Abstract
We report a case of microcephalic osteodysplastic primordial dwarfism syndrome with clinical profile (microcephaly, growth retardation, dysmorphic feature ,microdontia, cubitus varus and heel valgus) and genetic mutation on PCNT in whole exome sequencing.
Introduction
Microcephalic osteodysplastic primordial dwarfism type II is a rare recessively inherited syndrome characterized by severe intrauterine growth retardation (IUGR), severe postnatal growth retardation, short stature, skeletal abnormalities and an unusually small head size and blood vessel abnormalities; A number of affected individuals have developed dilation of the CNS arteries variously described as aneurysms and moyamoya disease. These vascular changes can be life threatening, even in early years because of rupture, CNS hemorrhage, and stroke. There is variability between affected individuals even within the same family [1,2].
MOPD II is caused by mutations inPCNT(21q22.3), encoding pericentrin, which anchors a wide range of centrosomal proteins and protein complexes during cell division. Disruption of pericentrin is thought to cause mitotic spindle defects, and impaired cell proliferation and contribute to the development of a wide variety of pathological conditions, including cancer and diabetes [3,4].
Case report
A 5-year-old child was presented to the Department of Pediatric endocrinology in Ramallah Governmental hospital, Palestine, seeking treatment for severe growth retardation since infancy. On detailed history, parents had 3rd degree consanguineous marriage as they are first cousins. Mother was 20 and father was 28 years old at marriage time and have five pregnancies; first and third-ranked pregnancies were aborted spontaneously but the second and fourth produce healthy term boys with a birth weight 3100g, 2600g respectively. Our patient is fifth in order of pregnancies. There was no history of any systemic disease during gestational period, the mother’s pre-natal ultrasound finding at 16 weeks of gestation showed symmetrical intrauterine growth restriction (IUGR) with oligohydramnios.
He was delivered vaginally at 37 weeks gestation with a birth weight of 800 grams, length of 34 cm and head circumference of 31 cm. He was admitted to NICU because of his extreme low birth weight, despite of his weight he didn't need any ventilator support till discharge from the hospital after two months and was discharged home with a weight of 1600 grams then he was followed with endocrine specialist to determine the cause of his growth restriction, Bone age was done and showed delayed bone age, although growth hormone level was within normal range, the patient was started on growth hormone. In the first year of life his response to treatment was well, but then the treatment showed no benefit despite of this the patient was continued on growth hormone till now. On detailed history regarding development, the child attained head holding at six months, sitting without support at year age, walking without support at four years, Say Ma, Ba at 1.5 years and say two world sentences at 3.5 years and his teeth was erupted at 2 years of age with upper molar teeth being the first to erupt. It is worth mentioning that was no family history of similar problem. Clinical examination showed a very thin boy, with incomprehensive language and squeaky voice; abnormally small and dysplastic dentition (Figure 1); a pleasant, sociable personality and small head that looked like triangle (Figure 2). His small forehead contrasted with down slanting eyes, and a large prominent nose. In addition to the dysmorphic face, we also noted cubitus varus deformity in both upper limbs (Figure 3), heel valgus deformity in both lower limbs (Figure 4), and wide base gait, there was no other skeletal deformities, the rest of exam was normal including neurological exam. He weighed 4kg, his height was 60cm and cranial perimeter was 37cm. After ruling out other causes of growth restriction, whole exome sequencing test was recommended to determine genetic mutation may contribute to observed phenotype , the test identified a homozygous 11bp deletion in PNCT gene chr 21:47786870-47786880,c.2981_2991del, resulting in frameshift at R994 ,this mutation is a damaging mutation cause what known as microcephalic osteodysplastic primordial dwarfism type 2 (Figure 5) .

Figure 1. Dysmorpcrhic feature; microcephaly, triangular head

Figure 2. Abnormal teeth (microdontia)

Figure 3. Upper limb deformity; Cubitus varus

Figure 4. Severe lower limb deformity; Heel Valgus
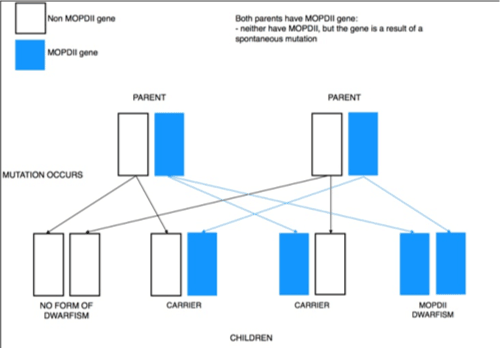
Figure 5. Autosomal recessive inheritance
References
- Rauch A, Thiel CT, Schindler D, Wick U, Crow YJ, et al. (2008) Mutations in the Pericentrin (PCNT) Gene Cause Primordial Dwarfism. Science 319: 816-819. [Crossref]
- Microcephalic osteodysplastic primordial dwarfism type II. Genetics Home Reference. 2011; Available at: http://ghr.nlm.nih.gov/condition/microcephalic-osteodysplastic-primordial-dwarfism-type-ii.
- Willems M, Geneviève D, Borck G, Baumann C, Baujat G, et al. (2010) Molecular analysis of pericentrin gene (PCNT) in a series of 24 Seckel/microcephalic osteodysplastic primordial dwarfism type II (MOPD II) families. J Med Genet 47: 797-802. [Crossref]
- Hall MN, Raff M, Thomas G (2004) Cell Growth: Control of Cell Size. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, pp. 167–192.