial cells. Infect Immun 81: 2678-2685. [Crossref]
- Nangaku M (1998) Complement regulatory proteins in glomerular diseases. Kidney Int 54:1419-28. [Crossref]
- Obrig TG, Louise CB, Lingwood CA, Boyd B, Barley-Maloney L, et al. (1993) Endothelial heterogeneity in Shiga toxin receptors and responses. J Biol Chem 268:15484-15488. [Crossref]
- Nolan DJ, Ginsberg M, Israely E, Palikuqi B, Poulos MG, et al. (2013) Molecular signatures of tissue-specific microvascular endothelial cell heterogeneity in organ maintenance and regeneration. Dev Cell 26: 204-219. [Crossref]
- Aird WC (2012) Endothelial cell heterogeneity. Cold Spring Harb Perspect Med 2: a006429. [Crossref]
2021 Copyright OAT. All rights reserv
- NOSE M, KATOH M, OKADA N, KYOGOKU M, OKADA H (1990) Tissue distribution of HRF20, a novel factor preventing the membrane attack of homologous complement, and its predominant expression on endothelial cells in vivo. Immunology 70:145–149. [Crossref]
- QUIGG RJ, MORGAN BP, HOLERS VM, ADLER S, SNEED AE III, et al. (1995) Complement regulation in the rat glomerulus: Crry and CD59 regulate complement in glomerular mesangial and endothelial cells. Kidney Int 48: 412-421. [Crossref]
- Hsu J, Serrano D, Bhowmick T, Kumar K, Shen Y, et al. (2011) Enhanced endothelial delivery and biochemical effects of α-galactosidase by ICAM-1-targeted nanocarriers for Fabry disease. J Control Release 149: 323-331. [Crossref]
- Proulx F, Seidman EG, Karpman D (2001) Pathogenesis of Shiga toxin-associated hemolytic uremic syndrome. Pediatr Res 50:163-171. [Crossref]
- Obata F, Obrig T (2010) Distribution of Gb(3) Immunoreactivity in the Mouse Central Nervous System. Toxins (Basel) 2:1997-2006. [Crossref]
- Amran MY, Fujii J, Suzuki SO, Kolling GL, Villanueva SY, et al. (2013) Investigation of encephalopathy caused by Shiga toxin 2c-producing Escherichia coli infection in mice. PLoS One 8: e58959. [Crossref]
- Trachtman H, Austin C, Lewinski M, Stahl RA. (2012) Renal and neurological involvement in typical Shiga toxin-associated HUS. Nat Rev Nephrol 8: 658-669. [Crossref]
- Wengenroth M, Hoeltje J, Repenthin J, Meyer TN, Bonk F, et al. (2013) Central nervous system involvement in adults with epidemic hemolytic uremic syndrome. AJNR Am J Neuroradiol 34:1016-1021. [Crossref]
- Tzipori S, Sheoran A, Akiyoshi D, Donohue-Rolfe A, Trachtman H (2004) Antibody therapy in the management of shiga toxin-induced hemolytic uremic syndrome. Clin Microbiol Rev17: 926-941. [Crossref]
- Trachtman H (2013) HUS and TTP in Children. Pediatr Clin North Am 60:1513-1526. [Crossref]
- Siegler RL (1994) Spectrum of extrarenal involvement in postdiarrheal hemolytic-uremic syndrome. J Pediatr 125: 511-518. [Crossref]
- Gallo EG, Gianantonio CA (1995) Extrarenal involvement in diarrhoea-associated haemolytic-uraemic syndrome. Pediatr Nephrol 9:117-119. [Crossref]
- Ullrich S, Bremer P, Neumann-Grutzeck C, Otto H, Rüther C, et al. (2013) Symptoms and clinical course of EHEC O104 infection in hospitalized patients: a prospective single center study. PLoS One 8: e55278. [Crossref]
- Melton-Celsa AR (2014) Shiga Toxin (Stx) Classification, Structure, and Function. Microbiol Spectr 2: EHEC-0024-2013. [Crossref]
- Karpman D, Håkansson A, Perez MT, Isaksson C, Carlemalm E, et al. (1998) Apoptosis of renal cortical cells in the hemolytic-uremic syndrome: in vivo and in vitro studies. Infect Immun 66: 636-644. [Crossref]
- Nevo Y, Ben-Zeev B, Tabib A, Straussberg R, Anikster Y, et al. (2013) CD59 deficiency is associated with chronic hemolysis and childhood relapsing immune-mediated polyneuropathy. Blood 121:129-135. [Crossref]
- Alegretti AP, Schneider L, Piccoli AK, Monticielo OA, Lora PS, et al. (2012) Diminished expression of complement regulatory proteins on peripheral blood cells from systemic lupus erythematosus patients. Clin Dev Immunol 2012: 725684. [Crossref]
- Chang JC (2017) Viral hemorrhagic fevers due to endotheliopathy-associated disseminated intravascular microthrombosis and hepatic coagulopathy: pathogenesis based on “two activation theory of the endothelium”. Clin Microbiol Infect Dis 2:1-6.
- Ray PE, Liu XH (2001) Pathogenesis of Shiga toxin-induced hemolytic uremic syndrome. Pediatr Nephrol 16: 823-839. [Crossref]
- Eisenhauer PB, Chaturvedi P, Fine RE, Ritchie AJ, Pober JS, et al. (2001) Tumor necrosis factor alpha increases human cerebral endothelial cell Gb3 and sensitivity to Shiga toxin. Infect Immun 69:1889-1894. [Crossref]
- Karpman D, Papadopoulou D, Nilsson K, Sjögren AC, Mikaelsson C, et al. (2001) Platelet activation by Shiga toxin and circulatory factors as a pathogenetic mechanism in the hemolytic uremic syndrome. Blood 97: 3100-3108. [Crossref]
- Hattori R, Hamilton KK, McEver RP, Sims PJ (1989) Complement proteins C5b-9 induce secretion of high molecular weight multimers of endothelial von Willebrand factor and translocation of granule membrane protein GMP-140 to the cell surface. J Biol Chem 264: 9053-9060. [Crossref]
- Nolasco LH, Turner NA, Bernardo A, Tao Z, Cleary TG, et al. (2005) Hemolytic uremic syndrome-associated Shiga toxins promote endothelial-cell secretion and impair ADAMTS13 cleavage of unusually large von Willebrand factor multimers. Blood 106: 4199-4209. [Crossref]
- Lo NC, Turner NA, Cruz MA, Moake J (2013) Interaction of Shiga toxin with the A-domains and multimers of von Willebrand Factor. J Biol Chem 288: 33118-33123. [Crossref]
- Chang JC (2016) A Thought on Possible Pathogenesis of Ebola Viral Hemorrhagic Disease and Potential Treatments: Could it be Thrombotic Thrombocytopenic Purpura-like Syndrome? Ther Apher Dial; 20: 93-98. [Crossref]
- http://emedicine.medscape.com/article/201181-overview#a4
- Fakhouri F, Roumenina L, Provot F, Sallée M, Caillard S, et al. (2010) Pregnancy-associated hemolytic uremic syndrome revisited in the era of complement gene mutations. J Am Soc Nephrol 21: 859-867. [Crossref]
- Hale GA, Bowman LC, Rochester RJ, Benaim E, Heslop HE, et al. (2005) Hemolytic uremic syndrome after bone marrow transplantation: clinical characteristics and outcome in children. Biol Blood Marrow Transplant 11: 912-920. [Crossref]
- Lesesne JB, Rothschild N, Erickson B, Korec S, Sisk R, et al. (1989) Cancer-associated hemolytic-uremic syndrome: analysis of 85 cases from a national registry. J Clin Oncol 7:781-789. [Crossref]
- Ito K, Komatsu Y (1993) Drug induced hemolytic uremic syndrome. Nihon Rinsho 51: 204-209. [Crossref]
- Valentijn KM, van Driel LF, Mourik MJ, Hendriks GJ, Arends TJ, et al. (2010) Multigranular exocytosis of Weibel-Palade bodies in vascular endothelial cells. Blood 116:1807-1816. [Crossref]
- Bernardo A, Ball C, Nolasco L, Choi H, Moake JL, et al. (2005) Platelets adhered to endothelial cell-bound ultra-large von Willebrand factor strings support leukocyte tethering and rolling under high shear stress. J Thromb Haemost 3: 562-570. [Crossref]
- Chauhan AK, Goerge T, Schneider SW, Wagner DD (2007) Formation of platelet strings and microthrombi in the presence of ADAMTS-13 inhibitor does not require P-selectin or beta3 integrin. J Thromb Haemost 5: 583-589. [Crossref]
- Valério E, Chaves S, Tenreiro R (2010) Diversity and impact of prokaryotic toxins on aquatic environments: a review. Toxins (Basel) 2: 2359-2410. [Crossref]
- Cheung V, Trachtman H (2014) Hemolytic uremic syndrome: toxins, vessels, and inflammation. Front Med (Lausanne) 1: 42. [Crossref]
- Murata A, Shimazu T, Yamamoto T, Taenaka N, Nagayama K, et al. (1998) Profiles of circulating inflammatory- and anti-inflammatory cytokines in patients with hemolytic uremic syndrome due to E. coli O157 infection. Cytokine10: 544-548. [Crossref]
- Tzipori S, Sheoran A, Akiyoshi D, Donohue-Rolfe A, Trachtman H (2004) Antibody therapy in the management of shiga toxin-induced hemolytic uremic syndrome. Clin Microbiol Rev 17: 926-941. [Crossref]
- Karpman D (2012) Management of Shiga toxin-associated Escherichia coli-induced haemolytic uraemic syndrome: randomized clinical trials are needed. Nephrol Dial Transplant 27: 3669–3674. [Crossref]
- Kim JJ, Goodship TH, Tizard J, Inward C (2011) Plasma therapy for atypical haemolytic uraemic syndrome associated with heterozygous factor H mutations. Pediatr Nephrol 26: 2073-2076. [Crossref]
- Scully M, Goodship T (2014) How I treat thrombotic thrombocytopenic purpura and atypical haemolytic uraemic syndrome. Br J Haematol 164: 759-766. [Crossref]
- Palma LM, Langman CB (2016) Critical appraisal of eculizumab for atypical hemolytic uremic syndrome. J Blood Med 7: 39-72. [Crossref]
- Kaplan BS, Ruebner RL, Spinale JM, Copelovitch L (2014) Current treatment of atypical hemolytic uremic syndrome. Intractable Rare Dis Res 3: 34-45. [Crossref]
- Lapeyraque AL, Malina M, Fremeaux-Bacchi V, Boppel T, Kirschfink M, et al. (2011) Eculizumab in severe Shiga-toxin-associated HUS. N Engl J Med 364: 2561-2563. [Crossref]
- Pape L, Hartmann H, Bange FC, Suerbaum S, Bueltmann E, et al. (2015) Eculizumab in Typical Hemolytic Uremic Syndrome (HUS) With Neurological Involvement. Medicine (Baltimore) 94: e1000. [Crossref]
- Delmas Y, Vendrely B, Clouzeau B, Bachir H, Bui HN, et al. (2014) Outbreak of Escherichia coli O104:H4 haemolytic uraemic syndrome in France: outcome with eculizumab. Nephrol Dial Transplant 29: 565-572. [Crossref]
- Kielstein JT, Beutel G, Fleig S, Steinhoff J, Meyer TN, et al. (2012) Collaborators of the DGfN STEC-HUS registry. Best supportive care and therapeutic plasma exchange with or without eculizumab in Shiga- toxin-producing E. coli O104:H4 induced haemolytic-uraemic syndrome: an analysis of the German STEC-HUS registry. Nephrol Dial Transplant 27: 3807-3815. [Crossref]
- Jokiranta TS (2017) HUS and atypical HUS. Blood 129: 2847-2856. [Crossref]
- Brady TM, Pruette C, Loeffler LF, Weidemann D, Strouse JJ, et al. (2016) Typical Hus: Evidence of acute phase complement activation from a daycare outbreak. J Clin Exp Nephrol 1:11. [Crossref]