Cardiomyopathies (CM) are potentially fatal heart muscle diseases and one of the leading causes of heart failure as well as indication for heart transplantation. Understanding their pathogenesis is clinically relevant to improve diagnosis, management and consequently minimize the current unacceptably high mortality rates. Metabolic storage diseases are an important aetiology of CM but the classical morphofunctional definition and classification relegates metabolic CM to simply an aetiology of dilated, hypertrophy or restrictive CMs thereby undermining dedicated research and a specific understanding of aetiologic-specific diagnosis and clinical management. However, over the past decade, the contribution of metabolic remodelling to the development of myocardial dysfunction has received substantial research attention. It is now appreciated that progressive pathologic changes in cardiac metabolism may precede myocardial dysfunction, indicating that metabolic remodelling is an important early event in the progression of CM. Consequently, research continues to identify metabolic storage diseases and critical metabolic pathways that may be able to prevent or ameliorate disease progression, and as a result, immensely improving our understanding of the pathophysiologic mechanisms of metabolic CM. This article aggregates current published evidence on metabolic storage disorders frequently implicated as an aetiology of CM with a particular focus on their pathophysiology, clinical presentation, diagnosis and clinical management approaches, as well as highlights grey areas that may benefit from additional research.
Abbreviations
ACMG: American College of Medical Genetics; AF: Atrial Fibrillation; AHA: American Heart Associations; ALT: Alanine Aminotransferase; AST: Aspartate Aminotransferase; ATP: Adenosine Tri-Phosphate
CK: Creatine Kinase; CK: Creatinine Kinase; CM: Cardiomyopathy; CNS: Central Nervous System; DCM: Dilated Cardiomyopathy; ECG: Electrocardiogram; ERT: Enzyme Replacement Therapy; ESC: European Society of Cardiology; GAA: Enzyme Acid Alpha-Glucosidase; GBE: Glycogen Branching Enzyme; GDE: Glycogen Debranching Enzyme; GSD: Glycogen Storage Disease; HCM: Hypertrophic Cardiomyopathy; HF: Heart Failure; IVS: Interventricular Septum; LDH: Lactate Dehydrogenase; LV: Left Ventricular; LVH: Left Ventricular Hypertrophy; LVM: Left Ventricular Mass; LVMI: Left Ventricular Mass Index; LVOT: LV Outflow Tract; MPS: Mucopolysaccharidosis; MRI: Magnetic Resonance Imaging; PCr: Phosphocreatine; PhK: Phosphorylase Kinase; PWT: Posterior Wall Thickness; RCM: Restrictive Cardiomyopathy; RV: Right Ventricular; RWT: Relative Wall Thickness; SCD: Sudden Cardiac Death: VA: Ventricular Arrhythmia; α-Gal A: α-galactosidase A
cardiac metabolism, metabolic storage diseases, metabolic cardiomyopathy
Cardiac metabolism is an energetic activity highly adaptive to fluctuations in energy supply and demand [1]. The human heart possesses the flexibility to metabolize many different classes of substrates such as carbohydrates, lipids, amino acids, and ketone bodies to generate adequate energy to maintain tissue and organ perfusion [1,2]. Substrate preference changes throughout the life cycle as well as under physiological and pathological conditions [3]. This metabolic flexibility is essential to support cardiac adaptability to environmental changes in the short-term but might be detrimental in the long-term because of increased likelihood of adverse cardiac events [3,4]. Primarily, metabolic CM may result from pathological conditions that cause a disturbance in cardiac energy production such as obesity, insulin resistance and diabetes mellitus Type 2, which are predominantly associated with an inflammatory pathophysiology. However, a completely different pathogenic model comprises cardiac manifestation of systemic metabolic storage diseases in the setting of deficiencies of various enzymes in a variety of metabolic pathways [4]. The absence or deficiency of certain enzyme leads to decreased enzyme activities in turn resulting in the deposition of incompletely degraded endogenous macromolecules within muscles and skeletal tissues [5,6]. The accumulation of these macromolecules within the myocardium impairs its function potentially resulting into cardiomyopathy (CM), which is defined as a heart muscle disease characterized by pathologic alterations in the structure and function of the myocardium in the absence of structural heart diseases or abnormal loading conditions [7,8]. With a growing incidence of metabolic storage disorders, there is a need for a comprehensive understanding of the causes, diagnosis and management of metabolic CM. However, because many several heterogeneous groups of storage diseases mostly glycogen and lysosomal storage diseases can cause CM, the development of a general diagnosis and treatment algorithm is too cumbersome to be clinically useful and a disease-specific diagnosis and treatment is preferred. Thus, this paper adopts a disease-specific approach to review published evidence on storage diseases that can cause metabolic CM with a particular emphasis on pathophysiology, clinical presentation, diagnosis and clinical management.
Causes of metabolic cardiomyopathy
Metabolic CM or cardiac involvement in systemic diseases can be broadly classified into two categories: (i) those conditions in which a structural or functional cardiac abnormality is associated with other anomalies, usually a recognizable syndrome; and (ii) those conditions in which the heart is involved in the disease process itself [9-13]. The first category involves disturbance in energy production due to known systemic diseases such as diabetes and obesity. The second category involves metabolic storage diseases, which is the focus of the present review. Generally, metabolic storage diseases represent a heterogeneous group of disorders in which certain enzymes are defective or insufficient. Biologically, enzymes are responsible for the breakdown of certain endogenous macromolecules within the cell for proper cellular function. Thus, a deficiency in certain enzymes reduces enzyme activity leading to macromolecules destined for enzymic degradation accumulating within the cells, which can become toxic causing the cell not to function properly or die. Although many vital organs can be affected, the effect on the myocardium impairs its normal function, and if left untreated, may progress to metabolic CM [7,14,15].
Most of metabolic storage diseases that can cause CM have a genetic basis, often manifesting early in childhood and affecting multiple organ systems including the heart [7]. These diseases are always progressive but the rate of progression may vary from one form of disease or affected individual to another. The involvement of the reticuloendothelial system has particular implications for haematologists as patients may present with hepatosplenomegaly and cytopenia. Systemic diseases acquired during adulthood such as diabetes mellitus or through lifestyle habits such as alcoholism can also cause metabolic CM but they manifest via different pathogenic pathways, usually involving a disturbance in energy production and inflammatory response [4]. Previously, data on metabolic storage disorders as a cause of CM were scarce. However, following the advent and success of enzyme replacement therapy (ERT) in the early 2000s invigorated the research on metabolic storage disorders because of the potential of the therapy to reverse and heal the disease, including significant improvement in cardiac function. The groups of metabolic disorders frequently implicated as a cause of metabolic CM in literature include glycogen storage diseases, mucopolysaccharidosis, sphingolipidoses, gangliosidosis and Refsum disease.
Glycogen storage diseases : Glycogen storage disease (GSD or glycogenosis) comprise several inherited diseases caused by abnormalities of the enzymes that regulate the synthesis or degradation of glycogen in muscles, liver, and other cell types [16]. In humans, GSD is a consequence of inborn errors of metabolism (genetically defective enzymes). Depending on the defective enzyme, the disease manifests as eleven distinct types (I, II, III, IV, V, VI, VII, IX, XI, XII and XIII) [16,17]. However, cardiac involvement features only in GSD II, III, IV and IX and may lead to morphologically different morphofunctional types of CM – dilated CM (DCM), hypertrophic CM (HCM) or restrictive CM (RCM).
Type II (Pompe Disease) : Glycogen synthesis in the fed state allows sustained glucose supply during fasting. Although glycogen stores are primarily in the liver, glycogen in the cardiomyocyte provides an immediate reserve of energy in times of high cardiac metabolic demand. In the setting of GSD II (also known as Pompe disease or acid maltase deficiency), there is an overload of glycogen within lysosome. GSD II is a fatal autosomal recessive genetic muscle disorder caused by deficiency of acid alpha-glucosidase (GAA) – a glycogen degrading lysosomal enzyme [18]. It is extremely heterogeneous, which varies regardless of age at onset, rate of disease progression and extent of organ involvement. Symptoms may manifest in the first few months of life but also may first appear in individuals in their sixties [19]. A review of 225 published cases classified GSD II into infantile and late (or adult) onset. Infantile onset is the most severe form presenting with prominent CM, hypotonia, hepatomegaly and death before 12 months of life. The late onset GSD II is less common and less severe, occurs at any age, lacks severe cardiac involvement, and is marked by progressive skeletal muscle dysfunction and a less ominous short-term prognosis [20].
Significant cardiac involvement is common in GSD II because glycogen accumulation within the cardiomyocyte affects both the structure and the function of the heart [21-23]. Typical cardiac issues in GSD II are CM, HF and arrhythmia. In infantile onset, glycogen accumulation imposes a very pronounced cardiac effect. Lysosomal glycogen accumulation may lead to significant cardiac hypertrophy (between +2 and +10 or higher z-scores for LV mass), which begins in utero and may remain significant even at 4 to 8 weeks of life. Cardiac response to glycogen accumulation can result in HCM and DCM. Generally, the late onset form has no clinically identifiable heart disease but systolic and diastolic dysfunction may manifest in these patients [24]. Electrocardiogram (ECG) abnormalities are universal because of conduction system defects due to the enlargement of specialized conducting tissues with glycogen. The topography of atrioventricular (AV) conducting system is also abnormal because of deforming effects of the hypertrophied summit of the ventricular system [25]. A shortened PR interval, increased QT dispersion (QTd) and large LV voltages are common in patients with GSD II [21-23,26]. These conduction abnormalities together with HCM are hallmarks of GSD II and place these patients at an elevated risk of tachyarrhythmias and sudden cardiac death (SCD) [27-29].
The American College of Medical Genetics (ACMG) diagnosis and management guidelines of GSD II [24] recommend chest x-ray (shows a massive cardiomegaly) and ECG – shows a short PR interval and very tall QRS complexes in infantile onset GSD II, which are rare in the late onset form thereby minimizing their diagnostic utility. In infantile onset, echocardiography shows HCM with or without LV outflow tract (LVOT) obstruction in the early stages of the disease. In late stages, patients exhibit impaired cardiac function and DCM. Electromyogram may be useful in patients with CM to document pre-symptomatic myopathy. Creatinine kinase (CK) elevation is a sensitive although non-specific marker for GSD II. Infantile onset has the greatest CK elevation (as high as 2000 UI/L) [30]. The levels of serum enzymes: aspartate aminotransferase (AST), alanine aminotransferase (ALT), or lactate dehydrogenase (LDH) may also be elevated [24,31]. Confirmatory diagnosis rests on a virtual absence of (infantile onset) or markedly reduced (late-onset) GAA activity in tissues such as cultured fibroblasts from skin biopsy, muscle biopsy, purified lymphocytes, mononuclear cells and lymphoid cell line [24]. Muscle biopsy in GSD II patients shows the presence of vacuoles that stain positive for glycogen. Quantitatively, muscle glycogen is elevated up to ten-fold above normal in infantile onset and to a lesser extent in late onset GSD II [24].
The current target of clinical management of GSD II is to decrease or eliminate glycogen accumulation in body tissues and organs, and possibly reverse the progression of GSD. However, before 2006, the prognosis for GSD II was poor, particularly for the infantile onset GSD II. Since then, the introduction of ERT in the form of recombinant human α-glucosidase has revolutionized the management of GSD II. Chronic ERT has had beneficial outcomes including a reversal of cardiac dysfunction and improvement in prognosis and quality of life [24]. Early initiation of treatment, before the fifth month of life, has more favourable outcomes including regression of LV hypertrophy and decreased b-type natriuretic peptide within six months of therapy. However, even in the era of ERT, the natural progression of GSD II remains unclear although serial assessment of cardiac function may play a key role in the longitudinal management of GSD II [24,31].
Type III (cori disease): GSD III (also known as debrancher deficiency or Cori disease) is primarily recognized for its hepatic manifestations. It is an autosomal recessive disease caused by a deficiency of glycogen debranching enzyme (GDE), which is a key enzyme in the degradation of glycogen. GSD III has two major types: GSD IIIa is more common (80%) and affects liver and muscle while GSD IIIb affects only the liver [32]. GSD III is typically diagnosed within the first year of life and symptoms of hepatic involvement are prominent. Infants commonly present with hypoglycemia, hepatomegaly, short stature, and dyslipidaemia. There are several reports of sudden death due to cardiac arrhythmias in GSD III patients [33-35]. Cardiac involvement in GSD III is variable but frequently may manifest as left ventricular hypertrophy (LVH) progressing to symptomatic HCM [36-40]. In rare instances, GSD III may present with life-threatening arrhythmias [41-43], cardiac symptoms, and may progress and culminate into SCD [24] or may require cardiac transplantation [44]. Elevated LV mass and wall thickness is common in GSD IIIa but rare in GSD IIIb, which occur despite preserved ventricular systolic function in both subtypes [45]. In a study of consecutive 37 GSD patients, Chong-Nguyen et al. [32] reported electrical LVH (43%), symptomatic HF (22%), LV dysfunction (3%), LVH (43%) and obstructive HCM (11%) progressing into HF with preserved ejection fraction increasing with age.
Diagnosis of myocardial involvement in GSD III lacks pathognomonic diagnostic features. Reports of small case series between 1989 and 1997 suggest that ventricular hypertrophy is a relatively common finding in GSD III although the exact incidence varies between 30% and 80% depending on the study, and reports on symptomatic CM were very rare [46]. Prognostic features of individuals who may have unfavourable prognosis are unknown and no correlation with myopathy or CK activity has been described [47-49]. There are some indications that ECG will become more common with age although LVH alone is insufficient to indicate clinically significant CM [47,50]. Limited evidence is available about heart rhythm abnormalities in GSD III with only a few series reporting ECG findings, which have shown cardiac hypertrophy [48,50-52]. Moreover, limited information is available about arrhythmias although isolated case reports suggest fatal arrhythmias may occur in GSD III [52]. Moses et al. [42] reported one patient in their series with symptomatic CM and significant LVH by echocardiograph with documented episodes of atrial fibrillation.
The American College of Medical Genetics (ACMG) diagnosis and management guidelines of GSD III [46] recommends the following diagnostic criteria for GSD III. Marked elevation of glycogen content in the affected tissues (3 to 5 times the normal levels) with structurally abnormal glycogen (shorter outer branches; indicated by decreased G-1-P to glucose ratio), deficiency of GDE enzyme in liver and/or muscle (confirms diagnosis) or targeted mutation analysis for prenatal diagnosis if specific knowledge on family mutation is available. Diagnosis for cardiac involvement includes echocardiography to assess for LVH, including ventricular wall thickness, ventricular mass and repeat tests every 12-24 months. Echocardiography evaluation of systolic function (fractional shortening or ejection fraction) should be performed periodically although assessment of diastolic function is useful because diastolic function may precede overt systolic dysfunction and could indicate the need for closer follow-up for potential CVD symptoms. Routine 12-lead ECG should also be performed to assess for arrhythmia and repeated every two years. Repeat echocardiograph and ECG are recommended in patients with significant LVH and/or clinical signs [46].
At present, management of GSD III focusses on dietary management. In infants and children, small frequent feedings, which includes complex carbohydrates and proteins excluding simple sugars) and avoidance of fasting is recommended. Bedtime snack, cornstarch therapy and/or continuous enteral feedings may be needed for overnight fast. Early introduction of cornstarch therapy in the first year of life is recommended is hypoglycaemia is present [46]. In adolescents and adults, high protein (25% of total calories), low complex carbohydrates (< 50% of total calories), avoidance of simple sugars and fasting. For patients with myopathy, bedtime snack (low-fat milk with protein powder of high protein formular for overnight enteral feeding. In GSD IIIb, dietary restriction is transitioned to a regular well-balanced diet [46]
Type IV (andersen disease): GSD IV (also known as Andersen disease) is an autosomal recessive disorder caused by mutations in the gene encoding glycogen branching enzyme (GBE) resulting in the deficiency of the GBE [53]. The GBE catalyses the last step in glycogen biosynthesis and its deficiency results in the accumulation of abnormal glycogen with fewer branching points in different tissues to various degrees [54]. Significant clinical variability exists among GSD IV patients. The age of onset ranges from neonatal to adult age. Patients are normal at birth but develop liver complications followed by progressive liver failure and death by ages 3 to 5 years and in non-progressive form of the disease, patients have mild liver disease and can reach adulthood without liver transplantation [55]. Cardiomyopathy and cardiac failure may manifest in the setting of myofibrillar damage resulting from cardiac amylopectin deposits. Cardiac and neuromuscular symptoms may accompany or predominate the clinical picture [53,54]. In multiple system involvement, GBE deficiency appears in both muscle and liver, including peripheral myopathy with or without CM and neuropathy [56,57]. In a review of two cases, Aksu et al. [58] described GSD IV with a different form of spectrum of CM ranging from DCM to HCM and from asymptomatic to decompensated HF. One case report has also described the development of RCM in a 54-year old woman with unspecified GSD [59]. The pathogenic and molecular mechanisms are not well- documented but it is suggested that the accumulation of the less soluble abnormal polysaccharide causes a foreign body reaction in the cardiomyocyte leading to osmotic swelling and cell death [60].
GSD IV is extremely heterogeneous in terms of tissue involvement, age of onset and clinical manifestation making diagnosis problematic. Diagnosis of GSD IV can be made prenatally using polymerase chain reaction (PCR)-based DNA mutation analysis and by evaluation of GBE activity of cultured amniocytes or chorionic. GBE deficiency can be demonstrated in liver biopsy specimens, erythrocytes, leukocytes and fibroblasts. Diagnosis for cardiac involvement rests on a series of tests to detect GDE deficiency and characterize cardiac problems associated with the disease. Tests include laboratory tests such as blood count including CK levels, blood glucose test and liver function tests, non-invasive cardiac imaging to assess cardiac structure and function using echocardiography and ECG tests to detect abnormalities such as prolonged QT interval [58].
Clinical management of GSD IV remains a challenge since there is no way to replace the deficient enzyme activity. Liver transplantation is the only known treatment strategy after compete heart, liver and muscle evaluation particularly for patients with classic and progressive disease. Some GSD IV patients have improved abnormal glycogen in affected extra hepatic organs such as heart or skeletal muscle after transplantation due to the development of systemic microchimerism that occurs after transplant, with lymphocytes and macrophages acting as migrating enzyme carriers. However, some patients succumb to HF despite successful liver transplantation [58-60].
Type IX (phosphorylase kinase): GSD IX is a disease caused by the deficiency in the enzyme phosphorylase kinase (PhK), which plays a major regulatory role in the breakdown of glycogen. Two types of PhK deficiency exist – liver PhK deficiency (characterized by early childhood onset of hepatomegaly and growth restriction, and fasting ketosis and hypoglycemia) and muscle PhK deficiency (very rare) characterized by exercise intolerance, myalgia, muscle cramps, myoglobinuria or progressive muscle weakness [61]. Liver PhK can be further sub-classified based on the involved gene: PhKA2 due to mutations in the X-linked glycogenosis, and PhKB and PhKG2 are autosomal recessive conditions [62]. Cardiac involvement is rare in GSD IX patients. In an analysis of 10 patients from unrelated families, seven of which had echocardiogram and three had ECG, Bali et al. [63] reported cardiac findings in only two patients – incomplete RBBB that did not affect cardiac function and a very small patent foramen ovale that did not require intervention. The basis of cardiac involvement in GSD IX-linked PhK gene mutation including diagnosis and clinical management is currently not well understood. Creating systematic registries and collecting longitudinal data may assist in better understanding of this rare but common GSD.
Mucopolysaccharidosis: Mucopolysaccharidosis (MPS) refer to a group of metabolic disorders caused by genetic mutation in eleven (11) distinct lysosomal enzymes involved in the sequential catabolism of glycosaminoglycans (GAGs: formerly mucopolysaccharides) resulting in the accumulation of incompletely degraded GAGs within virtually all organ tissues [64]. The involvement of CVD is a feature of all types of MPS where depositions in arterial walls produce lesions similar to atherosclerosis while deposition in cardiac valves leads to valvular stenosis or regurgitation. However, severe cardiac involvement occurs most frequently in MPS Types I, II, III and VI [17].
Type I (hurler’s syndrome): MPS Type I results from the absence or deficiency of the gene encoding the enzyme α-L-iduronidase presenting in three clinical forms: Hurler’s, Scheie’s and Hurler-Scheie syndrome. Clinical features begin to appear after 24 or 36 months of life. The most striking features are growth retardation, deafness, corneal clouding and sometimes glaucoma [64]. Clinical severity of the disease correlates with the level of enzymatic activity. The defect in the Hurler’s syndrome results in the absence of lysosomal α-L-iduronidase in fibroblasts (but some activity is present in the liver), which catalyses the degradation of heparan sulphate and dermatan sulphate to heparan and hyaluronic acid. Cardiac involvement appears in about 50% the patients [65]. Angina, cardiac murmur or systemic hypertension are frequent symptoms. The murmurs are variable and usually not loud. Left atrial enlargement may occur with severe mitral regurgitation and cardiomegaly is a common finding. There are no specific ECG features although LVH may be frequent and a long QT interval may manifest in some patients. Pathological findings in the heart include deposition in the myocardium, endocardium, sinus, and AV nodes. Coronary artery demonstrate severe minimal narrowing. Mitral valve is most frequently involved followed by aortic and tricuspid valves [65,66]. Type II (hunter’s syndrome)
Mucopolysaccharidosis Type II results from deficiency of iduronate sulfatase causing blockage of the degradation of dermatan sulphate, but unlike Type I, lacks corneal clouding possibly due to variable degree of blockage of the degradation of the mucopolysaccharide. Cardiac involvement in Type II produces aortic and mitral regurgitation or stenosis, ischaemic changes and evidence of myocardial dysfunction [64]. Echocardiography is a useful method for assessing cardiac involvement. The clinical course is extremely varied. Severely affected individuals die before the age of 15 years although there are reports of survival beyond the sixth decade of life. Death in younger patient is usually the result of neurological deterioration. Due to reduced reproductive fitness of Type II gene, a large minority must result from new mutations. Emergent treatment for Type II is recombinant human iduronate-2-sulfatase, which is well tolerated with demonstrated improvement in several outcome parameters including forced vital capacity urinary excretion of glycosaminoglycan, the size of the liver and spleen, and 6-minute walk distance [17,64].
Type III (sanfilippo’s syndrome): Mucopolysaccharidosis Type III disease results from a defect in any one of four enzymes (i) heparan N-sulphatase; (ii) N-acetyl-α-D-glucosaminidase; (iii) acetyl-CoA α-glucosaminide N-acetyltransferase; and (iv) N-acetyl-α-D-glucosamine-6-sulphate sulphatase responsible for the degradation of heparan sulphate and N-sulphated or N-acetylated α-linked glucosamine. Although there are four bio-distinct sub-types of the disease (designated A-D) although they present the same clinical features [64]. The onset of Type III is in the first few years of life with behavioural problems. Mental and neurological deterioration are severe and lead to death in the first two decades of life. Cardiac involvement is similar to that of other Mucopolysaccharidosis, with thickening of valvular leaflets [17].
Type IV (morquio’s syndrome): Mucopolysaccharidosis Type IV disease (or Morquio’s Syndrome) results from a defect in enzymes N-acetyl-galactosamine-6-sulfate sulfatase and ß-galactosidase responsible for the degradation of keratin sulphate. Since, keratan sulphate occurs in cartilage, intervertebral discs and the cornea, skeletal involvement with dwarfism, pectus deformities and bow-legged and corneal clouding are common manifestations. The heart is involved only in the severe type. Valves are primarily involved with the thickening of mitral and aortic leaflets. Concentric LVH and rarely asymmetrical septal hypertrophy have been described [64].
Type VI (maroteaux-lamy syndrome): Mucopolysaccharidosis Type VI disease (or Maroteaux-lamy syndrome) results from deficiency of enzyme arylsulfatase B causing the inability to hydrolyse the sulfate groups in dermatan sulfate. The clinical picture is similar to that of Type I. Affected infants can present with an acute cardiopathy [17]. Thickened mitral and aortic valvular leaflets necessitation valvular replacement has been noted in young adults as well as LV aneurysm. Death usually occurs in the third decade of life [64].
Type VII (sly’s syndrome): Mucopolysaccharidosis Type VII (or Sly’s syndrome) is a consequence of a defect in the enzyme β-glucuronidase inherited in autosomal recessive pattern characterized by coarse faces, corneal clouding, abdominal and inguinal, hernias, puffy hands and feet, hepatosplenomegaly and small thoracolumbar hump. CVD manifestations include hypertension, aortic aneurysm, aortic regurgitation, obstructive arterial disease and CM. Death as early as one month has been reported in a child with severe disease [17,64,65].
The pathophysiology of MPS is incompletely understood but the general assumption is that progressive GAGs infiltration and downstream effect may result in anatomical and functional alterations of the valves, coronary arteries, great vessels, conduction system and myocardium [66,67]. Cardiac histopathology and electron microscopy studies demonstrate increased GAGs content and infiltration of clear cells and granular cells within cardiac valves, endocardium, myocardial walls coronary arteries, aorta and the conduction system [68,69]. Light microscopy studies of cardiac tissue reveals the morphological consequence of GAGs storage – vacuolated cells with enlarged cytoplasm [64]. Sulfated GAGs are normal components of cardiac valves and the great vessels [70-72]. Alterations in GAGs metabolism may play an important role in the pathological processes such as myxomatous mitral valves [70,73] aortic aneurysm [74] and atherosclerotic vasculature [75]. Dermatan-sulfated GAGs are a prominent component of normal cardiac valve tissue, and may explain the association between MPS I, II and VT resulting in the accumulation of dermatan-sulfated GAGs and cardiac disease [76,77]. Cardiac valves in patients with MPS have GAG-laden cells (clear or Hurler’s cells), which in MPS I are activated valvular interstitial cells engaged in valve repair but ineffective. [78]. Although the effect of sulfated GAGs on the myocardium is not well known, it is hypothesized that they induce inflammation by activating the Toll-like receptor 4 pathway leading to upregulation of degradative processes [79].
Sphingolipidoses: Sphingolipidoses refers to a heterogeneous group of inherited lysosomal storage disorders in which enzymes or sphingolipid catabolism are absent or deficient leading to accumulation of sphingolipid in tissues. The clinical presentations are diverse primarily involving neurovisceral, visceral, or purely neurological systems, and rarely cardiovascular system. Of the many different sphingolipidoses, three have prominent myocardial involvement leading to the development of CM: Fabry’s disease, Gaucher disease and Niemann-Pick disease [4].
Fabry's disease (α-galactosidase A deficiency): Fabry disease (Anderson Fabry Disease or angiokeratoma corporis diffusum) is a lipid storage disorder resulting from deficient activity of the lysosomal enzyme α-galactosidase A (α-Gal A) leading to intracellular accumulation of globotriaosylceramide (a neutral glycosphingolipid) in many tissues and even in body fluids [80-82]. The disease is of X-linked recessive inheritance although heterozygous women can also show severe manifestations [83]. Cardiac involvement is present in both classic and atypical forms of the disease with concentric LVH as the hallmark. FD is a rare cause of CM with the proportion of FD in patients previously diagnosed with HCM ranging between 0.5% and 12% in case series [84]. Many patients are asymptomatic although about 60% may exhibit chest pain, dyspnoea or palpitations usually in the third decade of life in males and later on in heterozygous females [85]. Some patients may complain of angina mostly in combination with LVH, which might be due to increased oxygen demand but also in response to diffuse arteriopathy associated to cellular injury of the arterial walls [81].
Diagnostically, absence or very low levels in males of enzyme (α-Gal A) activity assay is sufficient to diagnose FD although genetic testing to look for pathogenic variants is highly recommended because benign polymorphisms can reduce enzyme levels. In atypical variants, biopsy (skin or renal) can make diagnosis [81]. ECG findings (LVH criteria and short PR interval) in FD is useful for raising clinical suspicion for FD as well as for risk stratification. Short PR interval, which can be present in children < 10 years of age, occurs in about 40% of FD patients [86,87]. Case reports show 17% incidence of AF on ambulatory monitoring and 8% cases of ventricular arrhythmia (VA) due to fibrosis and conduction abnormalities [88]. Although rare, SCD can be the first presentation due to VA or complete AV block. Heart valves involvement occurs in about 15% of FD patients with thickening, fibrosis and calcification but usually does not result in significant impairment [89]. The prevalence of aortic dilatation correlates with age, with a greater proportion in males (9.6%) [90], and often affects the sinus of Valsalva and the ascending aorta [91]. Hypertension is also prevalent (50%) contributing to increased CVD risk and ventricular hypertrophy [81].
Echo is essential in the diagnostic algorithm, staging and follow-up of FD patients. Significant LVOT obstruction is rare in case series although Calcagnino et al. [92] reported six patients with provoked LVOT significant gradient in a series of 14 symptomatic patients. Generally, LVEF is preserved in PD patients but depressed in advanced disease culminating in end stage HF. Diastolic dysfunction is impaired but restrictive physiology is rare. For both diastolic and systolic dysfunction, impairment may begin with increased wall thickness or fibrosis [93]. Cardiac MRI with LGE can be used to assess fibrosis – localized enhancement of the basal posterior LV wall accompanied by wall thinning in the late stages. More sensitive modalities such as strain echocardiography and MRI show markers of disease earlier than the ordinate echocardiographic findings. Global longitudinal strain (GLS) is low in FD without LVH [81].
Enzyme replacement therapy (ERT) is the first-line treatment for FD. Although there is no complete consensus on initiation indication and symptom or organ manifestation criteria, which vary among different guidelines. However, ERT treatment may be less effective when initiated after irreversible damage has occurred to the microcirculation. Treatment for cardiac symptoms may include angiotensin converting enzyme inhibitors (ACEI) or angiotensin receptor blockers (ARBs) to maintain urine protein to creatinine ration < 0.5 g/g and anticoagulant for AF; antianginal therapy (nitrates, β-blockers, calcium channel blockers, and antiplatelet drugs). Antiarrhythmic drugs can be used to convert patients in atrial fibrillation to sinus rhythm, and or pacemakers in patients with symptomatic bradycardia [94].
Gaucher disease (Glucosylceramidosis): Gaucher disease is the most common inherited disorder of glycolipid metabolism. It is an autosomal recessive disease resulting from deficiency of ß-glucocerbrosidase. The disease manifests in three types I to III based on the presence or absence of and the rate of neurological manifestations [80]. Types II and III present with neurological manifestations whereas Type I (the chronic non-neuronopathic form can be diagnosed at any age) presents with bone marrow disease and skeletal abnormalities. Cardiac manifestations are less common except in a rare homozygous D409H mutation in which calcification manifests in the aorta, and aortic and mitral valves as described in three siblings where two of them died after aortic valve replacement [95]. In Type I, cardiac involvement may present with myocardial infiltration or restrictive pericardial disease. Death may occur in early childhood, or particularly when the onset is late, there may be a normal life expectancy. The diagnosis of Gaucher’s disease rests on the findings of typical storage cells in bone marrow or by liver biopsy. Demonstration of enzymic deficiency in cultured skin fibroblasts or in leucocytes confirms diagnosis. Approaches to treatment include transplantation of organs and enzymic replacement therapy. Improved visceral involvement is a common outcome but neurological injury is generally non-responsive to exogenous enzymic therapy [95].
Niemann-pick disease (sphingomyelin lipidosis): Niemann–Pick disease (NPD) encompasses a heterogeneous group of autosomal recessively inherited diseases with varying degrees of lysosomal lipid storage and foam cell infiltration in tissues as well as overlapping clinical features including hepatosplenomegaly, pulmonary insufficiency and/or central nervous system (CNS) involvement [96,97]. Based on the clinical and biochemical study of 18 cases, Crocker [98,99] classified NPD into two distinct metabolic abnormalities. The first due to deficient activity of the enzyme acid sphingomyelinase (types A and D) and the second due to defective function in cholesterol transport (types C and D) but type D was undistinguishable from type C and mater shown to be an allelic of type C. Type A exhibits a severe neurovisceral disease while patients with type B has a chronic course with visceral involvement only. The primary organ systems affected in all ASM-deficient patients are the spleen, liver and lung [96,97]. Cardiac involvement is very rare [4,100]. A case of cardiac involvement with cardiomegaly with thickened LV wall and exceptional endocardial fibroelastosis has been described [101].
Gangliosidosis: Gangliosidosis is a lysosomal storage disease characterized by the accumulation of gangliosides (GM1 or GM2) or their related conjugates because of a defect in specific lysosomal hydrolases: acid ß-galactosidase (GM1 gangliosidosis) and hexosaminidase A or B, or both, or a deficiency of an enzymic activator (GM2 gangliosidosis).
GM1: The lack of enzymic activity for acid ß-galactosidase leads to the accumulation of GM1 gangliosidosis in the brain and other organs. Its wide variation in clinical picture resulted in a broad classification of infant, juvenile and adult forms [17]. The heart is frequently involved with a spectrum from congestive with systolic dysfunction to isolated valvar thickening. Cardiac involvement often includes cardiomegaly on chest x-ray, LVH on echo and congestive HF, diffuse nodular thickening of the mitral and tricuspid valves, and CM [102,103]. Microscopically, swollen histiocytes may contain periodic acid-Schiff (PAS) and alcian blue positive cytoplasmic granules, which also occur in slightly thickened aortic valve and myocardial cells contains storage material [104,105]. Atheromatous plaque may be present in coronary arteries. On ultrastructural study, myocardial cells show two types of deposits – membranous concentric bodies and membrane-bound vacuoles containing reticulogranular material [106].
GM2: GM2 gangliosidosis manifest in two forms – Type I (Tay-Sachs disease) due to deficiency in hexosaminidase A, and Type II (Sandhoff disease) with deficiency of hexosaminidase A and β-N-acetyl hexosaminidase. Type I patients often exhibit no clinical manifestations of CVD disease but show non-specific ECG changes and accumulations in cardiac tissues similar to that found in the brain [107]. Cardiomegaly and mitral regurgitation in Type II disease manifest and the changes are similar to those in GM1 gangliosidosis [81]. However, clinically relevant cardiac manifestation in GM2 is extremely rare with no published case of the disease leading to CM.
Refsum disease: Refsum disease is a rare autosomal recessively inherited condition due to a defect in the enzyme phytanoyl-CoA hydroxylase, which catalyses the breakdown of phytanic acid to pristanic acid using the CoA derivative as a substrate. The clinical phenotype of Refsum disease results from the accumulation of phytanic acid with increased levels in blood and other tissues including fat and neurons [108]. Classical clinical features of Refsum disease are (i) congenital abnormalities such as skeletal deformities; (ii) retinitis pigmentosa that develops slowly; and (iii) lesions, such as neuropathy, rash, and cardiac arrhythmias that can deteriorate or improve according to plasma phytanic acid level. The age of onset is variable, ranging from infancy to late adulthood. Symptoms first appear in the first and second decade of life, the initial presentation including weakness, instead gait and high blindness [109]. Cardiac involvement is rare. Conduction abnormalities especially advanced degree of AV block requiring pacemaker therapy are well known.
Diagnosis of Refsum disease should be considered in patients with retinitis pigmentosa. Elevated serum phytanic acid > 200µmol/L (normal is < 30 µmol/L) is pathognomonic for Refsum disease. Diagnosis of cardiac involvement should include ECG and cardiac ultrasound examination [110]. No curative therapy is available for Refsum disease. However, since the source of phytanic acid is almost exclusively exogenous from a wide range of food products such as dairy products, some meats and fish, diets low in phytanic acid produce clinical improvement. Although complete recovery is rare, treatment slows the progression of the disease [111]. The addition of plasmapheresis to the diet results in more rapid and significant control of phytanic acid levels. Plasmapheresis or lipapheresis can be used in the event of acute arrhythmias or extreme weakness because phytanic acid is transported on lipoproteins [112]. During plasmapheresis, cardiac monitoring should be continuous and plasma glucose concentration should be kept high to prevent onset or exacerbation of arrhythmias. Treatment for hypertension may help in delaying CM, which inevitably leads to arrhythmias. It is better to avoid amiodarone (antiarrhythmic drug) due to increased risk of hyperthyroidism – that may result in catabolism and increased phytanic acid release from tissues. If CM is non-response to medical therapy, cardiac transplantation can be lifesaving [112,113].
Meta-analysis of diagnosis and clinical management: In general, the process of diagnosing metabolic CM consists of two tests: (i) demonstrating the presence of the deficient (defective) causative enzyme; and (ii) demonstrating the evidence of pathologic alterations in cardiac conduction system, structure and/or function [114]. While the demonstrated presence of enzyme deficiency confirms the diagnosis of the underlying metabolic storage disease, the bulk of the evidence for the subsequent diagnosis of cardiac dysfunction is sporadic and the few available studies largely focus on individual diseases causing metabolic CM or the safety and/or the tolerability of treatment with very little mention of cardiac involvement [115-124]. In addition to insufficient research evidence, a wide heterogeneity of metabolic storage diseases that can cause CM in combination with a wide spectrum of non-specific clinical presentation renders a generalized diagnostic and treatment algorithm too cumbersome to have clinical relevancy. Despite these aetiologic and symptomatic variations, patients with metabolic CM may exhibit some common signs and symptoms helpful to raise clinical suspicion of the disease and prompt for additional cardiac tests to confirm diagnosis. Subsequent to diagnosis, treatment of metabolic CM typically targets the replacement of the defective (deficient) enzyme and relief symptoms of cardiac dysfunction. Thus, ERT is quickly emerging as a first-line therapy in metabolic storage diseases although evidence of its efficacy in preventing or reversing cardiac involvement is sporadic, and at most, not well understood. Thus, the present meta-analysis seeks to determine common cardiac changes as well as the clinical efficacy of ERT in patients with metabolic CM.
The search for relevant studies for inclusion in this meta-analysis was performed in PubMed using the following key search terms: “metabolic cardiomyopathy”, OR “metabolic storage diseases” OR “metabolic heart diseases” AND “diagnosis” OR “enzyme replacement treatment”. Studies were included if they (i) enrolled patients with metabolic storage disease and cardiac involvement; (ii) patients were diagnosed by demonstrating enzyme deficiency accompanied by the evidence of cardiac dysfunction and/or were treated by ERT; and (iii) reported treatment outcomes. The search yielded 464 studies out of which 20 articles published between 2001 and 2018 that met the inclusion criteria were included in the final analysis. Studies that did not evaluate cardiac function in both diagnosis and treatment outcomes, case reports and series with only two patients, conference papers and review articles were excluded in the present review.
Data for analysis were extracted in three main categories: (i) study characteristics – study design, inclusion criteria, disease duration, and treatment duration; (ii) patient characteristics – patient sample, age at disease onset, and sex proportion; and (iii) treatment outcomes – ECG, and echocardiographic and/or Cardiac MRI changes in cardiac structure and function. Study variables were described using standard summary statistics. Categorical variables were expressed as frequencies and percentages, and continuous variables as means and standard deviation (or range). Dichotomous data was expressed as weighted mean and 95% confidence interval (CI). The I2 statistics was used to quantify the percentage of total variation across studies that is not due to chance. Fixed effect model was used when I2 ≥ 50% while random effect model was used when I2 < 50%. Reported p-values are two-tailed, and a p-value < 0.05 was considered statistically significant (Table 1).
Table 1. A summary of studies on metabolic storage disease (ECG: Electrocardiogram; Echo: Echocardiograph; IVS: Intraventricular Septum; IVSd: Interventricular Septum Diameter in Diastole; LVH: Left Ventricular Hypertrophy; LVM: Left Ventricular Mass; LVMI: Left Ventricular Mass Index; LVPWd: Left Ventricular Posterior Wall Diameter in Diastole; MPS: Mucopolysaccharidosis; MRI: Magnetic Resonance Imaging; NS: Not Significant; NYHA: New York Heart Association; PWT: Posterior Wall Thickness; QRSd: QRS Duration; RBBB: Right Bundle Branch Block; RWT: Relative Wall Thickness; SR: Strain Rate)
1st author [Ref#] |
Year |
Design |
Patient no. |
Male n (%) |
Age (SD/ Range.) |
Underlying disease |
Therapy duration |
Cardiac test |
Main findings |
Kakkis [125] |
2001 |
Open label |
10 |
6(60) |
12.4 (5.13) |
MPS I |
52 weeks |
Echo |
All 10 patients reported an improvement by 1 to 2 NYHA functional classes. No objective data from echo findings to verify direct cardiac benefit. |
Schiffmann [126] |
2001 |
Double blind |
14 |
26(100) |
34 (2.26) |
Fabry Disease |
NR |
ECG |
Significant decrease in QRSd by 2.4±3.9 ms in treatment group vs. an increase of 3.6±1.17 ms in the placebo group and resolution of RBBB in one patient |
Weidemann [127] |
2003 |
Open label |
16 |
14(87.5) |
42 (3) |
Fabry Disease |
12 months |
Echo, MRI |
Peak systolic SR increased in the posterior wall after 12 months of ERT 2.8± 0.2 to 3.7± 0.3 s-1, p<0.05). End-systolic strain of the posterior wall 34± 3 to 45±4%, p<0.05. PWT decreased 13.8± 0.6 to 11.8± 0.6 mm, p<0.05) and LVMI decreased 201± 18 to 180± 21g, p<0.05. |
Spinelli [128] |
2004 |
Open label |
9 |
7(77.7) |
40.9 (12.9) |
Fabry Disease |
12 months |
ECG, Echo |
After 12 months of ERT: Non-significant decrease in LV PWT 13.3± 1.6 to 12.4± 1.5; IVRT: 86± 17 to 82± 16 ms; DT: 248±70 to 234± 60 ms but a significant decrease in IVS thickness in RWT and LVMI |
Ansong [23] |
2006 |
Open label |
19 |
14(73.7) |
5.5m |
GSD II |
6 months |
ECG, Echo |
PR interval lengthened from 83 to 107 ms (p<0.001), and QTd decreased from 83 to 53 ms (p<0.003). Significant decreases in LV voltage (67 [17– 83] to 48 [18 –77] mV, p<0.03) correlating with decrease in LV mass on 2D echo. No evident change in the QTc interval (429 to 413 ms, p=NS) |
Breunig [129] |
2006 |
Open label |
26 |
20(76.9) |
37.08 (9.5) |
Fabry Disease |
23 months |
Echo, MRI |
In patients with renal impairment, LV PWT did not change (14.1 to 13.4 mm) but in the absence of renal impairment, the outcome was more favorable (11.7±1 and 10.7± 0.7 mm, p=0.04) |
Ries [130] |
2006 |
Open label |
24 |
19(79.2) |
11.8 (5.5-18) |
Fabry Disease |
25 weeks |
ECG, Echo |
Non-significant decrease in LVH/h (indexed for height) for both boys (31.4±1.4 to 32.8±2.3) and girls (31.4± 1.4 to 32.8± 2.3). 3 children with high normal LVH/h demonstrated 15% decrease in LVH/H after 25 weeks of therapy |
Levine [131] |
2008 |
Open label |
8 |
4(50) |
4.2 (1.6)m |
Infantile GSD II |
12 months |
ECG, Echo, Chest X-ray |
LVM decreased from 191(157-565) to 87(53-124) p<0.001; PR interval increased from 90(80-100) to 120(100-140) p<0.001; and LV voltage decreased from 54(40-71) to 36(18-60) p<0.02 |
Imbriaco [132] |
2009 |
Open label |
11 |
8(72.7) |
35 (11) |
Fabry Disease |
45 months |
MRI |
At 45 months of ERT, LVM (188±60 to 153±47 g) and LV PWT (16±4 to 14±4 mm) significantly decreased, IVS 80±5 to 66±8 msec, apex 79±10 to 64±10 msec, and lateral wall 80±8 to 65±16 msec. |
Mehta [133] |
2009 |
Open label |
181 |
126(69.6) |
39.2 (12.3) |
Fabry Disease |
5 years |
Echo |
In patients with baseline LVH, LVMI significantly reduced after 5 years (71·4± 22·5 to 64·1± 18·7, p=0·0111) and increased in midwall fractional shortening 14·3±2·3 to 16·0±3·8% after 3 years (p=0·02). In patients without baseline hypertrophy, LVM index and MFS remained stable. |
Whybra [134] |
2009 |
Open label |
40 |
0(0) |
47 (17.9) |
Fabry Disease |
4 years |
Echo |
Mean LVM significantly decreased from 89.4± 29.3 to 66.5± 29.3, p<0.001 |
Strothotte [135] |
2010 |
Open label |
44 |
24(54.5) |
48.9 (12.9) |
Late onset GSD II |
12 months |
Serum CK |
Significant mean decrease of 10.5±14% in creatine kinase serum levels but no correlation with disease severity |
Forsha [136] |
2011 |
Double blind |
59 |
44(51) |
45 (38.5) |
Late onset GSD II |
12 months |
ECG, Echo |
At baseline, a short PR interval was present in 10%, 7% had decreased LV systolic function, and 5% had elevated LVM on echo (all in mild range). There was no change in cardiovascular status associated with ERT |
Angelini [137] |
2012 |
Open label |
74 |
33(44.6) |
43 (15.4) |
Juvenile/ GSD II |
NR |
Echo |
Nine (9) of 64 patients (14%) on echo showed a variable degree of LV and/or septal hypertrophy (one juvenile, eight adult cases) |
Rombach [138] |
2013 |
Open label |
57 |
30(52.6) |
NR |
Fabry Disease |
NR |
ECG, Echo |
LVMI increased during ERT in males (+1.2 p < 0.001) but remained stable in females (-0.3 p = 0.52) |
Weidemann [139] |
2013 |
Open label |
40 |
31(77.5) |
40 (9) |
Fabry Disease |
5.2 years |
Echo |
LVM decreased from 270± 87 to 224± 71; PWT from 13.2± 2.0 to 11.9± 1.8, EA from 1.3±0.4 to 1.2± 0.4 and DT from 225± 60 to 217± 63, 15 cardiovascular events and 7 deaths |
Hahn [140] |
2014 |
Open label |
23 |
13(56.5) |
2.8 (0-8.4) m |
Infantile GSD II |
NR |
Echo |
Reversal of cardiac hypertrophy in 96% of the patients and no reversal in 7% and sustained impaired contractility in 13% with 43% deaths |
Chien [141] |
2015 |
Open label |
10 |
NR |
NR |
Infantile GSD II |
63 months |
Echo |
The median pre-ERT LVMI was 122(70-186) g/m2 LVH improved dramatically during the first 3 months of ERT resolved in most patients 6 months after ERT initiation and remained stable thereafter. |
Kampmann [142] |
2015 |
Open label |
45 |
21(46.7) |
34.7 (12.8) |
Fabry Disease |
10 years |
Echo |
After 10 years ERT, LVMI decreased significantly by -13.55, range -23 to -4.05 g/m2 |
Lin [143] |
2018 |
Open label |
32 |
16(50) |
10.8 (1.1-29.1) |
MPS IV |
NR |
Echo |
Mean Z scores > 2 identified in 25%, 50%, 29%, and 69% of the LVMI, IVSd, LVPWd, and aortic diameter values. Diastolic dysfunction (E/A ratio < 1) in 4 patients (13%) but ejection fraction was normal (50–75%) in all the patients |
Study characteristics: This systematic review and meta-analysis included 20 studies consisting of 18 open label non-randomized prospective, retrospective or observational studies [23,125,127-135,137-143] and two randomized controlled double blind clinical trials [126,136]. In total, the 20 studies enrolled a population of 742 patients comprising of a substantial proportion of males (n=456; 61.5%). The age of the patients varied widely due to the inclusion of both paediatric and adult patients. The mean age was 29.04 years (SD=6.9; range 2.8 months to 48.9 years). Nearly two-thirds of patients had Fabry disease (n=453; 62.4%) [126-130,132-134,138,139,142], followed by late onset (adult) GSD II (n=196; 26.4%) [23,135,136,137], MPS (n=42; 5.7%) [125,143] and infantile GSD III (n=41; 5.5%) [131,140,141]. The primary diagnostic test for the causative metabolic storage disease performed in all the 20 studies was the demonstration of reduced enzymic activity or mutational analysis of the causative defective gene. Data about tests for cardiac function included routine and follow-up 12-lead ECG in seven (7) studies [23,126,128,130,131,136,138] and non-invasive cardiac imaging by echocardiography in 17 studies [23,125,127-131,133,134,136-143], MRI in two studies [129,132] and/or in one study each chest X-ray [131] and blood tests assessing for CK levels [135]. The mean duration for the disease varied substantially among the studies, ranging from 4 years to [130] to 15 years [136]. Treatment duration also varied widely among the studies, ranging from 25 weeks [130] to 10 years [142]. However, there were no sufficient data to assess treatment efficacy of ERT on hard clinical endpoints such as hospitalization, cardiac complications, and cardiac or all-cause death.
Diagnostic features: Diagnosis of the primary aetiologic agent (the culprit metabolic storage disease) varied based on the causative defective (or deficient) enzyme. However, diagnostic tests for abnormalities in cardiac function was dependent on ECG changes and on the findings of non-invasive cardiac imaging mostly by echocardiography or cardiac MRI. LVH was the most dominant diagnostic feature for cardiac involvement in patients with metabolic storage diseases demonstrated by both ECG and non-invasive imaging tests (echocardiography or cardiac MRI). In a pooled analysis of nine studies [127,129,132-134,136-138,142], 189 out of 509 patients have baseline LVH translating into an event rate of 47% (95% CI: 30% to 65%: Figure 1). Common ECG changes with insufficient data for pooled analysis reported in individual studies included LV voltage (67 mV [23] to 54 mV [131]), PR interval (83 ms [23] to 90 ms [131]), QT interval (429 ms [23] and 398 ms [136]). Diagnostic cardiac features demonstrated by individual studies on echocardiography or cardiac MRI included significantly increased LV mass (LVM), LV mass index (LVMI), posterior wall thickness (PWT) and isovolumetric relaxation time (IVRT) (p<0.05).
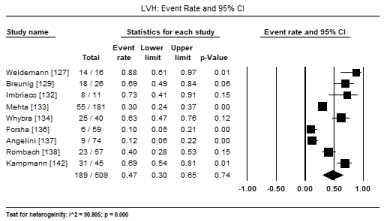
Figure 1. Forest plot for event rate for LV hypertrophy in metabolic CM patients.
Treatment outcomes: The present meta-analysis sought to find changes in cardiac structure and function after the initiation of ERT. The analysis finds ERT has favourable clinical outcomes in patients with CM secondary to Fabry disease or GSD II. Despite wide variations in both disease and treatment durations, pooled analysis of data from three studies [127,132,139] enrolling 67 patients revealed baseline LVM decreased significantly from 219.77 g (95% CI: 173.91-265.63) to 186.26 g (95% CI: 152.51-220.01). LVMI also decreased in two studies [133,134] enrolling 221 patients from a mean of 79.88 g/m2 (95% CI: 62.27-97.49) to 64.30 g/m2 (95% CI 61.69-66.91). In five studies [127-129,132,139] enrolling 102 patients, PWT decreased from a mean of 13.34 mm (95% CI 12.20-14.49) to 11.82 (95% CI 11.06-12.57). Finally, in two studies [127,128], IVRT increased from a mean of 103.9 ms (95% CI: 69.66-138.23) to 100.39 (95% CI: 65.12-135.66) (Table 2).
Table 2. Cardiac outcomes at pre- and post ERT treatment
Features |
Studies |
No. of patients |
Baseline |
95% CI |
Post-ERT |
95% CI |
LVM (g) |
127,132,139 |
67 |
219.77 |
173.91-265.63 |
186.26 |
152.51-220.01 |
LVMI (g/m2) |
133,134 |
221 |
79.88 |
62.27-97.49 |
64.30 |
61.69-66.91 |
PWT (mm) |
127,128,129,132,139 |
102 |
13.34 |
12.20-14.49 |
11.82 |
11.06-12.57 |
IVRT (ms) |
127,128 |
25 |
103.9 |
69.66-138.23 |
100.39 |
65.12-135.66 |
In addition to pooled analysis, individual studies suggest high frequency of cardiac hypertrophy that improved after ERT treatment. Lin et al. [143] assessed echocardiography changes in LV structure using z-scores and reported Z-scores > 2 in in 25%, 50%, 29%, and 69% of the LVMI, IVS diameter, LV PWT and aortic diameter values demonstrating high frequency of valvular heart disease and cardiac hypertrophy in patients with MPS. In addition to the standard 2D and m-mode echocardiography, one study [127] assessed changes in myocardial velocities using Doppler strain rate imaging. The study reported, at baseline, both peak systolic strain rate and systolic strain were significantly reduced in both radial and longitudinal direction in patients compared to healthy controls. After 12 months of treatment, peak systolic strain and end-systolic strain increased significantly in the posterior wall (2.8±0.2 to 3.7±0.3 s-1; p<0.05) and (34±3 to 45±4%; p<0.05) respectively accompanied by improvement in longitudinal function, LV PWT and LV mass. In addition to changes in cardiac structure, Strothotte et al. [135] assessed changes in CK serum levels in patients with late onset GSD II and reported a decrease of 10.5±14% but with no correlation with disease severity.
The present systematic review and meta-analysis synthesizing the literature on the diagnosis and treatment effects on patient with metabolic CM builds upon previous systematic reviews that evaluated general treatment outcomes including survival, six-minute walk test, cardiorespiratory function and quality of life, and/or safety of therapy [144-146]. The present review narrows the focus to the diagnosis of cardiac involvement and treatment outcomes on cardiac structure and function in patients with metabolic CM. The metabolic CM population comprises a subset of the DCM, HCM and RCM populations that is likely to be larger but may be under-diagnosed. Data on this sub-population in literature is rarely separated from metabolic diseases or other distinct morphological forms of CM despite their increasing prevalence and life threatening if undiagnosed and left untreated. The evidence identified in this review suggests that in addition to the demonstration of enzymic deficiency, ECG and non-invasive cardiac imaging are important diagnostic tools to assess for cardiac involvement and confirm the diagnosis of CM.
The present findings suggests that conduction abnormalities on ECG are frequent in patients with metabolic CM. Changes such as LV voltage, PR interval, QT interval and LV hypertrophy suggests cardiac involvement in patients with metabolic disease prompting for additional non-invasive imaging tests to confirm cardiac dysfunction. Echocardiography and cardiac MRI are common non-invasive cardiac imaging tests to confirm pathological changes in cardiac structure and function. Evidence of LVH is the most frequent observation demonstrated by significantly increased LVM and LVMI, and higher PWT. Diastolic dysfunction evaluated using Doppler echocardiography associated with cardiac involvement in metabolic diseases includes isovolumetric relaxation time (IVRT), which is significantly longer than in controls. However, deceleration time (DT) and E/A ratio do not significantly differ between controls as well as after ERT treatment. In metabolic CM patients, LVEF as a parameter to assess global ventricular function may be generally normal and not sensitive enough to detect impaired myocardial function. A previous study on Doppler myocardial imaging showed that myocardial velocities are reduced in hypertrophic and even in non-hypertrophic segments [147]. In Weidemann et al. [127] study, strain rate imaging is a more sensitive method to quantify regional myocardial function because it is less influenced by overall cardiac notion and tethering effects. In addition, systolic strain rate is more related to regional contractility and end-systolic strain rate more to stroke volume.
The present findings are consistent with expert consensus reports on metabolic diseases and cardiomyopathies. In the European consensus for starting and stopping ERT for GSD II, recommends diagnosis confirmation by enzyme analysis in leukocytes, fibroblasts or skeletal muscles and/or genetically by mutation analysis [117]. In addition to enzyme tests, the America Heart Association (AHA) 2016 scientific statement on current diagnostic and treatment strategies for specific DCM recommends echocardiography for assessing myocardial function in GSD II although cardiac MRI is feasible and provide quantification of LV mass, function and the presence of myocardial fibrosis. Furthermore, echocardiography may often misdiagnose metabolic CM as HCM because of similar echocardiographic features [114]. In Fabry disease, clinical suspicion is raised by presentation with end-organ dysfunction such as renal failure, cerebrovascular disease and CM. Echocardiography may be useful in evaluating cardiac function but myocardial biopsy confirms diagnosis [114]. The American College of Medical Genetics (ACMG) diagnosis and management guidelines of GSD II [24] indicates ECG changes such as short PR interval, and very tall QRS complexes is suggestive of infantile GSD II but rare in late onset GSD II. Although CK may be significantly elevated, it is non-specific to the diagnosis of metabolic CM. While current expert consensus suggest cardiac MRI may provide more diagnostic information compared to echocardiography, it suffers from limited availability. On the other hand, echocardiography has widespread use and improving its diagnostic utility and value may improve detection of metabolic CM. One of the included studies [127] suggested that myocardial strain and strain rate imaging might provide more accurate diagnostic findings than standard echocardiography but there is no sufficient evidence to confirm this diagnostic value. Further studies are warranted to confirm the diagnostic value of myocardial strain and strain rate imaging.
Although the present review focussed on the diagnosis of cardiac dysfunction in metabolic CM, it is important to mention the contemporary diagnostic strategies for the underlying metabolic disease that should prompt further non-invasive imaging assessment of cardiac function. The most common diagnostic method is enzyme activity assays – flourometry and Tandem mass spectrometry (TMS or MS/MS) methods – used in a wide range of metabolic disorders especially newborn screening for lysosomal storage diseases [10,114]. Fluorometry is an optical technique that works by evaluating the emission of fluorescence. The method measures the release of fluorescent 4-methylumbelliferone (4MU) after the breakdown of specific 4-MU synthetic substrate by specific enzymes. The measured fluorescence is directly proportional to the enzyme activity in dried blood sample. MS/MS method uses a single instrument using two or more mass analysers to measure molecular and atomic masses of whole molecules to quantify molecular species [10]. In addition to enzyme activity assays, molecular examination is useful for confirmation of enzyme activity assays and the determination of mutation in the probands molecular examination. Methods under molecular examination includes RFLP-PCR and ARMS-PCR (identifies known common point mutations in certain areas); mutation scanning (identifies gene variants without the location of the gene); DNA sequencing (identifies the precise position of the nucleotides in the genes); and next generation sequencing (investigates DNA sequencing in large regions, and very cost and time effective) [148-150].
The present evidence suggests that ERT is an effective therapy for patients with metabolic CM. The main pathogenic mechanism of metabolic CM is defective enzymes leading to progressive accumulation in body tissues of the specific substrate the enzyme degrades. The mechanism of ERT is exogenous infusion of recombinant enzymes to replace the defective ones leading to increased metabolism and reduction (or tissue clearance) of the accumulated substrates such as glycogen, mucopolysaccharide, sphingolipid or gangliosidosis. The therapy is effective in decreasing LVH demonstrated by a reduction of LV mass, LV mass index and LV posterior wall thickness to normal levels as well as improving regional myocardial function and diastolic function demonstrated by an increase in peak systolic strain and strain rate (radial function) accompanied by improvement in longitudinal function. However, ERT may be more effective when initiated early before irreversible damage has occurred in cardiac microcirculation and the myocardium.
The present findings are consistent with earlier reports on the therapeutic value of ERT in patients with metabolic diseases reducing LV hypertrophy and improving myocardial function. However, ERT is not an established therapy in all metabolic storage disorders. It is a standard therapy for patients with Gaucher’s Type I disease, where it has been associated with reduced hepatosplenomegaly and improved cardiac function. This success prompted investigations of ERT for several other metabolic disorders with promising clinical outcomes in Fabry’s disease, which involves the heart, kidney, gastrointestinal tract and peripheral nerves; GSD II that involves the heart, skeletal muscles and brain; and Hurler’s syndrome that involves the eyes, liver, joints, skeleton and the heart [94,95]. However, in Fabry disease, 12 months of ERT may not improve myocardial perfusion reserve although plasma Gb3 concentration decreases suggesting that individual variation in response to therapy was large depending on the degree of cardiac hypertrophy [121]. Nevertheless, response to ERT in the three metabolic disorders have proved generally encouraging although the degree and extent of benefit may vary considerably. In the present review, ERT has had promising results in Fabry disease, GSD II and MPS, with better results associated with starting ERT at a younger age.
Although early initiation of ERT may be associated with a greater promise of positive clinical outcomes including improving myocardial function, the exact time to initiate and stop therapy given a wide variation in treatment duration in the reviewed studies, ranging from 25 weeks [130] to 10 years [142] remains undefined. However, European consensus for starting and stopping enzyme replacement therapy in adult patients with GSD II based on systematic review of one clinical trial and 43 observational studies has recommended when to start and stop ERT for GSD II patients [117]. ERT should be initiated after definitive diagnosis of GSD, symptomatic (skeletal muscle weakness or respiratory muscle involvement), patient should commit to regular weekly treatment and monitoring, and absence of life-threatening illness in advanced stage. Criteria for stopping ERT include if the patient suffers from severe infusion-related reactions that cannot be properly managed, high antibody titres that significantly counteract ERT effect, presence of another life-threatening illness, or the absence of evidence that skeletal muscle or respiratory function in the first two years of treatment [117]. However, these criteria are subjective and their applicability to other metabolic storage disorders if not automatic. Further studies would be necessary to refine the criteria of starting and stopping ERT therapy in different types of metabolic storage diseases to improve therapeutic efficacy.
The search for relevant studies for inclusion in the present review revealed a paucity of studies specifically investigating metabolic cardiomyopathy. Most of the included studies enrolled patients with metabolic storage diseases (GSD II, Fabry disease and MPA) with general cardiac involvement. Most of the studies on other forms of metabolic storage diseases were case reports, conference papers, and expert opinions that were excluded. Even, in the included studies, there was a lack of randomized clinical trials (only two [126,136]) with a majority being open label studies. These findings suggest the need for additional research on metabolic cardiomyopathies to improve early diagnosis and prompt initiation of therapy for better results.
Despite the paucity of studies, the current evidence suggests that a combination of laboratory tests for deficient enzyme activity and demonstration of abnormalities in cardiac structure and function are useful in the diagnosis of metabolic CM and ERT increases enzymic activities and appears to improve cardiac function. Despite these findings, the heterogeneity of the underlying metabolic diseases only highlights diagnostic features that would raise clinical suspicion of metabolic CM and prompt for further specific tests to confirm diagnosis. The limited nature of the available evidence and the poor quality of the data (a majority of evidence is from open label studies with very limited randomized controlled trials) prevents any generalization regarding diagnosis and treatment effects of ERT on cardiac function.
There is also the lack of prognostic data to help clinicians to determine which patients will develop CM respond better to ERT. There are several critical features of ERT requiring attention and amelioration. There is a need to develop severity-score indices that could be used to explicitly quantify the benefit of ERT. There is a need also to develop to deliver therapeutic enzyme effectively to tissues such as cardiac muscle and kidney in Fabry’s disease, skeletal muscles in patients with GSD II, and joint tissues and structures in Hurler’s disease. To improve diagnosis and management of metabolic CM, additional large-scale prospective RCTs are warranted to confirm diagnostic features of metabolic CM, and long-term effect of ERT therapy on cardiac function and hard clinical end-points such as adverse cardiac complications, hospitalization and death.
Metabolic diseases affect the ability of the myocardium to function properly, and if left untreated, may lead to cardiomyopathy (CM), a potentially fatal myocardial disease. Pathologic conditions that can disturb cardiac metabolism include amino acid, lipids and mitochondria disorders although the most prevalent are metabolic storage disorders. The most common groups of metabolic storage disorders that cause metabolic CM include glycogen storage diseases (GSD: Types II, III, IV and IX); mucopolysaccharidosis (MPS: Types I-IV, VI, VII); Sphingolipidoses (Fabry disease, Gaucher’s disease and Niemann-Pick disease), gangliosidosis (GM: 1 and 2); and Refsum disease. The hallmark of the pathogenesis of storage diseases is a defect in certain enzymes responsible for the synthesis or degradation of certain endogenous macromolecules typically within muscle, liver or skeletal cells. These macromolecules includes glycogen (GSD), glycosaminoglycans (MPS), gangliosides (gangliosidosis), and phytanic acid (Refsum disease). The defect in these enzymes lead to their deficiency and the inability to degrade specific substrates resulting in their accumulation in body tissues including cardiomyocytes and coronary arteries with resultant anatomical and functional alterations of the valves, coronary arteries, conduction system and myocardium. Diagnosis of the underlying metabolic disease is established by the demonstration of the reduced enzyme activity in leukocytes, fibroblasts or skeletal muscle and/or demonstration of pathogenic mutations in the genes of the defective enzymes. On the other hand, a generalized algorithm for the diagnosis of cardiac involvement is not possible due to a wide heterogeneity of causative diseases, a wide spectrum of clinical presentation and a varied degree of myocardial involvement. However, metabolic CM shares common pathological changes such as LV hypertrophy, ECG changes (increased LV voltage, PR interval and QT interval) but non-specific and increased LV mass, posterior wall thickness and isovolumetric relaxation time on echocardiography or cardiac MRI. The standard treatment for metabolic disorders is enzyme replacement therapy (ERT), which has been demonstrated to be efficacious in decreasing or eliminating substrate accumulation in tissues as well as reducing cardiac hypertrophy and improving regional myocardial function in Gaucher’s disease, Fabry disease, GSD II disease and Hurler’s syndrome. The success has prompted investigations of ERT for other several metabolic diseases. To improve diagnostic and treatment, additional prospective studies on the value of myocardial strain and strain rate imaging, as well as prognostic and therapeutic value of ERT in patients with metabolic CM.
- Doenst T, Nguyen TD, Abel ED (2013) Cardiac metabolism in heart failure: Implications beyond ATP production. Circ Res 113: 709-724. [Crossref]
- Kolwicz SC, Jr., Purohit S, Tian R (2013) Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res 113: 603-616. [Crossref]
- Ritterhoff J, Tian R (2017) Metabolism in cardiomyopathy: every substrate matters. Cardiovasc Res 113: 411-421. [Crossref]
- Guertl B, Noehammer C, Hoefler G (2000) Metabolic cardiomyopathies Int J Exp Pathol 81: 349-372. [Crossref]
- Cai L, Tu BP (2012) Driving the cell cycle through metabolism. Ann Rev Cell Dev Biol 28: 59-87. [Crossref]
- Ward PS, Thompson CB (2012) Signalling in control of cell growth and metabolism. Cold Spring Harb Perspect Biol 4: a006783. [Crossref]
- Darras BT, Friedmann NR (2000) Metabolic myopathies: a clinical approach. Part I Pediatr Neurol 22: 87-97. [Crossref]
- Nishida K, Otsu K (2017) Inflammation and metabolic cardiomyopathy. Cardiovasc Res 113: 389-398. [Crossref]
- Vellodi A (2005) Lysosomal storage disorders. Br J Haematol 128: 413-431. [Crossref]
- Mokhtariye A, Hagh-Nazari L, Varasteh AR, Keyfi F (2019) Diagnostic methods for Lysosomal storage disease. Rep Biochem Mol Biol 7: 119-128. [Crossref]
- Beer M, Seyfarth T, Sandstede J, Landschutz W, Lipke C et al. (2002) Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with (31)p-sloop magnetic resonance spectroscopy. J Am Coll Cardiol 40: 1267-1274. [Crossref]
- Wisneski JA, Gertz EW, Neese RA, Gruenke LD, Craig JC (1985) Dual carbon-labeled isotope experiments using d-[6-14c] glucose and l-[1,2,3-13c3] lactate: A new approach for investigating human myocardial metabolism during ischemia. J Am Coll Cardiol 5: 1138-1146. [Crossref]
- Gertz EW, Wisneski JA, Stanley WC, Neese RA (1988) Myocardial substrate utilization during exercise in humans. Dual carbon-labelled carbohydrate isotope experiments. J Clin Invest 82: 2017-2025. [Crossref]
- Mandavia CH, Pulakat L, DeMarco V, Sowers JR (2012) Over-nutrition and metabolic cardiomyopathy. Metabolism 61: 1205-1210. [Crossref]
- Ren J, Pulakat L, Whaley-Connell A, Sowers JR (2010) Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. Journal of molecular medicine 88: 993-1001. [Crossref]
- Wolfsdorf JI, Weinstein DA (2003) Glycogen storage diseases. Rev Endocr Metab Disord 4: 95-102. [Crossref]
- Gilbert-Barness E (2004) Metabolic cardiomyopathy and conduction system defects in children. Ann Clin Lab Sci 34: 15-34. [Crossref]
- Chen YT, Amalfitano A (2000) Towards a molecular therapy for glycogen storage disease type II (Pompe disease). Mol Med Today 6: 245-251. [Crossref]
- Di Rocco M, Buzzi D, Taro M (2007) Glycogen storage disease type II: clinical overview. Acta Myol 26: 42-44. [Crossref]
- Winkel LP, Hagemans ML, van Doorn PA, Loonen MC, Hop WJ et al. (2005) The natural course of non-classic Pompe’s disease; a review of 225 published cases. J Neurol 252: 875-884. [Crossref]
- Gillette PC, Nihill MR, Singer DB (1974) Electrophysiological mechanism of the short PR interval in Pompe disease. Am J Dis Child 128: 622-626. [Crossref]
- Seifert BL, Snyder MS, Klein AA, O'Loughlin JE, Magid MS et al. (1992) Development of obstruction to ventricular outflow and impairment of inflow in glycogen storage disease of the heart: serial echocardiographic studies from birth to death at 6 months. Am Heart J 123: 239-242. [Crossref]
- Ansong AK, Li JS, Nozik-Grayck E, Ing R, Kravitz RM et al. (2006) Electrocardiographic response to enzyme replacement therapy for Pompe disease. Genet Med 8: 297-301. [Crossref]
- Kishnani PS, Steiner RD, Bali D, Berger K, Byrne BJ et al. (2006) Pompe disease diagnosis and management guideline. Genet Med 8: 267-288. [Crossref]
- Bharati S, Serratto M, DuBrow I, Paul MH, Swiryn S et al. (1982) The conduction system in Pompe's disease. Pediatr Cardiol 2: 25-32. [Crossref]
- Soliman OI, Van Der Beek NA, Van Doorn PA, Vletter WB, Nemes A et al. (2008) Cardiac involvement in adults with Pompe disease. J Intern Med 264: 333-339. [Crossref]
- Bulkley BH, Hutchins GM (1978) Pompe's disease presenting as hypertrophic myocardiopathy with Wolff-Parkinson-White syndrome. Am Heart J 96: 246-252. [Crossref]
- Francesconi M, Auff E, Ursin C, Sluga E (1982) Wolff-Parkinson-White syndrome combined with AV block 2 in an adult with glycogenosis (Type II)]. Wien Klin Wochenschr 94: 401-404. [Crossref]
- Tabarki B, Mahdhaoui A, Yacoub M, Selmi H, et al. (2002) Familial hypertrophic cardiomyopathy associated with Wolff-Parkinson-White syndrome revealing type II glycogenosis. Arch Pediatr 9: 697-700. [Crossref]
- Ausems MG, Lochman P, van Diggelen OP, Ploos van Amstel HK, et al. (1999) A diagnostic protocol for adult-onset glycogen storage disease type II. Neurology 52: 851-853. [Crossref]
- DiFiore MT, Manfredi R, Marri L, Zucchini A, et al. (1993) Elevation of transaminases as an early sign of late-onset glycogenosis type II. Eur J Pediatr 152: 784. [Crossref]
- Chong-Nguyen C, Fayssoil A, Laforet P, Gajdos V, Petit F et al. (2018) Hypertrophic cardiomyopathy in glycogen storage disease type III: Clinical features and long-term outcome. Arch Cardiovasc Dis 10: 198-199.
- Kotb MA, Abdallah HK, Kotb A (2004) Liver glycogenosis: are they a possible cause of polyneuropathy? A cross-sectional study. J Trop Pediatr 50: 196-202. [Crossref]
- Moses SW, Wanderman KL, Myroz A, Frydman M (1989) Cardiac involvement in glycogen storage disease type III. Eur J Pediatrics 148: 764-766. [Crossref]
- Lee PJ, Deanfield JE, Burch M, Baig K, McKenna WJ et al. (1997) Comparison of the functional significance of left ventricular hypertrophy in hypertrophic cardiomyopathy and glycogenosis type III. Am J Cardiol 79: 834-838. [Crossref]
- Austin SL, Proia AD, Spencer-Manzon MJ, Butany J, Wechsler SB et al. (2012) Cardiac pathology in glycogen storage disease type III. JIMD Rep 6: 65-72. [Crossref]
- Carvalho JS, Matthews EE et al (1993) Cardiomyopathy of glycogen storage disease type III. Heart Vessels 8: 155-159. [Crossref]
- Talente GM, Coleman RA, Alter C, Baker L, Brown BI et al (1994) Glycogen storage disease in adults. Ann Intern Med 120: 218-226. [Crossref]
- Lee P, Burch M, Leonard JV (1995) Plasma creatine kinase and cardiomyopathy in glycogen storage disease type III. J Inherit Metab Dis 18: 751-752. [Crossref]
- Akazawa H, Kuroda T, Kim S, Mito H, Kojo T et al. (1997) Specific heart muscle disease associated with glycogen storage disease type III: clinical similarity to the dilated phase of hypertrophic cardiomyopathy. Eur Heart J 18: 532-533. [Crossref]
- Miller CG, Alleyne GA, Brooks SE (1972) Gross cardiac involvement in glycogen storage disease type 3. Br Heart J 34: 862-864. [Crossref]
- Moses SW, Wanderman KL, Myroz A, Frydman M (1989) Cardiac involvement in glycogen storage disease type III. Eur J Pediatr 148: 764-766. [Crossref]
- Tada H, Kurita T, Ohe T, Shimomura K, Ishihara T et al (1995) Glycogen storage disease type III associated with ventricular tachycardia. Am Heart J 130: 911-912. [Crossref]
- Cochrane AB, Fedson SE, Cronin II DC (2007) Nesiritide as bridge to multiorgan transplantation: a case report. Transplant Proc 39: 308-310. [Crossref]
- Vertilus SM, Austin SL, Foster KS, Boyette KE, Bali DS et al (2010) Echocardiographic manifestations of Glycogen Storage Disease III: increase in wall thickness and left ventricular mass over time. Genet Med 12: 413-423. [Crossref]
- Kishnani PS, Austin SL, Arn P, Bali DS, Boney A et al. (2010) Glycogen storage disease type III diagnosis and management guidelines. Genet Med 12: 446-463. [Crossref]
- Lee P, Burch M, Leonard JV (1995) Plasma creatine kinase and cardiomyopathy in glycogen storage disease type III. J Inherit Metab Dis 18: 751-752. [Crossref]
- Hobson-Webb LD, Austin SL, Bali D, Kishnani PS (2010) The electrodiagnostic characteristics of Glycogen Storage Disease Type III. Genet Med 12: 440-445. [Crossref]
- Carvalho JS, Matthews EE, Leonard JV, Deanfield J (1993) Cardiomyopathy of glycogen storage disease type III. Heart Vessels 8: 155-159. [Crossref]
- Coleman RA, Winter HS, Wolf B, Gilchrist JM, Chen YT (1992) Glycogen storage disease type III (glycogen debranching enzyme deficiency): correlation of biochemical defects with myopathy and cardiomyopathy. Ann Intern Med 116: 896-900. [Crossref]
- Labrune P, Huguet P, Odievre M (1991) Cardiomyopathy in glycogen-storage disease type III: clinical and echographic study of 18 patients. Pediatr Cardiol 12: 161-163. [Crossref]
- Olson LJ, Reeder GS, Noller KL, Edwards WD, Howell RR, Michels VV (1984) Cardiac involvement in glycogen storage disease III: morphologic and biochemical characterization with endomyocardial biopsy. Am J Cardiol 53: 980-981. [Crossref]
- Andersen DH (1956) Familial cirrhosis of the liver with storage of abnormal glycogen. Lab Invest 5: 11-20. [Crossref]
- Brown BL, Brown DH (1966) Lack of an alpha-1,4-glucan: alpha-1,4-glucan 6-glycosyl transferase in a case of type IV glycogenosis. Proc Natl Acad Sci U S A 56: 725-729. [Crossref]
- McConkie-Rosell A, Wilson C, Piccoli DA et al. (1996) Clinical and laboratory findings in four patients with the non-progressive hepatic form of type IV glycogen storage disease. J Inherit Metab Dis 19: 51-58. [Crossref]
- Schroder JM, May R, Shin YS, Sigmund M, Nase-Huppmeier S (1993) Juvenile hereditary polyglucosan body disease with complete branching enzyme deficiency (type IV glycogenosis). Acta Neuropathologica.85: 419-430. [Crossref]
- Servidei S, Riepe RE, Langston C (1987) Severe cardiopathy in branching enzyme deficiency. J Paediatr 111: 51-56. [Crossref]
- Aksu T, Colak A, Tufekcioglu O (2012) Cardiac involvement in glycogen storage disease type IV: two cases and the two ends of a spectrum. Case Rep Med 2012: 764286. [Crossref]
- Patel P, Wan SH, Sinak L (2015) Glycogen storage disease induced restrictive cardiomyopathy. J Am Coll Cardiol 65: A609.
- Moses SW, Parvari R (2002) The variable presentations of glycogen storage disease type IV: a review of clinical, enzymatic and molecular studies. Curr Mol Med 2: 177-188. [Crossref]
- Kishnani PS, Goldstein J, Austin SL, Arn P, Bachrach B et al. (2019) Diagnosis and management of glycogen storage diseases type VI and IX: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med 21: 772-789. [Crossref]
- Ozen H (2007) Glycogen storage diseases: new perspectives. World J Gastroenterol 13: 2541-2553. [Crossref]
- Bali DS, Goldstein JL, Fredrickson K, Austin S, Pendyal S et al. (2017) Clinical and molecular variability in patients with PHKA2 variants and liver phosphorylase b kinase deficiency. JIMD Rep 37: 63-72. [Crossref]
- Braunlin EA, Harmatz PR, Scarpa M, Furlanetto B, Kampmann C et al. (2011) Cardiac disease in patients with mucopolysaccharidosis: presentation, diagnosis and management. J Inherit Metab Dis 34: 1183-1197. [Crossref]
- Wiseman DH, Mercer J, Tylee K, Malaiya N, Bonney DK et al. (2013) Management of mucopolysaccharidosis type IH (Hurler's syndrome) presenting in infancy with severe dilated cardiomyopathy: a single institution's experience. J Inherit Metab Dis 36: 263-270. [Crossref]
- Hinek A, Wilson SE (2000) Impaired elastogenesis in Hurler disease. Dermatan sulfate accumulation linked to deficiency in elastin binding protein and elastic fibre assembly. Am J Pathol 156:925-938. [Crossref]
- Martins AM, Dualibi AP, Norato D et al. (2009) Guidelines for the management of mucopolysaccharidosis type I. J Pediatr 155: S32-S46. [Crossref]
- Renteria VG, Ferrans VJ, Roberts WC (1976) The heart in the Hurler syndrome. Gross, histologic and ultrastructural observations in five necropsy cases. Am J Cardiol 38: 487-501. [Crossref]
- Hishitani T, Wakita S, Isoda T, Katori T, Ishizawa A, Okada R (2000) Sudden death in Hunter syndrome caused by complete atrioventricular block. J Pediatr 136: 268-269. [Crossref]
- Grande-Allen KJ, Griffin BP, Ratliff NB, Cosgrove DM, Vesely I (2003) Glycosaminoglycan profiles of myxomatous mitral leaflets and chordae parallel the severity of mechanical alterations. J Am Coll Cardiol 42: 271-277. [Crossref]
- Gupta V, Barzilla JE, Mendez JS et al. (2009) Abundance and location of proteoglycans and hyaluronan within normal and myxomatous mitral valves. Cardiovasc Pathol 18: 191-197. [Crossref]
- Latif N, Sarathchandra P, Taylor PM, Antoniw J, Yacoub MH (2005) Localization and pattern of expression of extracellular matrix components in human heart valves. J Heart Valve Dis 14: 218-227. [Crossref]
- Rabkin E, Aikawa M, Stone JR, Fukumoto Y, Libby P, Schoen FJ (2001) Activated interstitial myofibroblasts express catabolic enzymes and mediate matrix remodelling in myxomatous heart valves. Circulation 104: 2525-2532. [Crossref]
- Theocharis AD, Tsolakis I, Hjerpe A, Karamanos NK (2003) Versican undergoes specific alterations in the fine molecular structure and organization in human aneurysmal abdominal aortas. Biomed Chromatogr 17: 411-416. [Crossref]
- Nakashima Y, Wight TN, Sueishi K (2008) Early atherosclerosis in humans: role of diffuse intimal thickening and extracellular matrix proteoglycans. Cardiovasc Res 79: 14-23. [Crossref]
- Dangel JH (1998) Cardiovascular changes in children with mucopolysaccharide storage diseases and related disorders - clinical and echocardiographic findings in 64 patients. Eur J Pediatr 157: 534-538. [Crossref]
- Leal GN, de Paula AC, Leone C, Kim CA (2010) Echocardiographic study of paediatric patients with mucopolysaccharidosis. Cardiol Young 20: 254-260. [Crossref]
- Braunlin E, Tolar J, Mackey-Bojack S, Masinde T, Krivit W et al. (2011) Clear cells in the atrioventricular valves of infants with severe human mucopolysaccharidosis (Hurler syndrome) are activated valvular interstitial cells. Cardiovasc Pathol 20: 315-321. [Crossref]
- Simonaro CM (2010) Cartilage and chondrocyte pathology in the mucopolysaccharidoses: the role of glycosaminoglycan-mediated inflammation. J Pediatr Rehabil Med 3: 85-88. [Crossref]
- Nagueh SF (2014) Anderson-Fabry disease and other lysosomal storage disorders. Circulation 130: 1081-1090. [Crossref]
- Ruiz-Guerrero L, Barriales-Villa R (2018) Storage diseases with hypertrophic cardiomyopathy phenotype. Glob Cardiol Sci Pract 2018: 18. [Crossref]
- Arad M, Maron BJ, Gorham JM, Johnson Jr WH, Saul JP et al. (2005) Glycogen storage diseases presenting as hypertrophic cardiomyopathy. New Eng J Med 352: 362-372. [Crossref]
- Wang RY, Lelis A, Mirocha J, Wilcox WR (2007) Heterozygous Fabry women are not just carriers, but have a significant burden of disease and impaired quality of life. Genet Med 9: 34. [Crossref]
- Nakao S, Takenaka T, Maeda M, Kodama C, Tanaka A et al. (1995) An atypical variant of Fabry's disease in men with left ventricular hypertrophy. N Engl J Med 333: 288-293. [Crossref]
- Wu JC, Ho CY, Skali H, Abichandani R, Wilcox WR et al. (2010) Cardiovascular manifestations of Fabry disease: relationships between left ventricular hypertrophy, disease severity, and alpha-galactosidase A activity. Eur Heart J 31:1088-1097. [Crossref]
- Sheth KJ, Thomas JP (1982) Electrocardiograms in Fabry's disease. J Electrocardiol 15: 153-156. [Crossref]
- Senechal M, Germain DP (2003) Fabry disease: a functional and anatomical study of cardiac manifestations in 20 hemizygous male patients. Clin Genet 63: 46-52. [Crossref]
- Shah JS, Hughes DA, Sachdev B, et al. (2005) Prevalence and clinical significance of cardiac arrhythmia in Anderson-Fabry disease. Am J Cardiol 96: 842-846. [Crossref]
- Linhart A, Kampmann C, Zamorano JL, Sunder-Plassmann G, Beck M, Mehta A et al. (2007) Cardiac manifestations of Anderson-Fabry disease: results from the international Fabry outcome survey. Eur Heart J 28: 1228-1235. [Crossref]
- Barbey F, Qanadli SD, Juli C, Brakch N, Palacek T et al.(2010) Aortic remodelling in Fabry disease. Eur Heart J 31: 347-353. [Crossref]
- Von Scheidt W, Eng CM, Fitzmaurice TF, Erdmann E, Hubner G et al. (1991) An atypical variant of Fabry's disease with manifestations confined to the myocardium. N Engl J Med. 324: 395-359. [Crossref]
- Calcagnino M, O'Mahony C, Coats C, Cardona M, Garcia A et al. (2011) Exercise-induced left ventricular outflow tract obstruction in symptomatic patients with Anderson-Fabry disease. J Am Coll Cardiol 58: 88-89. [Crossref]
- Niemann M, Breunig F, Beer M, Hu K, Liu D et al. (2011) Tei index in Fabry disease. J Am Soc Echocardiogr 24: 1026-1032. [Crossref]
- Del Pino M, Andrés A, Bernabéu AÁ, de Juan-Rivera J, Fernández E, et al. (2018) Fabry nephropathy: An evidence-based narrative review. Kidney Blood Press Res 43: 406-421. [Crossref]
- Chabás A, Cormand B, Grinberg D, Burguera JM, Balcells S et al. (1995) Unusual expression of Gaucher’s disease: cardiovascular calcifications in three sibs homozygous for the D409H mutation. J Med Genet 2: 740-742. [Crossref]
- Schuchman EH, Desnick RJ (2009) Niemann-Pick Disease Types A and B. Mol Genet Metab 1479-1480. [Crossref]
- Vanier MT, Millat G (2003) Niemann–Pick disease type C. Clin Genet 64: 269-281. [Crossref]
- Crocker AC, Farber S (1958) Niemann–Pick disease: a review of eighteen patients. Medicine 37: 1-98. [Crossref]
- Crocker AC (1961) The cerebral defect in Tay–Sachs disease and Niemann–Pick disease. J Neurochem 7: 68-80. [Crossref]
- Linhart A, Elliott PM (2007) The heart in Anderson-Fabry disease and other lysosomal storage disorders. Heart 93:528-535. [Crossref]
- Westwood M. (1977) Endocardial fibroelastosis and Niemann-Pick disease. Br Heart J 39, 1394-1396. [Crossref]
- Lin HC, Tsai FJ, Shen WC, Tsai CH, Peng CT (2000) Infantile form GM1 gangliosidosis with dilated cardiomyopathy: a case report. Acta Paediatrica 89: 880-883. [Crossref]
- Simma B, Sperl W, Hammerer I (1990) GM1 gangliosidosis and dilated cardiomyopathy Klin Padiatr 202: 183-185. [Crossref]
- Gonatas NK, Gonatas J (1965) Ultrastructural and biochemical observations on a case of systemic late infantile lipidosis relationship to Tay-Sachs disease and gargoylism. J Neuropathol Exp Neurol 24: 318-340. [Crossref]
- Hadley RN, Hastrom JWC (1971) Cardiac lesions in a patient with familial neurovisceral lipidosis (generalized gangliosidosis). Am J Clin Pathol 55: 237-240. [Crossref]
- Gilbert EF, Varakis I, Opitz JM et al. (1975) Generalized gangliosidosis type II (juvenile GM gangliosidosis). A pathological, histochemical and ultrastructural study. Z Kinderheilkd 20: 151-180. [Crossref]
- Rodriquez-Torres R, Schneck L, Kleinberg W (1971) Electrocardiographic and biochemical abnormalities in Tay-Sachs disease. Bull NY Acad Med 47: 717-730. [Crossref]
- Jansen GA, Wanders RJ, Watkins PA, Mihalik SJ (1997) Phytanoyl-coenzyme A hydroxylase deficiency-the enzyme defect in Refsum's disease. N Engl J Med 337: 133-134. [Crossref]
- Poulos A, Pollard AC, Mitchell JD, Wise G, Mortimer G (1984) Patterns of Refsum's disease. Phytanic acid oxidase deficiency. Archives of disease in childhood 59: 222-229. [Crossref]
- Jansen GA, Oftnan R, Ferdinandusse S, Ijlst L, Muijsers AO et al. (1997) Refsum disease is caused by mutations in the phytanoyl–CoA hydroxylase gene. Nature Genetics 17: 190-193. [Crossref]
- Baldwin EJ, Gibberd FB, Harley C, Sidey MC, Feher MD et al. (2010) The effectiveness of long-term dietary therapy in the treatment of adult Refsum disease. J Neurol Neurosurg Psychiatry 81: 954-957. [Crossref]
- Wierzbicki AS, Sankaralingam A, Lumb PJ, Hardman TC, Sidey MC et al. (1999) Transport of phytanic acid on lipoproteins in Refsum disease. J Inherit Metab Dis 22:29-36. [rossref]
- Quinlan CD, Martin EA (1970) Refsum's syndrome: report of three cases. J Neurol Neurosurg Psychiatry 33: 817-823. [Crossref]
- Bozkurt B, Colvin M, Cook J, Cooper LT, Deswal A et al. (2016) Current diagnostic and treatment strategies for specific dilated cardiomyopathies: a scientific statement from the American Heart Association. Circulation 134: e579-e646. [Crossref]
- Joanne M, Skye N, Tracy M (2019) The effectiveness of enzyme replacement therapy for juvenile‐onset Pompe disease: A systematic review. J Inherit Metab Dis 42: 57-65. [Crossref]
- Angelinin C, Semplicini C. (2018) Enzyme replacement therapy for the treatment of Pompe disease Enzyme replacement therapy for the treatment of Pompe disease. Curr Neurol Neurosci Rep 12: 70-75. [Crossref]
- Van der Ploeg AT, Kruijshaar ME, Toscano A, Laforêt P, Angelini C et al. (2017) European consensus for starting and stopping enzyme replacement therapy in adult patients with Pompe disease: a 10‐year experience. Eur J Neurol 24: 768-e31. [Crossref]
- Brady RO, Schiffmann R (2004) Enzyme-replacement therapy for metabolic storage disorders. Lancet Neurol 3: 752-756. [Crossref]
- Da Silva EM, Strufaldi MW, Andriolo RB, Silva LA (2011) Enzyme replacement therapy with idursulfase for mucopolysaccharidosis type II (Hunter syndrome). Cochrane Database Syst Rev 2011: [Crossref]
- Shemesh E, Deroma L, Bembi B, Deegan P, Hollak C etal. (2015) Enzyme replacement and substrate reduction therapy for Gaucher disease. Cochrane Database Syst Rev 2015(3). [Crossref]
- Kalliokoski RJ, Kantola I, Kalliokoski KK, Engblom E, Sundell J et al. (2006) The effect of 12‐month enzyme replacement therapy on myocardial perfusion in patients with Fabry disease. J Inherit Metab Dis 29: 112-118. [Crossref]
- Kishnani PS, DiRocco M, Kaplan P, Mehta A, Pastores GM et al. (2009) A randomized trial comparing the efficacy and safety of imiglucerase (Cerezyme) infusions every 4 weeks versus every 2 weeks in the maintenance therapy of adult patients with Gaucher disease type 1. Mol Genet Metab 96: 164-170. [Crossref]
- Schiffmann R, FitzGibbon EJ, Harris C, DeVile C, Davies EH et al. (2008) A. Randomized, controlled trial of miglustat in Gaucher's disease type 3. Ann Neurol 64: 514-522. [Crossref]
- Pastores GM, Rosenbloom B, Weinreb N, Goker-Alpan O, Grabowski G et al. (2014) A multicenter open-label treatment protocol (HGT-GCB-058) of velaglucerase alfa enzyme replacement therapy in patients with Gaucher disease type 1: safety and tolerability. Genet Med 16: 359. [Crossref]
- Kakkis ED, Muenzer J, Tiller GE, Waber L, Belmont J et al. (2001) Enzyme-replacement therapy in mucopolysaccharidosis I. New Eng J Med 344: 182-188. [Crossref]
- Schiffmann R, Kopp JB, Austin III HA, Sabnis S, Moore DF et al. (2001) Enzyme replacement therapy in Fabry disease: a randomized controlled trial. Jama 285: 2743-2749. [Crossref]
- Weidemann F, Breunig F, Beer M, Sandstede J, Turschner O et al. (2003) Improvement of cardiac function during enzyme replacement therapy in patients with Fabry disease: a prospective strain rate imaging study. Circulation 108: 1299-1301. [Crossref]
- Spinelli L, Pisani A, Sabbatini M, Petretta M, Andreucci MV et al. (2004) Enzyme replacement therapy with agalsidase β improves cardiac involvement in Fabry's disease. Clin Genet 66: 158-165. [Crossref]
- Breunig F, Weidemann F, Strotmann J, Knoll A, Wanner C (2006) Clinical benefit of enzyme replacement therapy in Fabry disease. Kidney Int 69: 1216-1221. [Crossref]
- Ries M, Clarke JT, Whybra C, Timmons M, Robinson C et al. (2006) Enzyme-replacement therapy with agalsidase alfa in children with Fabry disease. Pediatrics 118: 924-932. [Crossref]
- Levine JC, Kishnani PS, Chen YT, Herlong JR, Li JS (2008) Cardiac remodeling after enzyme replacement therapy with acid α-glucosidase for infants with Pompe disease. Pediatr Cardiol 29: 1033-1042. [Crossref]
- Imbriaco M, Pisani A, Spinelli L, Cuocolo A, Messalli G et al. (2009) Effects of enzyme-replacement therapy in patients with Anderson–Fabry disease: a prospective long-term cardiac magnetic resonance imaging study. Heart 95: 1103-1107. [Crossref]
- Mehta A, Beck M, Elliott P, Giugliani R, Linhart A et al. (2009) Enzyme replacement therapy with agalsidase alfa in patients with Fabry's disease: an analysis of registry data. Lancet 374: 1986-1996. [Crossref]
- Whybra C, Miebach E, Mengel E, Gal A, Baron K et al. (2009) A 4-year study of the efficacy and tolerability of enzyme replacement therapy with agalsidase alfa in 36 women with Fabry disease. Genet Med 11: 441. [Crossref]
- Strothotte S, Strigl-Pill N, Grunert B, Kornblum C, Eger K et al. (2010) Enzyme replacement therapy with alglucosidase alfa in 44 patients with late-onset glycogen storage disease type 2: 12-month results of an observational clinical trial. J Neurol 257: 91-97. [Crossref]
- Forsha D, Li JS, Smith PB, van der Ploeg AT, Kishnani P et al. (2011) Cardiovascular abnormalities in late-onset Pompe disease and response to enzyme replacement therapy. Genet Med 13: 625-631. [Crossref]
- Angelini C, Semplicini C, Ravaglia S, Bembi B, Servidei SE et al. (2012) Observational clinical study in juvenile-adult glycogenosis type 2 patients undergoing enzyme replacement therapy for up to 4 years. J Neurol 259: 952-958. [Crossref]
- Rombach SM, Smid BE, Bouwman MG, Linthorst GE, Dijkgraaf MG et al. (2013) Long term enzyme replacement therapy for Fabry disease: effectiveness on kidney, heart and brain. Orphanet J Rare Dis 8: 47. [Crossref]
- Weidemann F, Niemann M, Störk S, Breunig F, Beer M et al. (2013) Long‐term outcome of enzyme‐replacement therapy in advanced F abry disease: evidence for disease progression towards serious complications. J Intern Med 274: 331-341. [Crossref]
- Hahn A, Praetorius S, Karabul N, Dießel J, Schmidt D et al. (2014) Outcome of patients with classical infantile pompe disease receiving enzyme replacement therapy in Germany. JIMD Rep 20: 65-75. [Crossref]
- Chien YH, Lee NC, Chen CA, Tsai FJ, Tsai WH et al. (2015) Long-term prognosis of patients with infantile-onset Pompe disease diagnosed by newborn screening and treated since birth. J Paediatr 166: 985-991. [Crossref]
- Kampmann C, Perrin A, Beck M (2015) Effectiveness of agalsidase alfa enzyme replacement in Fabry disease: cardiac outcomes after 10 years’ treatment. Orphanet J Rare Dis 10: 125. [Crossref]
- Lin HY, Chen MR, Lin SM, Hung CL, Niu DM et al. (2018) Cardiac features and effects of enzyme replacement therapy in Taiwanese patients with Mucopolysaccharidosis IVA Orphanet J Rare Dis 13: 148. [Crossref]
- Schoser B, Stewart A, Kanters S, Hamed A, Jansen J et al. (2017) Survival and long-term outcomes in late-onset Pompe disease following alglucosidase alfa treatment: a systematic review and meta-analysis. J Neurol 264: 621-630. [Crossref]
- Connock M, Juarez-Garcia A, Frew E, Mans A, Dretzke J et al.(2006) A systematic review of the clinical effectiveness and cost-effectiveness of enzyme replacement therapies for Fabry's disease and mucopolysaccharidosis type 1. Health Technol Assess 10: iii-iv. [Crossref]
- Joanne M, Skye N, Tracy M (2019) The effectiveness of enzyme replacement therapy for juvenile onset Pompe disease: A systematic review. J Inherit Metab Dis 42:57–65. [Crossref]
- Pieroni M, Chimenti C, Ricci R, Sale P, Russo MA et al. (2003) Early detection of Fabry cardiomyopathy by tissue Doppler imaging. Circulation 107: 1978-1984. [Crossref]
- Taylor CF (2009) Mutation scanning using high resolution melting. Biochem Soc Trans 37: 433-437. [Crossref]
- Grada A, Weinbrecht K (2013) Next-generation sequencing: methodology and application. J Invest Dermatol 133: e11. [Crossref]
Metzker ML (2010) Sequencing technologies – the next generation. Nat Rev Genet 11: 31-46. [Crossref]