Abstract
Our main aim is to make a contribution to the establishment of sources of oxidants as the key factors in understanding the role that oxidants and oxidative stress play in the pathogenesis of neurodegeneration, i.e. in the progression of Alzheimer’s disease (AD). Our original observation pointed out the main difference between the hemoglobin and methemoglobin degradation, did from the heme oxygenation when a hemoglobin results in ferrous (Fe2+) iron, but methemoglobin catabolism produces ferric (Fe3+) iron. The methemoglobin plays a role of the carrier, the donor of cytotoxic and redox-active ferric (Fe3+) iron, and it also acts as the originator of neurodegenerative diseases. The abundant and permanent source of redox-active ferric (Fe3+) iron which, without Ferrous-Ferric inversions, has “in situ” direct impact on endothelial small vessels in the brain accumulates and increases the rate of capillary endothelial cell apoptosis and possibly crosses into brain parenchyma to the astrocytes, glia, neurons, and other neuronal cells. Our understanding of the transport and neuronal accumulation of ferric (Fe3+) iron points to how microvessels are organized into a well-structured neurovascular unit with harmful consequences to the brain. Our previously conducted research found that the neonatal jaundice incidence (p=0.034), heart murmur at a later age (p=0.011) and mild disorders in children and adults such as dyslalia and learning/memory impairments (p=0.002) were significantly higher than in children and adults of control mothers without pregnancy methemoglobinemia.
The consequence are performed as initial brain iron harmful effects from the mother-fetal pregnancy methemoglobinemia complication, and according to our hypothesis in humans could be followed with the neuronal death, the disease aging process, and leading finally to the severe disorders as AD, PD and other neurodegenerative diseases.
Key words
Hemoglobin and Methemoglobin catabolism, brain capillary, ferric iron deposition, blood brain barrier, apoptosis, neurovascular unit, neurodegenerative brain disease.
Introduction
It is still not known why iron levels are abnormally high in some regions of the brain in neurodegenerative disorders and it is also not clear whether iron accumulation in the brain is the initial event that causes neuronal death or is a consequence of the disease process. According to the previous research, we consider that, at least in some neurodegenerative disorders, brain iron misregulation is the initial cause of neuronal death and that this misregulation might be the linked to either genetic or non-genetic factors.
Ferric-iron brain accumulation as a cause of neurodegenerative brain disease: A new insight to understanding the mechanism of iron transport
Our research focused on people who continuously inhale strong oxidants such as NOx (NO andNO2) and it showed that these oxidants can reversibly oxidize oxyhemoglobin (Fe2+) to methemoglobin (Fe3+). Irreversible methemoglobinemia can occur because of the disruption of the oxidant/antioxidant balance, supported by the synergic SO2 metabolites, leading to the degradation of antioxidant thiols [1]. Methemoglobin by itself and heme have prooxidant properties and induce structural and functional changes in the vascular endothelium [2,3]. These changes include cell growth arrest, senescence, morphological alterations and cell apoptosis, and they lead to both vessel trombosis and endothelial cell denudation under the influence of redox-active ferric iron (Fe3+), which is a product of heme-oxygenase, responsible for methemoglobin-heme degradation [4]. In the blood, Nitric oxide and Superoxide form Peroxinitrites (ONOO), that convert oxyhemoglobin into methemoglobin, and the methemoglobin released from heme induces endothelial cytolysis [5]. The toxicity of H2O2 is also dependent upon its reaction with ferrous iron to form hydroxyl radicals by the Fenton reaction. The ferrous iron needed for this reaction is formed by the reduction of cellular ferric iron by superoxide ions [6]. We would like to emphasize the difference between physiological hemoglobin catabolism and pathological methemoglobin catabolism, because their different final products, ferrous and ferric iron, have distinct characteristics. Ferrous iron has the potential for catalyzing and generating highly cytotoxic hydroxyl radicals as the Fenton reactions (ferrous iron + H2O2 ---> ferric iron + OH + OH). Ferric iron is then reduced back to ferrous iron, peroxide radical and proton by the same hydrogen peroxide.
The substantial difference between the intracellular ferric iron originates from Fenton reaction, and ferric iron originates from methemoglobin catabolism influence a level of methemoglobin cellular uptake which increases the methemoglobinemia leading to ferric-iron-induced oxidative stress injury.
We consider that during methemoglobin catabolism, the final ferric iron product is a significantly added source of ferric iron derived from the Fenton reaction and its continuous formation has an impact upon the brain’s neurovascular unit.
According to the suggestions made in the previous research, we consider that, at least in some disorders, brain iron misregulation is the initial cause of neuronal death and that this misregulation might be the result of either genetic or non-genetic factors [7], and is considered to have autoimmune determinants [8]. Our previous research results suggest that methemoglobin plays a particularly important role as the carrier and donor of the cytotoxic and redox-active ferric (Fe3+) iron, and determines how iron is transported intracellularly (Figures 1-4).
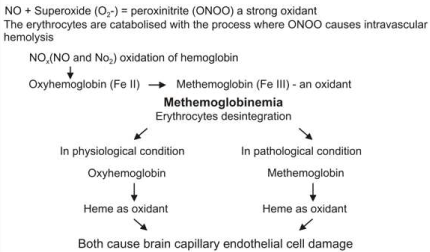
Figure 1. The hemoglobin catabolism

Figure 2. The heme oxygenase

Figure 3. Focal positivity for feric iron binding in neurons showing patchy accumulations in brain of person died of Alzhimer disease in low magnification (x4HPF A and x10 HPF B).
Source: Stifter Sanja, Department of Pathology, University of Rijeka School of Medicine.

Figure 4. Free iron positivity in the cerebral parenchyma of a 77 year old patient with Alzheimer's disease A) Focal distribution of positive glia and neurons; B) accumulation of strong iron-positive perivascular regions. Staining: Perl's Prussian Blue reaction for ferric iron- Magnification 40x.
Source: Stifter Sanja, Department of Pathology, University of Rijeka School of Medicine.
Neuroscience has traditionally focused on the neurons of the central and peripheral nervous systems, and it is now becoming clear that neurons, glia and microvessels are organized into a well-structured neurovascular unit, while recent studies have highlighted the importance of brain endothelial cells in this modular organization [9]. Iron is essential for the normal cell function, but it also generates toxic Reactive Oxygen Species (ROS) that adversely affect vascular endothelium of the blood-brain barrier. The exposure of human neuroblastoma IMR-32 cells to Ferric ammonium citrate (FAC) was investigated as a model of neuronal iron overload and neurodegeneration. The consequences of the exposure of cells to Ferric ammonium citrate showed that the exposure is associated to the increased oxidant cell levels, the activation of redox-sensitive signals, and apoptosis [10]. Leung G, Moody AR has demonstrated that ferric heme is significantly more pro-oxidant than the ferrous heme. In addition, they have shown that this ferric heme has a significantly higher relaxivity than its ferrous counterparts. In tandem, this evidence suggests that the MR imaging–detected T1 (longitudinal relaxation time) high signal intensity within the vessel wall is an endogenous biomarker of an intraplaque environment that is highly pro-oxidant and proatherogenic. MR imaging measures showed a T1 relaxivity that was 10 times higher for ferric than for ferrous forms of hemoglobin. These results support the hypothesis that ferric methemoglobin-generated T1 high signal intensity reflects a pro-oxidant environment that, in the setting of vessel wall disease, might be proatherogenic [11].
This justifies the study of the oxidant effect of methemoglobin catabolic products on vital organs and the CNS, resulting in their dysfunction. The research MRI data showed that extracellular methemoglobin generates significantly more lipid oxidation than the intracellular products; however, methemoglobin in both these environments has similar measures of r1 (longitudinal relaxivity of relaxation rate). Therefore, the T1 high signal intensity, due to methemoglobin, is not solely restricted to an environment that causes lipid oxidation. Thus, the ability of this high signal intensity to reflect the plaque at risk may be diminished. However, it is known that in the absence of any chemical modifications, ferric heme substantially degrades the integrity of the RBC membrane, and the eventual fate of a static RBC is lysis. Thus, the intracellular methemoglobin is destined to rapidly become extracellular, thereby adding to the oxidative drive [12]. The cellular and intercellular iron transport mechanisms in the central nervous system (CNS) are still poorly understood, meanwhile the accumulating evidence suggests that impaired iron metabolism is the initial cause of neurodegeneration [13]. Brar S, et al. concluded that the development of Parkinsonism during the course of AD appears to be associated with the accumulation of iron, which in turn may contribute to the pathogenesis of neurologic decline [14] .
Results
Our results point out the consequence of the toxic environmental oxidants, caused by brain damage with a view to the role of methemoglobin catabolism in pregnancy as the source of ferric (Fe3+) iron form concentrated in various brain regions. Methemoglobin and hemolysis both occur as a result of oxidative stress, but the prevalent difference between them is that methemoglobin is a reversible phenomenon (oxidant–antioxidant balance) whereas hemolysis, which occurs as a result of oxidative stress on the erythrocyte membrane, is an irreversible event. Methemoglobinemia can additionally exacerbate an existing anemia, stimulating hypoxia that may be additionally dangerous. Our prospective study of methemoglobin in pregnancy revealed a significant rise the level of methemoglobin >1.5 g/L (r= 0.72, p < 0.01) in the air-polluted exposure period, which can be explained on the basis of an oxidant–antioxidant imbalance, resulting in methemoglobinemia [15]. Methemoglobinemia and stillbirths recorded throughout exposure period are significantly higher than those recorded in the control period (p = 0.0205) and the frequencies of reproductive loss were significantly lower in the control than in the exposure period (p < 0.05) [16].
As we have found no evidence of the consequences of mother methemoglobinemia on the fetus, the second objective was to direct attention to methemoglobin as an early biomarker of the environmental toxic of oxidative stress effects, which puts pregnancy at risk and may later impair the health of newborns, children and adolescents. The conducted proper research found that neonatal jaundice incidence (p=0.034), heart murmur at a later age (p=0.011) and mild disorders in children and adults such as dyslalia and learning/memory impairments (p=0.002) were significantly higher than in children and adults of control mothers without pregnancy methemoglobinemia [17]. Lavezzi AM, Mohorovic L, et al. recently presented the findings confirmed by pathohistological technique that under the adverse conditions, ferric iron positivity in capillary endothelial cells of the blood-brain barrier in the fetus rises, also resulting in preterm birth, stillbirth or early neonatal death [18]. The application of the Blue Prussian method highlighted the accumulations of blue granulations, indicative for non-heme Fe3+ positive reactions, in the brainstem and cerebellum of 12 (33%) of the 36 victims, while there were none in the control group. In the positive cases, iron deposits were widespread in the brain parenchyma or localized in specific areas showing a variable extent and intensity (Figures 5 and 6)
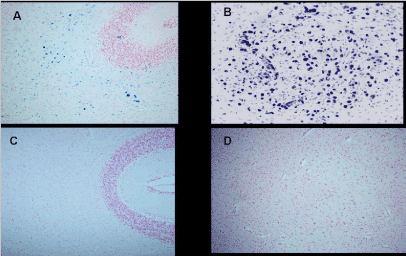
Figure 5. Cerebellum-Ferric iron positivity in: A) subcortical region and B) parenchyma in the dentate nucleus area in a 6 month-old victim of SIDS (case no.9). In C) and D) equivalent ironnegative regions in an age-matched control case. Staining: Perls' Prussian Blue reaction for ferric iron-Magnification A)C): 10x; B)D): 20×.
Source:Lavezzi AM, Mohorovic L Alfonsi G, Coma MF, Matturri L, Brain iron accumulation in unexplained fetal and infant death victims with smoker mothers. The possible involvement of maternal methemoglobinemia. Lino Rossi Research Center for the study and prevention of unexpected perinatal death and SIDS, Department of Surgical, Reconstructive and Diagnostic Sciences, University of Milan, Italy.
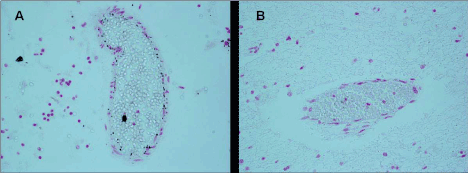
Figure 6. Blood-brain barrier vessels- A) Iron positivity in endothelial cells of a victim of SIUD aged 36 gestational weeks (case no.3). B) Iron negativity in an age-matched control case. Staining: Perls' Prussian Blue reaction for ferric iron-Magnification A) B): 20×.
Source:Lavezzi AM, Mohorovic L Alfonsi G, Coma MF, Matturri L, Brain iron accumulation in unexplained fetal and infant death victims with smoker mothers.The possible involvement of maternal methemoglobinemia. Lino Rossi Research Center for the study and prevention of unexpected perinatal death and SIDS, Department of Surgical, Reconstructive and Diagnostic Sciences, University of Milan, Italy.
Discussion
On the basis of our observation, we point out the specific cellular methemoglobin and heme catabolism when the last product is the ferric iron which, together with cytotoxic and paramagnetic property, has a notable role “in-situ”. We suggest that ferric iron and ferric iron-induced oxidative stress constitute a common mechanism involved in the development of neurodegeneration, and we also suggest the initial cause of neuronal death as a result of environmental toxic factors. The experiments showed that ferric and ferrous iron can enter cells via different pathways, however they do not indicate which pathway is dominant in humans [19].
Heme, the major functional form of iron, is synthesized in the mitochondria. Smith MA, et al. suggests that iron is able to participate oxidation in in-situ and it readily catalyzes an H2O2-dependent oxidation, and they indicate that iron accumulation could be an important contributor towards the oxidative damage of Alzheimer’s disease [20]. According to our point of view, our work supports the above statement about the importance of disturbed heme metabolism when the heme oxygenase-1, which is an enzyme that catalyzes the conversion of methemoglobin and heme to ferric iron, is increased in Alzheimer’s disease, suggesting the increased heme turnover as a source of redox-active iron. Perry G, et al. have found that while mitochondrial DNA and cytochrome C oxidase activity are increased in Alzheimer’s disease, the number of mitochondria is decreased, indicating the accelerated mitochondria turnover and suggesting mitochondrial dysfunction as a potentially inseparable component of the initiation and progression of Alzheimer’s disease [21]. It was also discovered that oxidative damage occurs primarily within the cytoplasm rather than in mitochondria. This leads to the hypothesis that mitochondria, as a source, provide hydrogen peroxide, which, as an intermediate, once in the cytoplasm, will be converted into highly reactive hydroxyl radicals through Fenton reaction in the presence of metal ion and cause damage to the cytoplasm [22]. Cell apoptosis is initiated by extracellular and intracellular signaling pathways, the death receptor- and the mitochondria-mediated pathway.
Various pathologies can result from oxidative stress-induced apoptotic signaling, consequently leading to ROS increases and/or antioxidant decreases, the disruption of intracellular redox homeostasis, and irreversible oxidative modifications of lipid, protein, or DNA [23].
Furthermore, iron participates in diverse pathologic processes by way of the Fenton reaction, which catalyzes the formation of reactive oxygen species (ROS). To test the hypothesis that this reaction accelerates apoptosis, Jacob AK,et al used human umbilical vein endothelial cells (HUVECs) as surrogates for the microvasculature in vivo. HUVECs were loaded with Fe [III] (ferric chloride and ferric ammonium citrate) and executed apoptosis after a heat shock stimulus [24]. Brain iron is a major contributor to magnetic resonance imaging (MRI) contrast in the normal gray matter. Non-heme brain iron is present mainly in the form of ferritin. The quantitation of non-heme brain ferric iron indicated by MRI helps in the diagnosis and monitoring of different neurological diseases [25]. Most of the brain’s non-heme iron is believed to be present as a storage pool consisting of ferritin or hemosiderin and also as a product of methemoglobin catabolism [26]. However, the concentration of transferrin–bound iron is always far too small to affect MRI. This fact suggests that the role of methemoglobin catabolism as the source of ferric iron (FeIII) form concentrated in various brain regions should be considered. Nowadays, non-heme-bound Fe3+ is quantified by using Magnetic Resonance Imaging (MRI), thanks to its paramagnetic properties. It is believed that most non-heme bound iron is deposited in the form of ferritin, hemosiderin, or methemoglobin catabolic products, whereas transferrin-bound iron concentration is always low and cannot be detected by MRI [27]. Recent research results indicate a ferrihydrite-magnetite core-shell ferritin structure. It was also found that the magnetite in the ferritin iron core is not a source of free toxic ferrous iron, as it had been previously believed. Therefore, the presence of magnetite in the ferritin cores of patients with Alzheimer's disease is not the cause of their increased brain ferrous iron (II) concentration [28].
Conclusion
Abundant in the source of cytotoxic and redox-active ferric (Fe3+) iron which without Ferrous- Ferric inversions, “in situ” as a cause of iron–induced cellular oxidative stress, have a direct and specific impact on the brain endothelial small vessels, and increase the rate of endothelial cell apoptosis and so make possible the accumulation of methemoglobin, heme and ferric iron, in brain parenchyma. In conclusion, we point out the importance of methemoglobinemia not only as the biomarker and precursor of environmental oxidants’ noticeable effects, but also as a carrier and donor of the redox-active ferric iron form. We identify ferric iron as the originator of the accumulation of brain parenchyma that has an important role in crossing the brain’s microvessels to the neurons (neurovascular unit damage), causing neuronal death, continuous ageing process, and finally leading to severe neurodegenerative disorders such as AD, PD, MS and other diseases. Nevertheless, as to the relation between environmental oxidants and the pathogenesis of neurodegenerative diseases, need further research.
References
- Mohorovic L, Materljan E, Micovic V, Malatestinic D, Stifter S, and Lavezzi AM (2012) Impacts of Exogenously Derived Nitrogen Oxide and Sulfur Compounds on the Structure and Function of the Vascular Endothelium Link Pregnancy Hypertension with Later Life Hypertension. J Hypertens Open Access 1: 103. [crossref]
- Balla J, Vercellotti GM, Jeney V, Yachie A, Varga Z, et al. (2007) Heme, heme oxygenase, and ferritin: how the vascular endothelium survives (and dies) in an iron-rich environment. Antioxid Redox Signal 9: 2119-2137. [crossref]
- Jeney V, Balla J, Yachie A, Varga Z, Vercellotti GM, et al. (2002) Pro-oxidant and cytotoxic effects of circulating heme. Blood 100: 879-887. [crossref]
- Maisels MJ, Kring E (2006) The contribution of hemolysis to early jaundice in normal newborns. Pediatrics 118: 276-279. [crossref]
- Denicola A, Souza JM, Radi R (1998) Diffusion of peroxynitrite across erythrocyte membranes. Proc Natl Acad Sci U S A 95: 3566-3571. [crossref]
- Starke PE, Farber JL (1985) Ferric iron and superoxide ions are required for the killing of cultured hepatocytes by hydrogen peroxide. Evidence for the participation of hydroxyl radicals formed by an iron-catalyzed Haber-Weiss reaction. J Biol Chem 260: 10099-10104. [crossref]
- Qian ZM, Shen X (2001) Brain iron transport and neurodegeneration. Trends Mol Med 7: 103-108. [crossref]
- Singh AV, Zamboni P (2009) Anomalous venous blood flow and iron deposition in multiple sclerosis. J Cereb Blood Flow Metab 29: 1867-1878. [crossref]
- Abbott NJ, Rönnbäck L, Hansson E (2006) Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci 7: 41-53. [crossref]
- Salvador GA, Oteiza PI (2011) Iron overload triggers redox-sensitive signals in human IMR-32 neuroblastoma cells. Neurotoxicology 32: 75-82. [crossref]
- Leung G, Moody AR (2010) MR imaging depicts oxidative stress induced by methemoglobin. Radiology 257: 470-476. [crossref]
- Baysal E, Monteiro HP, Sullivan SG, Stern A (1990) Desferrioxamine protects human red blood cells from hemin-induced hemolysis. Free Radic Biol Med 9: 5-10. [crossref]
- Mills E, Dong XP, Wang F, Xu H (2010) Mechanisms of brain iron transport: insight into neurodegeneration and CNS disorders. Future Med Chem 2: 51-64. [crossref]
- Brar S, Henderson D, Schenck J, Zimmerman EA (2009) Iron accumulation in the substantia nigra of patients with Alzheimer disease and parkinsonism. Arch Neurol 66: 371-374. [crossref]
- Mohorovic L (2003) The level of maternal methemoglobin during pregnancy in an air-polluted environment. Environ Health Perspect 111: 1902-1905. [crossref]
- Mohorovic L, Micovic V (2012) The importance of “first-blood circulation stage”, a new insight into the pathogenesis of clinical manifestations of preeclampsia. Advances in Bioscience and Biotechnology 3: 945-950. [crossref]
- Mohorovic L1, Materljan E, Brumini G (2008) Are Neonatal Jaundice, Heart Murmur, Dyslalia and Learning/ Memory Impairments Consequences of Mother Exposure to Environmental Oxidants? [crossref]
- Lavezzi AM, Mohorovic L, Alfonsi G, Corna MF, Matturri L (2011) Brain iron accumulation in unexplained fetal and infant death victims with smoker mothers--the possible involvement of maternal methemoglobinemia. BMC Pediatr 11: 62. [crossref]
- Conrad ME, Umbreit JN, Moore EG, Hainsworth LN, Porubcin M, et al. (2000) Separate pathways for cellular uptake of ferric and ferrous iron. Am J Physiol Gastrointest Liver Physiol 279: G767-774. [crossref]
- Smith MA, Harris PL, Sayre LM, Perry G (1997) Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc Natl Acad Sci U S A 94: 9866-9868. [crossref]
- Perry G, Taddeo MA, Petersen RB, Castellani RJ, Harris PL, et al. (2003) Adventiously-bound redox active iron and copper are at the center of oxidative damage in Alzheimer disease. Biometals 16: 77-81. [crossref]
- Marlatt M, Lee HG, Perry G, Smith MA, Zhu X (2004) Sources and mechanisms of cytoplasmic oxidative damage in Alzheimer's disease. Acta Neurobiol Exp (Wars) 64: 81-87. [crossref]
- Circu ML, Aw TY (2010) Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med 48: 749-762. [crossref]
- Jacob AK, Hotchkiss RS, DeMeester SL, Hiramatsu M, Karl IE, et al. (1997) Endothelial cell apoptosis is accelerated by inorganic iron and heat via an oxygen radical dependent mechanism. Surgery 122: 243-253. [crossref]
- Vymazal J, Brooks RA, Patronas N, Hajek M, Bulte JW, et al. (1995) Magnetic resonance imaging of brain iron in health and disease. J Neurol Sci 134 Suppl: 19-26. [crossref]
- Schenck JF, Zimmerman EA (2004) High-field magnetic resonance imaging of brain iron: birth of a biomarker? NMR Biomed 17: 433-445. [crossref]
- Rouault TA, Cooperman S (2006) Brain iron metabolism. Semin Pediatr Neurol 13: 142-148. [crossref]
- Gálvez N, Fernández B, Sánchez P, Cuesta R, Ceolín M, et al. (2008) Comparative structural and chemical studies of ferritin cores with gradual removal of their iron contents. J Am Chem Soc 130: 8062-8068. [crossref]