Characteristic differences have been observed at genetic and metabolic levels between the mitochondria of normal and cancerous cells. These include altered expression and activity of enzymes involved in aerobic and anaerobic respiration, oxidation-reduction pathway and mitochondrial DNA (mtDNA) translation, transcription. While these phenomena have been widely described, the mechanism involved for development of mtDNA mutations and their functionality in cancer, drug resistance and disease progression is still unknown.
We have here presented the review of the known mitochondrial DNA alterations in human oral cancer & precancer and discussed the possible mechanisms of emergence and their clinical relevance. The aim of this review was to understand the role of mtDNA mutation in oral carcinogenesis and explore the potential use of mitochondrial mutations as biomarkers for prediction of oral precancer & cancer, and, as potential targets for anti-cancer agents.
Mitochondria, Mitochondrial DNA, Mutations, Oral Cancer, OSCC, Oral Precancer, Oral potentially malignant disorder, OPMD, biomarker
DNA: Deoxyribonucleic Acid; RNA: Ribonucleic Acid; mtDNA: Mitochondrial DNA; OSCC : Oral squamous cell carcinoma; OPMD: Oral potentially malignant disorders; nt: Nucelotide; rRNA: Ribosomal RNA; tRNA: Transfer RNA; ATP: Adenosine Tri Phosphate; ROS: Reactive Oxygen Species; Bcl-2: B Cell lymphoma 2; BAX: BCL2 Associated X; HIF: Hypoxia Inducible Factor; PI3K: Phosphoinositide 3-kinase; Akt: RAC-alpha serine/threonine-protein kinase; NADH: Reduced Nicotinamide adenine dinucleotide; NOX1: Nicotinamide adenine dinucleotide phosphate oxidase; ND5: NADH dehydrogenase 5; PET: Positron emission tomography; FDG: 2-18F-2-deoxyglucose; DLCs: Delocalized lipophilic cations
Oral squamous cell carcinoma (OSCC) is the sixth most common cancer in the world accounting for 3% of all new cancers in the Western world [1] and up to 40% of all cancers in other parts of the world, such as India [2]. It is the second leading cause of mortality in developed countries [3]. OSCC is believed to progress through defined serially consecutive stages of oral premalignant lesions: hyperplasia to epithelial dysplasia of varying degree, to carcinoma in situ, and invasive cancer.
Oral potentially malignant disorders (OPMD) are clinical presentations that may have a potential to become cancer [4], and often precede oral cancer. Clinically, OPMD most frequently present as leukoplakia and erythroplakia [5]. The cancer risk for OPMD varies from as low as 0.13% to as high as 50% [6-8].
Once invasive cancer is formed, the prognosis becomes poorer, with 5-year survival rates of 40-50% in the Western world and even lower in India (20-43%) [1,2,8]. Despite refinement of surgical techniques and adjuvant therapies, the survival rate has not improved in the last decades. High local recurrence and formation of second primary malignancies are the major causes of this high mortality rate [8-10]. Survivors frequently have to endure serious cosmetic and/or functional compromise. The key to improving the dismal mortality and morbidity of OSCC is to prevent its conversion from OPMD and its progression. During the last decade, molecular techniques have developed rapidly, and the availability of new technologies has led to a growing interest in developing new approaches that could be used to identify high-risk lesions so that intervention can be targeted to such lesions more effectively and prognosis is improved. As a result, there has been a recent emphasis on the study of OPMD and early phases of carcinogenesis for screening and management [7,11,12].
It is well documented that despite similar exposure to carcinogens, only a proportion of exposed individuals develop cancer. This suggests that some individuals are resistant to the exposure. On the other hand, there is an emerging population of cancer patients who lack the exposure to known agents [12-14]. In the latter case, an inherent susceptibility for mutagens exposure can play a role in the carcinogenesis. This susceptibility can be used in determining the cancer risk. Future lies in identifying markers of susceptibility that associate with clinical, histological and molecular markers. However, this aspect is little understood. Current research suggests that it is multifactorial and multigenic, acting both synergistically and consequentially. Example may be cited for cytochrome p450 genes involved in the metabolism of carcinogens. Some of them code for Phase I enzymes that activate precarcinogens to moeties that can damage DNA. These enzymes are checked by the Phase II enzymes through chemical conjugation. The subtle equilibrium between these such events in consonance with actual level of carcinogen exposure determines an individual's risk of developing cancer [15-17]. Mitochondrial metabolism is an important component of such a homeostasis system [18-20]. There have been numerous reports implicating mitochondrial defects in several cancers [21-23]. The majority of the mitochondrial defects rally around mitochondrial DNA damage [24-28].
Mitochondria under electron micrographs have been visualized as static, ‘cigar-shaped’ organelles, bounded by the two distinct membranes, outer and inner separated by intermembrane space (Figure 1). Visualized by fluorescence microscopy after staining with rhodamine 123 dye, mitochondria appear as a long filamentous dynamic network that show changes in size, form and location [29,30]. Mitochondria have their own genome, double stranded circular 16.6 kb DNA, with self-transcription, translation, and protein assembly machinery, providing them genomic independence [30,31]. MtDNA is less than 1% of total cellular DNA, but its gene products are essential for normal cell function. The inner mitochondrial membrane harbours the enzymes forming the respiratory chain in oxidative phosphorylation. The complete respiratory chain inside mitochondria contains 87 polypeptides in which 13 are mitochondria coded. Each mitochondrion contains generally less than 10 copies of its genome [31,32]. Approximately 1500 genes constitute the mitochondrial genome. 37 genes are in the 16 569 nt mtDNA and the remainder belong the nuclear DNA. The mtDNA genes code a 12S and 16S rRNA, 22 tRNAs, and 13 essential OXPHOS polypeptide subunits [30,32]. Mt DNA is tightly organized and clever in arrangement such that it performs duty as spacer between the genes for proteins, and code tRNAs [33].
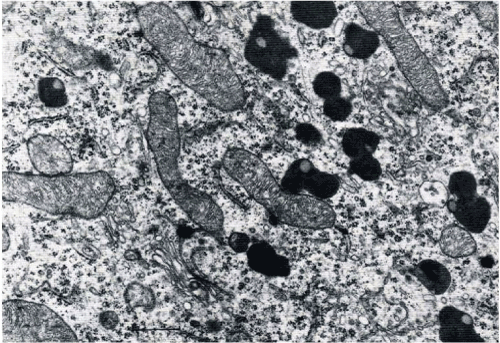
Figure 1. Mitochondrial profile and polymorphic pigment granule Scale 0.5um
Mt DNA is 100 times more mutations prone than nuclear DNA [34,35] due to lack of histone protection [30,31,34,35], limited repair capacity [34,35] and close proximity to the electron transport chain, and the active site of superoxide radicals generation. Mt DNA mutations/deletions are reported at a less than 1% frequency [33-35]. It increases with advancing age and is prone to large scale deletions in the flanking repeats regions [34-36].
The following theories have been proposed for origin of mitochondrial mutations:
- Slip mismatching.
- Illegitimate elongation of the D-loop strand.
- Free radical induced deletions and mutations.
- Clonal expansion of mutations in the stem cells.
The first two theories fail to explain for deletions that occur where there are no repeats [37-39]. Moreover they also cannot explain point mutations. Today, free radical induced mtDNA mutation theory seems more relevant [39,40] as it postulates on the imbalance between free radical generation and scavenging systems. During electron flow along electron transport system, few flowing electrons escape or leak and react with molecular oxygen to form superoxide radicals (O2-). This leakage occurs mainly at complexes I and III [39-41]. Superoxide is detoxified by manganese superoxide dismutase located inside the mitochondrial matrix generating partially stable H2O2. H2O2 remains inside mitochondria until it diffuses out to the cytosol and the nucleus. Reduced transition metals provide H2O2 an additional electron generating reactive free hydroxyl radical (.OH). Inside mitochondria, glutathione peroxidase slowly reduces H2O2 to water by while the same is done in the cytosol by peroxisomal enzyme catalase. Disturbance in homeostasis between free radical generation system leads to large scale deletions across susceptible regions in mtDNA.
Mitochondrial activities have an profound effect on cellular physiology beyond ATP production. In fact interrelated events occur, as shown in Figure 2. Reactive oxygen species (ROS), produced inside mitochondria, are involved in the regulation of many physiological processes [21,39-43]. However, if produced excessively, mitochondrial ROS can damage enzymes, lipids and mutagenize the mtDNA [19-21,39,43]. Mitochondria also regulate intracellular Ca2+ homeostasis, and are key to the regulation of cell death pathways [44-47] via mitochondrial permeability transition pore (mtPTP) [21,44-47].
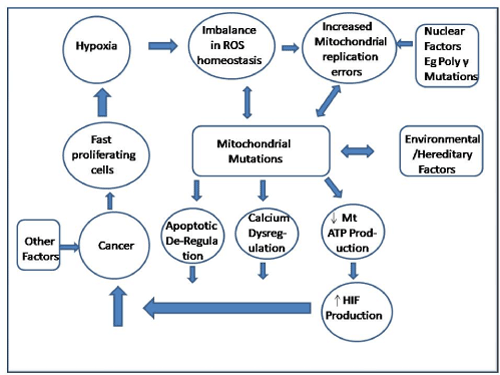
Figure 2. Mitochondria Events during Carcinogenesis
Researchers suggest a role for oxidative stress in oral carcinogenesis [18,21,48-50]. Oxidative stress is a disturbance in the balance between the production and consumption resulting in a relative excess of ROS. Mitochondria are a major source of production of ROS because of their involvement in oxidative phosphorylation. By nature, ROS are unstable. They damage nuclear and mitochondrial DNA [35,36]. The mitochondrial genome is prone to such damage that can cause mitochondrial dysfunction [30,38,42]. In addition to producing mutations, such damage may also change the expression levels of genes that induce cell growth (proliferation & differentiation) and effect lipid peroxidation, protein alteration, and membrane rupture [43,46].
Conditions for increased ROS generation vary greatly from psychological to genetic and environmental. More than 19 carcinogenic products are present in cigarette smoke, which are pyrolytic in nature. Their mechanism of action upsets the delicate balance of ROS generation and consumption cycle. This imbalance in the homeostasis of free radical levels may lead to mtDNA mutations upsetting electron transport components, and in turn may compromise the normal electron flow resulting in increased electron leakage and increased generation of superoxide radicals, which are the progenitors of other free radicals or ROS. Literature shows that mtDNA mutations and increased oxidative stress are interrelated and have been reported in various cancers [23-28,47-53]. The fact that mitochondria are involved in free radical induced cell death supports free radical theory. A strong association exits between apoptosis and mitochondria. Moreover, it is known that the mitochondria initiate the early apoptotic events [21,25,54] evident by involvement of two members of the Bcl-2 family. Bcl-2, the anti-apoptotic factor, maintains inner mitochondrial membrane electrochemical gradient by controlling influx and efflux of Ca2+ into and out of the mitochondria, whereas BAX, the pro-apoptotic member, is involved in the disrupting of aforementioned membrane electrochemical gradient [55-59].
Mitochondria are DNA containing cell organelles having semi-autonomous existence. It is involved in cellular respiration, food metabolism, free radical homeostasis, apoptosis, and cell death. Recent literature provides substance to Warburg hypothesis that defective mitochondrial functioning contributes to the carcinogenesis. Warburg effect postulates that most cancer cells rely on aerobic glycolysis. Proponents of Warburg effect point out that this effect may be the consequence of mitochondrial damage, or cellular adaptation to low-oxygen levels inside tumors, or a result of cancer deranging mitochondrial functioning to save cancerous cells from apoptosis. Mitochondrial involvement in cellular energy production, free radical homestasis, aging, and apoptosis point out their importance in tumorigenesis [60-62]. Though profound literature exists for nuclear DNA changes there recent shift to effects of mutations within mtDNA.
Mt DNA mutations in oral cancer were first reported in 2000 [49] and range from insertion, deletion, chain termination mutations to missense mutations. There are number of reports citing considerable changes in both quantity (mitochondrial copy number) and quality (nucleotide changes) of mt DNA contents [63-69]. It is reported that the actual mtDNA copy number in certain cancers correlate with the specific site of mutation inside cancer [70-73]. For example, mutations in the D-loop region, which control mtDNA replication, would result in decreased mtDNA copy number [74-76]. On the other side, mtDNA mutations in genes of respiratory chain proteins would result in an increased mtDNA copy number. These effects are a compensatory response to mitochondrial dysfunction [60,77]. Besides, different haplogroups associated with mitochondria, which are ethnicity related, produce different susceptibility to environmental factors [78-80]. This is evident by presence of numerous polymorphisms with different mutational load in various ethnic groups for the same disease. In the majority of cases, multiple mitochondrial mutations are presentdue to accumulation of several mtDNA mutations as according to clonal expansion model, mutant somatic mitochondrial genome replicates at a higher rate than the wild type [81-82]. Though the mtDNA mutations occur throughout mitochondrial genome, yet they are more in frequency in the protein coding genes, rRNA genes and in the D-loop region. Since majority of these mutations are T to C and G to A base transitions, causative factor most likely is ROS generating mutagen. Mt DNA mutations can be somatic and germline. A number of studies have been done to define germline mutations and polymorphisms, which are mentioned in Table 1. Generally germline mutations may underline the inherited susceptibility to develop oral cancer through precancer. However, our efforts were largely targeted at defining somatic mitochondrial mutations, which are acquired during one’s lifetime due to carcinogen exposure.
Table 1. Mitochondrial DNA mutations in oral cancer
Mutation |
Reference |
Description |
Mitochondrial C-tract Alterations |
Ha et al, 2002 |
Study Parameters
- Number of cases examined = 93
- Type of cases = Premalignant lesions of the head and neck
- Area Examined= Poly-cytosine tract (C-tract) of the displacement loop.
|
Results
- C-tract alteration found in Thirty four of the 93 (37%) patients.
- Increased incidence from histologically benign hyperplasia (22%) to in situ squamous carcinoma (62%: P < 0.01).
|
Inferences
- Mitochondrial DNA alterations occur in the premalignant lesions in thehead and neck at the early stage.
- Their incidence increases with histologicalseverity.
- C-tract alterations could be used as markers of histopathological progression.
|
Mitochondrial DNA Mutations at
- D-loop 66-71 del G
- D-loop T204C
- D-loop G207A
- D-loop A207G
- D-loop C222T
- D-loop C246T
- D-loop 303-309 polycytosine changes
- D-loop C313A
- D-loop C318T
- D-loop C489T
- ND2 G4510T
- ND2 A4986C
- ND2 A5026G
- COX III 9485 del C frameshift
- ND3 C10245T
- ND4 T11794C
- D-loop T16320C
- D-loop C16419A
|
Tan et al., 2003, 2004 |
Study Parameters
- Number of cases examined =17
- Type of cases= Oral squamous cell carcinoma and oral adenoid cystic carcinoma along with paired adjacent control tissues.
- Area Examined= Complete mtDNA
|
Results
- mtDNA mutations present in 14 of 18 (77.8%) tumour samples.
- Total 26 somatic mtDNA mutations reportedincluding 6 in mRNA coding regions.
- Of these 3 missense mutations (C14F, H186R, T173P), 1frameshift mutation (9485delC)
- 8that is 44%tumor samples had C tract alterations
|
Inferences
- Somatic mtDNA mutations occur in oral cancer.
- Missense and frameshift mutations in evolutionary conserved regions of mtDNA
- These mutations can have functional relevance in the pathogenesis.
|
Mitochondrial DNA 4,977-bp Deletion |
Shieh et al., 2004 |
Study Parameters
- Number of cases examined =12
- Type of cases= Paired Oral squamous cell carcinoma, pre cancer and blood lymphocytes.
- Area Examined = mtDNA
|
Results
- Frequency of deleted mtDNA 4,977-bp increased from precancer to cancer.
- Decrease in the proportion of deleted mtDNA from precancer to cancer lesions.
|
Inferences
- Cytoplasmic segregation of the mutant mtDNA during cell division promotes oral carcinogenesis.
|
Mitochondrial DNA D-Loop
mutations at
- nt 146
- nt152
- nt 186
- nt 4917 (ND2)
|
Prior et al., 2006 |
Study Parameters
- Number of cases examined =30
- Type of cases = Paired OSCC tumour and non-tumour tissue (tumour negative lymph node) from neck dissections
- Area examined = mtDNA D loop
|
Results
- 3 Mitochondrial D-Loop mutations reported in tumour samples.
- 7 different types of mutation in the D Loop region between nt 8 and 429.
- Increased frequency of ND2 gene mutation from normal to cancer.
|
Inferences
- The mtDNA mutation hotspotbetweennt 152 and 186 as potential biomarkers for oral SCC
- Mutations at nt 4917 is a potential mutational hotspot in oral SCC
|
Mitochondrial DNA mutations
in ND4 gene at
- nt 11125
- nt 11203
- nt 11288
- nt 11306
- nt11479
- nt 11719
- nt 11812
|
Allegra et al., 2006 |
Study Parameters
- Number of cases examined =10
- Type of cases = head and neck squamous cell carcinoma linesand compared with normal DNA isolated from fibroblasts of the same patients
- Area examined = mtDNA ND4 gene
|
Results
- 8 somatic mutations in the ND4 gene.
- 5 ND4 gene polymorphisms.
- 5 silentpolymorphisms.
- Of the 8 somatic mutations, 3 altered the amino acid sequence.
|
Inferences
- Synomymous mutations might affect enzyme function.
- mtDNAmutations and polymorphisms once developed in the tumour, become the stable clonal markers.
- Theycould serve as biomarkers for cancer.
|
Mitochondrial DNA D-Loop
mutationsat
- nt 213
- nt 214
- nt 239
- nt 303-309
- nt408
- nt 514
|
Lievre et al., 2006 |
Study Parameters
- Number of cases examined =109
- Type of cases =OSCC
- Area examined = mtDNAD Loop gene
|
Results
- 25 somatic D-Loop mutations reported
- These were identified in 23 of the 109 (21%) tumours.
- The majority of the mutations were located in the Polycytosine tract (19 out of 25, 76%)
- These were insertions or deletions of one (n=15) to several (n=4) base pairs.
|
Inferences
- D-Loop mutations could be considered as a biomarker for the early detection.
|
Mitochondrial DNA mutations in
- D-loop
- ND2
- ND5
- COIII
- CYTB
- ATP6
- 12S rRNA
- 16S rRNA
- tRNA
- ND4L (mitochondrially encoded NADH dehydrogenase subunit 4L gene)
|
Zhou et al., 2007 |
Study Parameters
- Number of cases examined =83 HNSCC
- Type of cases =OSCC
- Area examined = mtDNA
|
Results
- mtDNA mutations in 41 of 83 (49%) tumors.
- A total of 228 mutations when compared with matched normal blood leukocyte.
- Mutations were found in the D-loop and coding region.
- The majority of tumors contained from 1 to 4 mutations.
|
Inferences
- More nonsynonymous amino acid-changing mutations in ND2, ND5, COIII, CYTB, and ATP6 genes.
- Sequencing of margins with dysplasia and its comparison with the tumor, suggest mtDNAmutation role in tumor progression.
|
Mitochondrial DNA mutations in
- Polycytosine Tract
- ND2
- ND5
- COIII
- CYTB
- ATP6
- rRNA
|
Pietka et al., 2008 |
Study Parameters
- Type of cases =OSCC
- Area examined = mtDNA
|
Results
- 37% of patients with premalignant lesions and 62% with carcinoma in situ containedmtDNA mutations.
- Mutations in COIII, CYTB, and ATP6 in 17% of patient.
|
Inferences
- Increasing trend/frequency of mtDNA mutations from precancer to cancer.
- Majority of mutations in polycytosine tract.
- Both content and number of mtDNAincreased with severity of lesion.
|
A total of 37 somatic mtDNA mutations :
- 17 in the noncoding regions mtDNA, comprising:
- 9 in tRNA at nucleotide positions : 1606, 1643, 5537, 5570, 5700, 5799, 7538, 10457, 15984
- 1 in the D-Loop at nucleotide position 5756
- 4 in12SrRNA at nucleotide positions 705, 912, 1323, 1444
- 3 in 16SrRNA at nucleotide positions 2069, 2188, 2517
- 20 mutations was found in the coding regions
- 1 in ND2 at nucleotide position 4689
- 4 in ND5 (Complex I) at nucleotide positions 12447, 13322, 13808, 13814
- 3 in CYTB (Complex III) at nucleotide positions 14803, 15249,15640
- 8 in COX-I at nucleotide positions 6027, 6027, 6380, 6542, 6794, 7029, 7242, 7284
- 3 in COX-II at nucleotide positions 7767, 7865, 8170
- 1 in COXIII (Complex IV) at nucleotide position 9320
|
Dasgupta et al., 2010 |
Study Parameters
- Number of cases examined = 50
- Type of cases =HNSCC including OSCC
- Area examined = mtDNA
|
Results
- mtDNA mutations were mostly heteroplasmicwith nucleotide transitions (A↔G; T↔C).
- Most of the noncoding mutations were in tRNAs(53%).
- 75% of the reported coding mtDNA mutations were nonsynonymous affecting Complex IV with significantly higher mutational load.
|
Inferences
- Analysis of mtDNA mutation for molecular assessment of histologically negative margins and as well for monitoring cancer progression in locoregional recurrences.
- Study could not show functional effect of mutations via expression study.
- Relatively very low number of mutations in D-loop, which is contrary to literature.
|
Mitochondrial D-loop mutation |
Liu et al., 2012 |
Study Parameters
- Number of cases examined = 59
- Type of cases =OSCC
- Area examined = mtDNA D loop
|
Results
- Somatic mutations of the D-loop in 38 (64.4%) patients
- Most of mutations in the poly-C tract.
|
Inferences
- Patients with D-loop mutations have better survival (2 year disease specific survival rate: 73.4 vs. 45.0%, P = 0.0374).
|
A total of 645 somatic mtDNA mutations in whole of mitochondrial DNA. There distribution is:
- 355 in D-Loop
- 181 novel mutations (71 synonymous and 110 nonsynonymous) in protein-coding genes.
|
Lai et al., 2013 |
Study Parameters
- Number of cases examined = 300
- Type of cases = OSCC
- Area examined = mtDNA
- Followup time=69 months
|
Results
- 329 unique nucleotide positions reported.
- 85%, 47.1%, 13.8%and 8.3% of total cases had somatic mutationspresent in the D-loop region, protein coding genes, the rRNAsand tRNAs respectively.
- 91 pathogenic mutations reported
|
Inferences
- Highest mutation frequency (mutations/nucleotides) in D-loop region.
- Inside D loop, D310 polycytidine stretch region (165/645), 514–523, 568–573, 956–965, 5895–5899 and 16180–16195 were 10 most commonly mutated sites. reported by authors which showed strong.
- Pathogenic mtDNA mutations are a potential prognostic marker for OSCCs.
- TP53 R72Ppolymorphism was associated with pathogenic mtDNA mutations.
|
Total of 26 somatic mutations in mtDNA comprising at nucleotide positions:
- nt146
- nt150
- nt185
- nt195
- nt789
- nt3168
- nt3669
- nt4136
- nt4580
- nt4703
- nt4883
- nt6755
- nt8925
- nt9061
- nt12007
- nt13542
- nt13869
- nt14697
- nt15229
- nt15299
- nt15721
- nt16294
- nt16311
- nt16325
- nt16463
- nt16519
|
Mondal et al., 2013 |
Study Parameters
- Number of cases examined = 10
- Type of cases =OSCC
- Area examined = mtDNA
|
Results
- 9 somaticmutations in the non coding region that is hypervariable regions, HVR1 and HVR2.
- 17 somaticmutations in the coding region.
- 3 novel mutations inside D loop at nucleotide positions 16294, 16325 and 16463
- Complex I mutations at nucleotide positions 4136 and 13542 were frequent.
- 3 novel mutations in Complex III at nucleotidepositions 15229, 15299 and 15721 occurred
|
Inferences
- Majority (47%) of coding somatic mutations were in Complex I.
- Only 1 mutation in ND1 region was synonymous.
- Majority of mutations were base transitions T > C (38.4%)
- D loop and complex I is hotspot for mutations.
|
Mitochondrial mutations:
- 4977 bp deletion
- 50 bp deletion (covering nt298–347/307–356)
- Poly Catnt303–309
- nt146
- nt152
- nt199
- nt204
- nt215
- nt234
- nt257
- nt16129
- nt16140
- nt16148
- nt16153
- nt16165
- nt16189
- nt16234
- nt16294
- nt16311
- nt16338
|
Datta et al., 2015 |
Study Parameters
- Number of cases examined = 74 oral precancer& 117 cancer
- Type of cases =Leukoplakia& OSCC
- Area examined = mtDNA
|
Results
- 10-foldless ∼5 kb deletion in cancer tissues as compared to those in controls.
- Number of mutations in D – Loop greater in cancer than precancer.
- Mostof the mutations (19/30) were single base substitutions.
- Remaining mutations (11/30) were repeatalterations in the C-tract (at 303–309np).
|
Inferences
- 5 kb deletion and D loop mutations can be used in differentiating oral precancer, cancer and healthy tissues.
- However, the study could be more authentic if the samples belonged to same patients that is from precancer to cancer stage.
- Then functional study of importance of loss of 5kb in oral precancer and cancer is still not proven. Lastly, the sample size was far too less for such advocacy.
|
Mitochondrial Mutations:
- Length heteroplasmy within the polycytosine stretches of HVS-I(16188 -16195) and HVS-II (310)
- specific variants on 124 different nucleotide positions.
- C64Y
- A183R
- G2916R
- C5297Y
- A5894R
- G6762R
- G9565R
- T9865Y
- G10310R
- G12868R
- G12736R
- T13897Y
|
Kloss-Brandstätter et al.,2015 |
Study Parameters
- Number of cases examined = 85 samples 28 cases
- Type of cases = OSCC
- Area examined = mtDNA
|
Results
- 9% of somatic mutations intRNAs, 18.3% in ribosomal RNAs, 52.2% in protein-coding regions, and 22.6% in the control region.
- Tumor samples had aexhibited higher length heteroplasmyas compared to their corresponding benign tissue samples
|
Inferences
- Majority of reported mutations were synonymous.
- Detection of low levels of heteroplasmy in tumors might help in early identification and monitoring of neoplastic progression.
- Clonal expansion of mitochondrial DNA mutations especiallymtDNAheteroplasmies occur during progression.
- Limitations of the study were descrepancies in the mutational pattern of mtDNAheteroplasmies in different areas within the same tumor.
- Another was difference in presence of heteroplasmic mutations below 10% in benign tissues(79.3%) and in tumor tissues (63.9%) wasstatistically not relevant.
|
Mitochondrial Mutations:
- nt66
- nt146
- nt150
- nt199
- nt205
- Polycytosine tract
- nt369
- nt392
- nt414
- nt467
- nt514
- nt567
- nt2288
- nt3882
- nt3970
- nt6392
- nt9053
- nt9824
- nt10230
- nt10310
- nt10398
- nt13135
- nt13152
- nt 16257
- nt16261
- nt 16304
- nt 16316
- nt16340
- nt16524
|
Lin et al., 2015 |
Study Parameters
- Number of cases examined = 120
- Type of cases = OSCC
- Area examined = mtDNA D Loop
|
Results
- Majority of mutations were in D-loop (62.5%).
- Inside D loop hotspot for mutation was polycytidinestretch over np 303 of mtDNA(85.3%).
- Most of reportedmtDNA mutations were heteroplasmic (64.0%).
|
Inferences
- Somatic mutations in D-loop of mtDNAwere associated with a better survival in oral squamous cell carcinoma patients.
- Limitation is very little similarity of mutations between blood/plasma and tissue samples (23.4/26.7%).
|
Mitochondrial Mutations:
|
Liu et al. 2015 |
Study Parameters
- Number of cases examined = 130
- Type of cases =Head and Neck including OSCC
- Area examined = mtDNA D Loop
|
Results
- D-loop mutations between plasma/blood and tumor tissues of subjects (27.1% and 25.5%).
- Majority of mutations reported were in polycytosine tract and hetroplasmic in nature.
|
Inferences
- Greater number of somatic D loop mutations in OSCC cases than other head and neck cancer types.
- Patients with mtDNA D loop somatic had better prognosis than those without.
- Authors reported poor correlation for mtDNA
|
9 mutations in D-Loop:
- 1 point mutation,
- 2 base deletions,
- 3 insertion mutations and
- 3 heterozygous mutations
|
Yuan et al., 2015 |
Study Parameters
- Number of cases examined = 30
- Type of cases =OSCC
- Area examined = mtDNA D Loop
|
Results
- Total 9 mutations were reported including one point mutation, two base deletions, three insertion mutations and three heterozygous mutations.
- 8 cases out of 30 had mutations in the D-loop region.
|
Inferences
- Authors reported 27% mutation rate in D-loop.
- Polymorphic site at 313 was also identified.
- Heterozygous mutations were rare and might be involved in pathogenesis.
|
Mitochondrial Mutations comprising:
- 271 SNPs,
- 7 novel SNPs (or SNVs),
- 15 somatic mutations
|
Chattopadhyay et al, 2016 |
Study Parameters
- Number of cases examined = 38
- Type of cases =OSCC
- Area examined = mtDNA
|
Results
- Authors reported majority of mutations and SNPs in D-loop (76 SNPs and 1 somatic).
- Second in number was CyB (36 SNPs) followed by ATP6 (24 SNPs), ND5 (17 SNPs and 5 somatic), ND4 (18 coding and 2 somatic) and other non-coding and coding DNA sequences.
|
Inferences
- Bioinformatic tools predicted that of total reported DNA variations 53 and 8 non-synonymous SNPs and somatic mutations, respectively may have deleterious effects on the protein.
- Limitation of tudy was very low sample size.
- The strength of paper was that it used matched samples for cases and controls.
|
Total of 164 Somatic Mutations in mtDNA comprising:
- 56 nonsynonymous mutations
- 3 frame-shift mutations
- 4 stopgain mutations
- 12synonymous somatic mutations
- 46 non-coding (rRNAs, tRNAs orthe D – loop)
|
Palodhi et al, 2019 |
Study Parameters
- Number of cases examined = 89
- Type of cases =OSCC
- Area examined = mtDNA
|
Results
- D –loop region had thirty-two mutation.
- ATP8 was found to be the least mutated mitochondrial gene. \
- Significantly higher proportion of nonsynonymous somatic mutations were reported in mitochondrial respiratory complex I (P=2.3×10−8) and complex IV genes (P=5.5×10−7).
- The dN/dS ratio of somatic mutations was found to exceed 1 for all complexes (Complex I=7.4, Complex III=3.5, Complex IV=13.3, Complex V=3.3).
|
Inferences
- Variable ratio of nonsynonymous to synonymous mutations (NS/S) between somatic and germline mutation data,the implication for which is unexplained.
- Positive selection for nonsynonymous somatic mutations in mtDNA for OSCC – GB tumors.
- The limitation of this study that it is difficult to generalize the results to OSCC in general and often OSCC-BG does not occur in isolation.
|
Synonymous mutation at
DNA polymorphisms in
- nt 73, 263, 523, 524, 16223 (D-loop)
- nt 4769, 10,398,10873,11719,12705 (Complex I)
- nt14766, 15301, 15326 (Complex III)
- nt7028, 9540 (Complex IV )
- nt8860, 8701 (Complex V)
- nt750, 1438, 2706 (rRNA)
|
Shu et al., 2019 |
Study Parameters
- Number of cases examined = 8
- Type of cases =OSCC of Tongue
Area examined = mtDNA |
Results
- Total of 21 polymorphisms reported which included 6 in the non-coding region (D-loop), 5 in Complex I, 3 in Complex III, 2 in Complex IV, 2 in Complex V and 3 in rRNA.
- 2 out of 8 cases displayed mitochondrial microsatellite instability (mtMSI) which was localized in the D310 region.
- 1 synonymous mutation in the ND5 gene (T>C transition at nucleotide position 13,830)
- No muitations in the tRNA gene.
|
Inferences
- Majority of nucleotide variations in D Loop and Complex I.
- Present study has used a small sample size and no healthy controls.
- Polymorphisms and mtMSI may be a hotspot of genome alteration in tongue cancer.
|
Due to free radical production, the respiratory chain protein genes (Figure 3) are more prone to oxidative damage [18,19,39]. Increased oxidative damage causes decrease in oxidative phosphorylation resulting in low ATP production, which, in turn, further causes mtDNA damage [35,41]. Interestingly, mtDNA mutations have been reported even in noncancerous oral mucosa exposed to known risk factors like betel quid [83] and tobacco [84] pointing out their early involvement. Mucosa harbouring premalignant changes has also been found to contain mtDNA mutations with the prevalence increasing from hyperplasia to carcinoma in situ [72,73].
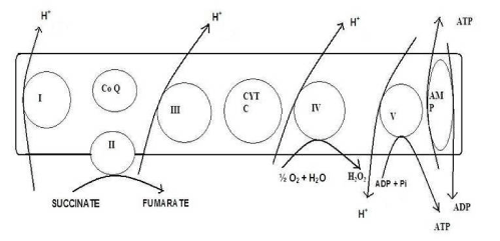
Figure 3. Inner Mitochondrial Membrane showing components as well as events of Electron Transport Chain & Oxidative Phosphorylation
Several reports have associated metabolic aberrations in cancerous cells with their mitochondrial bioenergetic function. The facts that normal and malignant cells have different preferences for respiratory substrates, have different rates of electron and anion transport, and the different capacity for calcium concentration support the statement [85]. Certain enzymes involved in oxidative phosphorylation are known to have decreased activity in cancer cells. For example, the enzymatic activity velocity for ATPase in mitochondria [86] and submitochondrial particles [86] is considerably lower than that in normal liver. The same is true for Mitochondrial cytochrome c oxidase activity when compared with epithelial cell [87]. Another aspect is physiological effects for example, mitochondrial membrane potential. This has been reported to be higher in carcinoma cells than in normal epithelial cells [88]. The biggest hurdle as of now is that there are no common metabolic aberrations to all cancer cells identified till date.
The characteristic feature of cancer cells is rapid proliferation and growth, which results in hypoxia because local vasculature is unable to supply an adequate amount of oxygen. Strikingly, such hypoxic conditions cause cellular death in non-malignant cells. Cancerous cells escape hypoxia mediated death by lowering p53 expression or p53 mutation or through Hypoxia Inducible Factor (HIF) dependent mechanisms [89]. Hypoxia upregulates the glycolytic pathway in tumour cells because mitochondria cannot compensate for extra energy requirement even after induction of HIF-1 [90,91], Phosphoinositide 3-kinase (PI3K) and its downstream target Akt (also known as protein kinase B) [92]. Thus, it appears that mitochondrial functioning has little role in shunting of pathway for increased ATP demand in tumour cells. If during subsequent metabolic events some abnormality either in function or structure arises in mitochondria therefore it appears that even incompetent mitochondria would not adversely affect the growth of tumour cells. However, such a conclusion is wrong because, firstly, normal mitochondrial functioning is still required for basic energy demands for normal metabolic activities and, secondly, recent data show glycolysis accounts for approximately 60% of total ATP production in majority of cancer cells [93,94]. It means that though most cancer cells perform respiration but in them the rate of oxidative phosphorylation is reduced because of increased glycolysis and lactate production. Therefore, this shunting of energy pathway has a hidden purpose of supplying more substrates for increased DNA synthesis and growth of tumour cells. Lactate dehydrogenase inhibition or the pyruvate dehydrogenase activation inducing tumour cells to oxidize pyruvate in the TCA cycle with consequent mitochondrial respiration stimulation proves [93-95] that mitochondrial activity is not fundamentally impaired in cancer cells. However, the issue of mitochondrial mutations present in oral tumours is complex and at present it is debatable whether they are just bystanders in the process of tumorigenesis.
It has been reported that tumour prognosis correlates clinically with degree of glycolysis. Researchers have defined tumour aggressiveness as the ratio between the activities of the mitochondrial enzyme ATP synthase and the glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase [93]. This definition is a biochemical one according to which mitochondrial function in cancerous cells can be downregulated by low oxygen levels, variations in metabolic fluxes and gene expression. It has been postulated that mitochondrial DNA mutations together with defects in respiratory enzyme complexes increased ROS production. In support of this hypothesis it has been reported that defective activity of mitochondrial respiratory chain leads to: 1) NADPH oxidase (NOX1) over expression which produces superoxide [41,96,97] 2) increased nuclear DNA damage and hypermutagenesis [98]; and 3) apoptosis resistance [45]. Such cellular phenotype changes contribute to development of cancer. However, mitochondrial dysfunction during metabolic transformation of cancerous cells is still debated [61] and a more detailed analysis of molecular events in the process of tumorigenesis is required to assign characteristic role to mitochondrial mutations.
Mitochondrial DNA lacks introns and therefore all mutations that occur are in the coding region and therefore of biological consequence [46,99]. MtDNA mutations can be either pathogenic, neutral or beneficial. In the germ line, mtDNA variants are in heteroplasmy, and when they reach ‘critical threshold’ they affect the cellular phenotype. Within mature cells somatic mtDNA mutations accumulate over time leading to gradual loss of cellular function [20,31]. Again after crossing a critical, yet unknown, point this activates the mtPTP apoptotic cell death. As a result, the accumulated mtDNA mutations behaves like signal of the biological clock [20,21,23,73]. The interaction between the accumulated somatic mtDNA mutations and the partially inherited mitochondrial defects synergistically increases the effect of defect, and, which accounts for the late-onset and progressive course of mitochondria-associated diseases [97-99]. Selection of an beneficial ntDNA mutation increases its frequency and copies in the population. As recombination does not occur inmitochondria, all sequence variants belonging to one particular mutation gets enriched, producing a phenomenon known as ‘Hitchhiking’ [80-82]. The linked variants on an individual mtDNA molecule gets collected and form mtDNA haplotype. Over time descendants of an original mutant mtDNA assume more variations thereby generate a group of related haplotypes, called haplogroup. Haplogroups form region-specific branches of the mtDNA tree. It can be said that Mitochondrial DNA mutations arise in oral cancer in either of two ways, first in the female germ line (oncogenic germline mutations) or secondly, in the mtDNAs of the tissues (tumor-specific somatic mutations) [27,28,100]. Thus it may be proposed that mitochondrial mutations may confer selective advantage, which is again very debatable and leads to crucial analysis of Warburg’s hypothesis and its subsequent effects [73,82,99-101].
It may be possible that there are some intracellular mechanisms which may diminish mtDNA mutations effects on phenotype and physiology. However, until today, they are merely speculative. The issue of recombination of mtDNA molecules between two mitochondriah has been studied [79,102-104] where isolated two types of respiration-deficient cell lines having pathogenic mtDNA mutations were made hybrids through their fusion with normal cells. This restored normal morphology and respiratory enzyme activity of hybrids, pointing to successful exchange of genetic mitochondrial material. The authors proposed that such a mechanism may be the possible reason of preservation of intracellular progressive accumulated mtDNA lesions with age. But, such a phenomenon has not beenreported in mammalian cells. The most likely explanation for this appears that due to maternal inheritance mtDNA sequence of individuals tends to be relatively uniform. The question is, firstly, whether this process exists in mammals in normal course of action or is an incidental phenomenon and, secondly, how relevant it is in the context of oral cancer where it may serve to provide a protective shield so that normal mitochondrial function occurs even in the presence of mtDNA mutations.
The opinion that the mitochondrial mutations have no relevance in causation of oral cancer is controversial. Firstly, the argument does not find support in the numerous reported associations between tumour mutations and population polymorphisms. Secondly, our knowledge of the functionality and biology of the mtDNA and mRNA is still limited. It is possible that nonsynonymous nucleotide changes in the mRNA might alter some yet unknown and unidentified functions of the mRNA. It is accepted that some of the ‘tumor-specific’ variants reported are false positive and overzealous sequencing reports, but the numerous associations reported across globe and population cannot be completely overlooked and claimed misleading and product of statistical error. The biological relevance of the associations between ‘tumor-specific somatic mutations’ and population variants is supported by a review of present synonymous mutations. The most compelling evidence is a population variant seen for the nt 13708 G to A missesnse mutation in ND5 (A458T) reported in a breast cancer tumor [105]. Such compelling evidence is still lacking in the case of oral cancer. However, the list of reported mtDNA mutations (Table 1) in oral cancer is too large to be ignored. Then, if they are involved, the questions are:
(a) Is there a common mechanism of the reported mitochondrial DNA mutations in causation of oral cancer given the heterogeneity in the tissues of origin?
(b) Which mtDNA mutations really have population and individual significance?
(c) Which mutations are initiators and which are propagators?
With regard to these questions, although it is accepted that all mutations in mitochondria may have different paths of action and effects, yet the common endpoint of all must be suppression of apoptosis and increased stimulus for growth. Investigations in this respect are to be carried out in order to single out relevant mutations and formulate a common pathway at which therapeutic interventions can be applied.
The use of mitochondrial DNA mutations/alterations as bio-marker is rapidly expanding in metabolic disorders, human phylogeny, and human forensic sciences. Broadly, biomarkers are groupedas biomarkers of progression, susceptibility, and/or the treatment responses. As mitochondria DNA is highlyprone to oxidative insult, it can be used as early biomarker of exposure and progression. Literature shows that, firstly, the majority of mitochondrial are base transitions from T to C and G to A and secondly, the D-loop region seems to be the most frequent site of somatic mutations across most tumor types. Besides, preneoplastic lesions have shown nucleotide variations suggesting their presence early in process and their role in tumor progression.
Mutant mtDNA in tumor cells, being small in size and more in number [23,24,49,106], is readily detectable in urine, blood and saliva samples of different cancer patients [26,27,43]. Despite this no single site of mutation or type of mutation has been found common across the wide spectrum of cancer patients. Available sequencing technologies like microarray-based, next generation, ion based and biochip based technology provide a reliable and rapid method to detect all mutations of the entire mitochondrial genome. Recently, sequencing of the entire mitochondrial genome have been done with different sequencing technologies but characterization of the multiple deletions associated with tumors is still required to detect the mutation load on an individual basis.. Mitochondrial genome has mutations throughout in recurrent fashion across both primary tumor tissues and corresponding body fluids. Thus, mtDNA mutation analysis may provide a molecular tool for the early detection and prognosis of cancer. Relatively simple diagnostic tests for detecting mtDNA mutations based on latest technologies have exciting predictive potential for cancer detection and prognosis. Dysfunctioning mitochondria can be tested through Warburg effect which has found recent medical application in form positron emission tomography (PET). Here high aerobic glycolysis rate in malignant tumors is visualized by uptake of 2-18F-2-deoxyglucose (FDG) (a radioactive modified hexokinase substrate) [91]. This feature is relevant clinically to diagnose and monitor treatment responses of cancers with drugs.
Boon for oral cancer related diagnostics is their easy anatomical access where samples from developing premalignant and malignant lesions can be easily taken to determine the levels of DNA adducts and oral cancer progression risk. Measuring free radical levels in a cell can be a useful biomarker for mtDNA damage [47] and mitochondrial functioning. Despite a strong association between free radical levels and mtDNA deletions, it is difficult to say whether mitochondrial mutations or deranged metabolic conditions are predecessor. Moreover, accuracy of techniques for measuring free radical is variable. Then such association studies data are correlative therefore they must be presented only after due verification and consensus [43,101].
Studies have reported differences in molecular compositions of the mitochondrial inner membrane between normal and cancer cells. Elevated cholesterol levels, variable total and individual phospholipids levels have been reported in inner mitochondrial membrane of cancerous cells [107]. Similarly, number of differences has been reported in the appearance and/or relative abundance of several proteins as well between cancerous and control cells. Gene expression profiles between normal and cancer cells have been reported to be different. Prominent genes include proteins like Bcl-2 and Bcl-XL, the peripheral benzodiazepin receptor (PBR), the PBR-associated protein Prax-1, mitochondrial creatine kinase [108,109] and BAX, a proapoptotic, inner mitochondrial membrane protein [110,111]. Defective protein expression here is due to defective mitochondrial DNA and therefore this can be said to have functional relevance. Therefore proteomic based biomarker based mitochondrial functioning can be a useful aid for diagnosis.
Mitochondrial mutations that occurred in early stage of oral cancer could confer a survival or proliferative advantage to the malignant clone. But on the other side there are irrespective of the genomic region involved, many mtDNA mutations are identical to adaptive mutations found in the population [20,83,112]. It has been reported that mitochondrial DNA content increases progression of dysplastic features with from mild to severe [71]. This feature along with mtDNA mutations can be utilized to identify early stages of oral cancer, assessing risk from carcinogen, and in establishing clonality of separate lesions. In one study involving 190 tumour-specific somatic mtDNA mutations including oral cancer, 72% were mtDNA sequence variants found in the general population, which included 52% of the tumour somatic mRNA missense mutations, 83% of the tRNA mutations, 38% of the rRNA mutations, and 85% of the control region mutations [23,113]. It is possible that some of the associations are the result of sequencing errors. Mitochondrial DNA mutations in tumours may be characterized as: (1) severe mutations which can inhibit OXPHOS, increase ROS production and promote tumour cell proliferation and (2) milder mutations that confer selective advantage to tumours cells for adapting to new environments. Therefore, this parallel phylogeny between normal adaptation and malignancy again implies a functional significance to these alterations that provides a survival advantage. Moreover, analysis of several tumour-specific somatic missense mutations with population counterparts appears to provide legitimacy to such associations. However, there are still questions to be answered. Firstly, in case of disappearance of a mutation would others follow? Secondly, what is exact demarcation of effects between each type of mutation? Thirdly, which type of mutation has a greater role in tumour initiation and progression [46,114].
Differences in mtDNA structure and function between normal and cancer cells offer the potential for use as targets for novel and site-specific anti-cancer agents [115]. In one study, delocalized lipophilic cations (DLCs) were used because they accumulated selectively in carcinoma cells due to increased mitochondrial membrane potential. These compounds were partially efficacious in killing carcinoma cell both in vitro and in vivo [116] when used in photochemotherapy (PCT), a treatment modality in which selectively taken up or retained such photoreactive drug or photosensitizer inside malignant cells is light activated [117,118]. It has been tried for treatment for cancers of many organs whose tissue is accessible to transmitted light directly through it natural or mechanically via fiber optic endoscopes. Another treatment modality uses mitochondrial protein-import machinery for transferring such molecules inside mitochondria. Direct green fluorescent protein delivery inside mitochondria for its live staining is an pertinent example of the case [119,120]. Short peptides which contain “homing” motif targeting specific cell types and a pro-apoptotic sequence become toxic when they are internalized by disruptive mitochondrial membrane [121]. Another novel chemotherapy strategy employs membrane permeabilization and consequent apoptosis by targeting specific mitochondrial membrane proteins [118,122]. Researchers are also focussing on developing mitochondriotropic drug and DNA delivery system through conventional liposomes which become mitochondria-specific due to attachment of known mitochondriotropic residues. Such mitochondria-specific liposomes can specifically deliver drugs or mtDNA and destroy dysfunctional mitochondria or replenish mitochondria with healthy copies of the genome [123].
Mitochondria plays a key role in tumorigenesis as mitochondrial dysfunction either becoming the driving cause of tumorigenesis or as a ‘second hit’ in the process of cancer metabolic transformation [46, 114]. In both the cases, however, metabolic reprogramming increases the cancer cells’ survival and proliferation advantage by increasing ATP production in an oxygen independent manner and providing building blocks for macromolecule biosynthesis. Regardless of the cause of metabolic shift, cancer cells become dependent on distinctive metabolic pathways and/or isoenzymes. Identifying and targeting these changes would provide the platform of cancer therapeutics.
The dynamic nature of mitochondria makes it an attractive molecular marker of cancer. MtDNA contains only 16 kb, which can easily be deciphered using high-throughput methods. An in-depth analysis is needed for delineating the functional significance of specific mtDNA alterations in oral carcinogenesis. It is possible that many mutations change the apoptotic and energy homeostasis in the cell, causative and promoting agent in carcinogenesis. Moreover, some of them may also alter the autophagic mechanisms of the cell. Proteomic studies for identifying the biochemical consequences of mtDNA mutations, is difficult because controlling gene expression in vivo is still intriguing. The fact that mtDNA mutations occur in significant frequency in cancer is irrefutable. The obvious next steps include cataloguing the location and type of mutations in different pathologic stages of oral cancer, recording resultant functional derangements, and realizing the therapeutic tools to target mitochondria.
Not applicable
Not applicable
All data generated or analysed during this study are included in this published article [and its supplementary information files].
The authors declare that they have no competing interests
Authors are thankful to Indian Council for Medical Research &Dept of Health Research for funding.
Rahul Pandey conceptualized and wrote the manuscript, Divya Mehrotra contributed to writing in manuscript and supervised the work, Carlo Catapano Rajiv Sarin and Devendra Parmar reviewed the manuscript and suggested modifications.
- Greenlee RT, Hill-Harmon MB, Murray T, Thun M (2001) Cancer statistics, 2001. CA Cancer J Clin 51: 15-36. [Crossref]
- Stewart BW, Kleihues P (2003) World Cancer Report. WHO International Agency for Research on Cancer, Lyon. pp: 1-351.
- AxellT, Pindbor JJ, SmithCJ, van der Waal I. Oral white lesions with special reference to precancerous and tobacco- related lesions: conclusions of an international symposium held in Uppsala, Sweden, May 18-21, 1994. International collaborative group on oral white lesions. J. Oral. Pathol. Med 25: 49-54. [Crossref]
- Pindborg JJ, Reichart PA, Smith CJ, van der Waal I (1997) Histological typing of cancer and precancer of the oral mucosa, 2 ed Springer, Hiedelberg.
- Shikhani AH, Matanoski GM, Jones MM, Kashima HK, Johns ME (1986) Multiple primary malignancies in head and neck cancer. Arch Otolaryngol Head Neck Surg 112: 1172-1179. [Crossref]
- Silverman S Jr, Gorsky M, Kaugars GE (1996) Leukoplakia, dysplasia, and malignant transformation. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 82: 117. [Crossref]
- Silverman S Jr, Gorsky M, Lozada F (1984) Oral leukoplakia and malignant transformation. A follow-up study of 257 patients. Cancer 53: 563-568. [Crossref]
- Cooper JS, Pajak TF, Rubin P, Tupchong L, Brady LW, et al. (1989) Second Malignancies in patients who have head and neck cancer: incidence, effect on survival and implications based on the RTOG experience. Int J Radiat Oncol Biol Phys 17: 449-456.
- Day GL, Blot WJ (1992) Second primary tumors in patients with oral cancer. Cancer 70: 14-19. [Crossref]
- Lippman SM, Hong WK (1989) Second malignant tumors in head and neck squamous cell carcinoma: the overshadowing threat for patients with early-stage disease. Int J Radiat Oncol Biol Phys 17: 691-694.
- Lumerman H, Freedman P, Kerpel S (1997) Oral epithelial dysplasia and the development of invasive squamous cell carcinoma. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod 79: 321-29. [Crossref]
- Koch WM, Lango M, Sewell D, Zahurak M, Sidransky D (1999) Head and neck cancer in nonsmokers: a distinct clinical and molecular entity. Laryngoscope 109: 1544-1551. [Crossref]
- Wey PD, Lotz MJ, Triedman LJ (1987) Oral cancer in women nonusers of tobacco and alcohol. Cancer 60: 1644-1650. [Crossref]
- Feigelson HS, Ross RK, Yu MC, Coetzee GA, Reichardt JK, et al. (1996) Genetic susceptibility to cancer from exogenous and endogenous exposures. J Cell Biochem Suppl 25: 15-22. [Crossref]
- Fouret P, Monceaux G, Temam S, Lacourreye L, St Guily JL (1997) Human papillomavirus in head and neck squamous cell carcinomas in nonsmokers. Arch Otolaryngol Head Neck Surg 123: 513-516. [Crossref]
- Moll UM, Schramm LM (1998) p53--an acrobat in tumorigenesis. Critical Reviews in Oral Biology & Medicine 9: 23-37.
- Capuano F, Guerrieri F, Papa S (1997) Oxidative phosphorylation enzymes in normal and neoplastic cell growth. J Bioenerg Biomembr 29: 379-384. [Crossref]
- Lightowlers RN, Chinnery PF, Turnbull DM, Howell N (1997) Mammalian mitochondrial genetics, heredity, heteroplasmy and disease. Trends Genet 13: 450-455.
- Park CB, Larsson NG (1997) Mitochondrial DNA mutations in disease and aging. Proc Soc Exp Biol Med 216: 283-290.
- Greaves LC, Reeve AK, Taylor RW, Turnbull DM (2012) Mitochondrial DNA and disease. J Pathol 226: 274-286. [Crossref]
- Carew JS, Huang P (2002) Mitochondrial defects in cancer. Mol Cancer 1: 9. [Crossref]
- Singh KK, Kulawiec M, Still I, Desouki MM, Geradts J, et al. (2005) Inter-genomic cross talk between mitochondria and the nucleus plays an important role in tumorigenesis. Gene 354: 140-146. [Crossref]
- Brandon M, Baldi P, Wallace DC (2006) Mitochondrial mutations in cancer. Oncogene 25: 4647-4662.
- Penta JS, Johnson FM, Wachsman JT, Copeland WC (2001) Mitochondrial DNA in human malignancy. Mutat Res 488: 119-133. [Crossref]
- Chatterjee A, Mambo E, Sidransky D (2006) Mitochondrial DNA mutations in human cancer. Oncogene 25: 4663-4674. [Crossref]
- Copeland WC, Wachsman JT, Johnson FM, Penta JS (2002) Mitochondrial DNA alterations in cancer. Cancer Invest 20: 557-569. [Crossref]
- Czarnecka AM, Kukwa W, Krawczyk T, Scinska A, Kukwa A, et al. (2010) Mitochondrial DNA mutations in cancer--from bench to bedside. Front Biosci (Landmark Ed) 15: 437-460. [Crossref]
- Seoane M, Mosquera-Miguel A, Gonzalez T, Fraga M, Salas A, et al. (2011) The mitochondrial genome is a "genetic sanctuary" during the oncogenic process. PLoS One 6: e23327. [Crossref]
- Johnson LV, Walsh ML, Chen LB (1980) Localization of mitochondria in living cells with rhodamine 123. Proc Natl Acad Sci U S A 77: 990-994. [Crossref]
- Anderson S, Bankier AT, Barrell BG, de Bruijn MHL, CoulsonAR, et al. (1981) Sequence and organisation of the human mitochondrial genome. Nature 290: 457-465.
- Shoffner JM, Wallace DC (1992) Mitochondrial genetics: principles and practice. Am J Hum Genet 51: 1179-1186.
- Mercer TR, Neph S, Dinger ME, Crawford J, Smith MS, et al. (2011) The human mitochondrial transcriptome. Cell 146: 645-658.
- Attardi G (1985) Animal mitochondrial DNA: an extreme example of genetic economy. Int. Rev. Cytol 93: 93-145.
- Pesole G, Gissi C, DeChirico A, Saccone C (1999) Nucleotide substitution rate of mammalian mitochondrial genomes. J Mol Evol 48: 427-434.
- Beckman KB, Ames BN (1999) Endogenous oxidative damage of mt DNA. Mutat Res 424: 51-58. [Crossref]
- Bohr VA, Dianov GL (1999) Oxidative DNA damage processing in nuclear and mitochondrial DNA. Biochimie 81: 155-160. [Crossref]
- Clayton DA (2000) Transcription and replication of mitochondrial DNA. Hum Reprod 15 Suppl 2: 11-17. [Crossref]
- Croteau DL, Bohr VA (1997) Repair of oxidative damage to nuclear and mitochondrial DNA in mammalian cells. J Biol Chem 272: 25409-25412. [Crossref]
- Ozawa T (1995) Mechanism of somatic mitochondrial DNA mutations associated with age and diseases. Biochim Biophys Acta 1271: 177-189. [Crossref]
- Staniek K, Gille L, Kozlov AV, Nohl H (2002) Mitochondrial superoxide radical formation is controlled by electron bifurcation to the high and low potential pathways. Free Radic Res 36: 381-387. [Crossref]
- Saybasili H, Yuksel M, Haklar G, Yalcin AS (2001) Effect of mitochondrial electron transport inhibitors on superoxide radical generation in rat hippocampal and striatal slices. Antioxid Redox Signal 3: 1099-1014.
- Shokolenko I, Venediktova N, Bochkareva A, Wilson GL, Alexeyev MF (2009) Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res 37: 2539-2548. [Crossref]
- Yakes MF, Van Houten B (1997) Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci U S A 94: 514-519.
- Rizzuto R, Simpson AW, Brini M, Pozzan T (1992) Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature 358: 325-327.
- Wilson MR (1998) Apoptosis: unmasking the executioner. Cell Death Differ 5: 646-652. [Crossref]
- Gogvadze V, Orrenius S, Zhivotovsky B (2008) Mitochondria in cancer cells: what is so special about them? Trends Cell Biol 18: 165-173. [Crossref]
- Liu CY, Lee CF, Hong CH, Wei YH (2004) Mitochondrial DNA mutation and depletion increase the susceptibility of human cells to apoptosis. Ann N Y Acad Sci 1011: 133-145. [Crossref]
- Prior SL, Griffiths AP, Baxter JM, Baxter PW, Hodder SC, et al. (2006) Mitochondrial DNA mutations in oral squamous cell carcinoma. Carcinogenesis 27: 945-950. [Crossref]
- Fliss MS, Usadel H, Caballero OL, Wu L, Buta MR, et al. (2000) Facile detection of mitochondrial DNA mutations in tumors and bodily fluids. Science 287: 2017-2019. [Crossref]
- Saranath D, Bhoite LT, Deo MG (1993) Molecular lesions in human oral cancer: the Indian scene. Eur J Cancer B Oral Oncol 29B: 107-112. [Crossref]
- Poetsch M, Petersmann A, Lignitz E, Kleist B (2004) Relationship between mitochondrial DNA instability, mitochondrial DNA large deletions, and nuclear microsatellite instability in head and neck squamous cell carcinomas. Diagnostic Molecular Pathology 13: 26-32.
- Lee HC, Yin PH, Yu TN, Chang YD, Hsu WC, et al. (2001) Accumulation of mitochondrial DNA deletions in human oral tissues -- effects of betel quid chewing and oral cancer. Mutat Res 493: 67-74. [Crossref]
- Gasparre G, Porcelli AM, Bonora E, Pennisi LF, Toller M, et al. (2007) Disruptive mitochondrial DNA mutations in complex I subunits are markers of oncocytic phenotype in thyroid tumors. Proc Natl Acad Sci U S A 104: 9001-9006. [Crossref]
- Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, et al. (2008) ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 320: 661-664. [Crossref]
- Kroemer G (1997) The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat Med 3: 614-620. [Crossref]
- Lee HC, Wei YH (2007) Oxidative stress, mitochondrial DNA mutation, and apoptosis in aging. Exp Biol Med (Maywood) 232: 592-606. [Crossref]
- Reed JC (1997) Double identity for proteins of the Bcl-2 family. Nature 387: 773-776. [Crossref]
- Reed JC (1994) Bcl-2 and the regulation of programmed cell death. J Cell Biol 124: 1-6. [Crossref]
- Salas A, Yao YG, Macaulay V, Vega A, Carracedo A, et al. (2005) A critical reassessment of the role of mitochondria in tumorigenesis. PLoS Med 2: e296.
- Wallace DC (2005) A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet 39: 359-407.
- Shay JW, Werbin H (1987) Are mitochondrial DNA mutations involved in the carcinogenic process? Mutat Res 186: 149-160. [Crossref]
- Dasgupta S, Koch R, Westra WH, Califano JA, Ha PK, et al. (2010) Mitochondrial DNA mutation in normal margins and tumors of recurrent head and neck squamous cell carcinoma patients. Cancer Prev Res (Phila) 3: 1205-1211.
- Tan DJ, Chang J, Chen WL, Agress LJ, Yeh KT, et al. Novel heteroplasmic frameshift and missense somatic mitochondrial DNA mutations in oral cancer of betel quid chewers. Genes Chromosomes Cancer 37: 186-194.
- Shieh DB, Chou WP, Wei YH, Wong TY, Jin YT (2004) Mitochondrial DNA 4,977-bp deletion in paired oral cancer and precancerous lesions revealed by laser microdissection and real-time quantitative PCR. Ann N Y Acad Sci 1011: 154-167.
- Mondal R, Ghosh SK (2013) Accumulation of mutations over the complete mitochondrial genome in tobacco-related oral cancer from northeast India. Mitochondrial DNA 24: 432-439.
- Palodhi A, Ghosh S, Biswas NK, Basu A, Majumder PP, et al. (2019) Profiling of genomic alterations of mitochondrial DNA in gingivobuccal oral squamous cell carcinoma: Implications for disease progress. Mitochondrion 46: 361-369.
- Shu HY, Li HC, Xie WQ, Ni B, Zhou HY (2019) Mitochondrial DNA variations in tongue squamous cell carcinoma. Biomed Rep 10: 23-28. [Crossref]
- Dasgupta S, Koch R, Westra WH, Califano JA, Ha PK, et al. (2010) Mitochondrial DNA mutation in normal margins and tumors of recurrent head and neck squamous cell carcinoma patients. Cancer Prev Res (Phila) 3: 1205-1211. [Crossref]
- Yuan RT, Sun Y, Bu LX, Jia MY (2015) Gene mutations in the D-loop region of mitochondrial DNA in oral squamous cell carcinoma. Mol Med Rep 11: 4496-500.
- Kim MM, Clinger JD, Masayesva BG, Ha PK, Zahurak ML, et al. (2004) Mitochondrial DNA quantity increases with histopathologic grade in premalignant and malignant head and neck lesions. Clin Cancer Res 10: 8512-8515.
- Ha PK, Tong BC, Westra WH, Sanchez-Cespedes M, Parrella P, et al. (2002) Mitochondrial C-tract alteration in premalignant lesions of the head and neck: a marker for progression and clonal proliferation. Clin. Cancer Res 8: 2260-2265.
- Lai CH, Huang SF, Liao CT, Chen IH, Wang HM, et al. (2013) Clinical significance in oral cavity squamous cell carcinoma of pathogenic somatic mitochondrial mutations. PLoS One 8: e65578.
- Lin JC, Wang CC, Jiang RS, Wang WY, Liu SA (2015) Impact of somatic mutations in the D-loop of mitochondrial DNA on the survival of oral squamous cell carcinoma patients. PLoS One 10: e0124322. [Crossref]
- Liu SA, Jiang RS, Wang WY, Lin JC, et al. (2015) Somatic mutations in the D-loop of mitochondrial DNA in head and neck squamous cell carcinoma. Head Neck 37: 878-883. [Crossref]
- Liu SA, Jiang RS, Chen FJ, Wang WY, Lin JC (2012) Somatic mutations in the D-loop of mitochondrial DNA in oral squamous cell carcinoma. Eur Arch Otorhinolaryngol 269: 1665-1670. [Crossref]
- Ono T, Isobe K, Nakada K, Hayashi JI (2001) Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat Genet 28: 272-275. [Crossref]
- Tishkoff SA, Verrelli BC (2003) Patterns of human genetic diversity: implications for human evolutionary history and disease. Annu Rev Genomics Hum Genet 4: 293-340.
- Thyagarajan B, Padua RA, Campbell C (1996) Mammalian mitochondria possess homologous DNA recombination activity. J Biol Chem 271: 27536-27543.
- Mishmar D, Ruiz-Pesini E, Golik P, Macaulay V, Clark AG, et al. (2003) Natural selection shaped regional mtDNA variation in humans. Proc Natl Acad Sci U S A 100: 171-176.
- Kloss-Brandstätter A, Weissensteiner H, Erhart G, Schäfer G, Forer L, et al. (2015) Validation of Next-Generation sequencing of entire mitochondrial genomes and the diversity of mitochondrial DNA mutations in oral squamous cell carcinoma. PLoS One 10: e0135643.
- Zhou S, Kachhap S, Sun W, Wu G, Chuang A, et al. (2007) Frequency and phenotypic implications of mitochondrial DNA mutations in human squamous cell cancers of the head and neck. Proc Natl Acad Sci U S A 104: 7540-7545. [Crossref]
- Elliott HR, Samuels DC, Eden JA, Relton CL, Chinnery PF (2008) Pathogenic mitochondrial DNA mutations are common in the general population. Am J Hum Genet 83: 254-260.
- Pai CY, Hsieh LL, Lee TC, Yang SB, Linville J, et al. (2006) Mitochondrial DNA sequence alterations observed between blood and buccal cells within the same individuals having betel quid (BQ)-chewing habit. Forensic Sci Int 156: 124-130. [Crossref]
- Pedersen PL (1978) Tumor mitochondria and the bioenergetics of cancer cells. Prog Exp Tumor Res 22: 190-274. [Crossref]
- Pedersen PL, Morris HP (1974) Uncoupler-stimulated adenosine triphosphatase activity. Deficiency in intact mitochondria from Morris hepatomas and ascites tumor cells. J Biol Chem 249: 3327-3334.
- Plas DR, Thompson CB (2005) Akt-dependent transformation: there is more to growth than just surviving. Oncogene 24: 7435-7442.
- Johnson LV, Walsh ML, Bockus BJ, Chen LB (1981) Monitoring of relative mitochondrial membrane potential in living cells by fluorescence microscopy. J Cell Biol 88: 526-535.
- Moreno-Sánchez R, Rodríguez-Enríquez S, Marín-Hernández A, Saavedra E (2007) Energy metabolism in tumor cells. FEBS J 274: 1393-1418. [Crossref]
- Wang GL, Semenza GL (1993) Characterization of hypoxia-inducible factor 1 and regulation of DNA binding activity by hypoxia. J Biol Chem 268: 21513-21518. [Crossref]
- Busk M, Horsman MR, Kristjansen PE, van der Kogel AJ, Bussink J, et al. (2008) Aerobic glycolysis in cancers: implications for the usability of oxygen-responsive genes and fluorodeoxyglucose-PET as markers of tissue hypoxia. Int J Cancer 122: 2726-2734.
- Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, et al. (2004) Akt stimulates aerobic glycolysis in cancer cells. Cancer Res 64: 3892-3899.
- Cuezva JM, Krajewska M, de Heredia ML, Krajewski S, Santamaría G, et al. (2002) The bioenergetic signature of cancer: a marker of tumor progression. Cancer Res 62: 6674-6681. [Crossref]
- Capuano F, Varone D, D'Eri N, Russo E, Tommasi S, et al. (1996) Oxidative phosphorylation and F(0)F(1) ATP synthase activity of human hepatocellular carcinoma. Biochem. Mol. Biol. Int. 38: 1013-1022.
- Cuezva JM, Ostronoff LK, Ricart J, López de Heredia M, Di Liegro CM, et al. (1997) Mitochondrial biogenesis in the liver during development and oncogenesis. J Bioenerg Biomembr 29: 365-377. [Crossref]
- Hileman EO, Achanta G, Huang P (2001) Superoxide dismutase: an emerging target for cancer therapeutics. Expert Opin Ther Targets 5: 697-710.
- Park SY, Chang I, Kim JY, Kang SW, Park SH, et al. (2004) Resistance of mitochondrial DNA-depleted cells against cell death: role of mitochondrial superoxide dismutase. J Biol Chem 279: 7512-7520.
- Bohr VA, Anson RM (1995) DNA damage, mutation and fine structure DNA repair in aging. Mutat Res 338: 25-34. [Crossref]
- Williams RS (2002) Another surprise from the mitochondrial genome. N Engl J Med 347: 609-612. [Crossref]
- Mithani SK, Taube JM, Zhou S, Smith IM, Koch WM, et al. (2007) Mitochondrial mutations are a late event in the progression of head and neck squamous cell cancer. Clin Cancer Res 13: 4331-4335. [Crossref]
- Pietka G, Kukwa W, Bartnik E, SciÅska A, Czarnecka AM (2008) [Mitochondrial DNA mutations in the pathogenesis in the head and neck squamous cell carcinoma]. Otolaryngol Pol 62: 158-164. [Crossref]
- Dujon B, Slonimski PP, Weill L (1974) Mitochondrial genetics IX: a model for recombination and segregation of mitochondrial genomes in Saccharomyces cerevisiae. Genetics 78: 415-437.
- Lunt DH, Hyman BC (1997) Animal mitochondrial DNA recombination. Nature 387: 247. [Crossref]
- Belliard G, Vadel F, Pelletier G (1979) Mitochondrial recombination in cytoplasmic hybrids of Nicotina tabacum by protoplast fusion. Nature 281: 401-403.
- Parrella P, Xiao Y, Fliss M, Sanchez-Cespedes M, Mazzarelli P, et al. (2001) Detection of mitochondrial DNA mutations in primary breast cancer and fine-needle aspirates. Cancer Res 61: 7623-7626.
- Zeviani M, Antozzi C (1997) Mitochondrial disorders. Mol Hum Reprod 3: 133-148. [Crossref]
- Garcea R, Canuto RA, Guatero B, Biocca M, Feo F (1980) Phospholipid composition of inner and outer mitochondrial membranes isolated from Yoshida hepatoma AH-130. Cancer Lett 11: 133-139.
- O'Gorman E, Beutner G, Dolder M, Koretsky AP, Brdiczka D, et al. (1997) The role of creatine kinase in inhibition of mitochondrial permeability transition. FEBS Lett 414: 253-257. [Crossref]
- Galiègue S, Jbilo O, Combes T, Bribes E, Carayon P, et al. (1999) Cloning and characterization of PRAX-1. A new protein that specifically interacts with the peripheral benzodiazepine receptor. J Biol Chem 274: 2938-2952.
- Brimmell M, Mendiola R, Mangion J, Packham G (1998) BAX frameshift mutations in cell lines derived from human haemopoietic malignancies are associated with resistance to apoptosis and microsatellite instability. Oncogene 16: 1803-1812. [Crossref]
- Rampino N, Yamamoto H, Ionov Y, Li Y, Sawai H, et al. (1997) Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science 275: 967-969. [Crossref]
- Zhang C, Lee A, Liu VW, Pepe S, Rosenfeldt F, et al. (1999) Mitochondrial DNA deletions in human cardiac tissue show a gross mosaic distribution. Biochem Biophys Res Commun 254: 152-157. [Crossref]
- Hertweck KL, Dasgupta S (2017) The Landscape of mtDNA Modifications in Cancer: A Tale of Two Cities. Front Oncol 7: 262. [Crossref]
- Kirches E (2017) MtDNA As a Cancer Marker: A Finally Closed Chapter? Curr Genomics 18: 255-267. [Crossref]
- Datta S, Chattopadhyay E, Ray JG, Majumder M, Roy PD, et al. (2015) D-loop somatic mutations and ∼5 kb "common" deletion in mitochondrial DNA: important molecular markers to distinguish oral precancer and cancer. Tumour Biol 36: 3025-3033. [Crossref]
- Modica-Napolitano JS, Nalbandian R, Kidd ME, Nalbandian A, Nguyen CC (2003) The selective in vitro cytotoxicity of carcinoma cells by AZT is enhanced by concurrent treatment with delocalized lipophilic cations. Cancer Lett 198: 59-68. [Crossref]
- Modica-Napolitano JS, Joyal JL, Ara G, Oseroff AR, Aprille JR (1990) Mitochondrial toxicity of cationic photosensitizers for photochemotherapy. Cancer Res 50: 7876-7881. [Crossref]
- Modica-Napolitano JS, Singh KK (2002) Mitochondria as targets for detection and treatment of cancer. Expert Rev Mol Med 4: 1-19. [Crossref]
- Modica-Napolitano JS, Aprille JR (1987) Basis for the selective cytotoxicity of rhodamine 123. Cancer Res 47: 4361-4365. [Crossref]
- Modica-Napolitano JS, Koya K, Weisberg E, Brunelli BT, Li Y, et al. (1996) Selective damage to carcinoma mitochondria by the rhodacyanine MKT-077. Cancer Res 56: 544-550. [Crossref]
- Ellerby HM, Arap W, Ellerby LM, Kain R, Andrusiak R, et al. (1999) Anti-cancer activity of targeted pro-apoptotic peptides. Nat Med 5: 1032-1038. [Crossref]
- Costantini P, Jacotot E, Decaudin D, Kroemer G (2000) Mitochondrion as a novel target of anticancer chemotherapy. J Natl Cancer Inst 92: 1042-1053. [Crossref]
- Fantin VR, Leder P (2006) Mitochondriotoxic compounds for cancer therapy. Oncogene 25: 4787-4797.
- Boddapati SV, Tongcharoensirikul P, Hanson RN, D'Souza GG, Torchilin VP, et al. (2005) Mitochondriotropic liposomes. J Liposome Res 15: 49-58. [Crossref]