Heart failure secondary to massive pulmonary embolism (MPE) is a commonly underdiagnosed and potentially life-threatening complication of deep vein thrombosis (DVT), with death usually occurring due to right ventricular failure in the setting of sustained hypotension and cardiogenic shock. Early diagnosis and prompt institution of PE specific therapy greatly improves clinical management of MPE. However, MPE presents with non-specific signs and symptoms that may delay diagnosis and institution of definitive treatment. Even with the availability of pharmacologic, catheter-based and surgical intervention, mortality remains high. This review paper discusses the current understanding of MPE from data from recent studies and registries as well as highlights areas requiring clarification by ongoing and future research.
deep vein thrombosis, heart failure, acute pulmonary embolism, major pulmonary embolism, venous thromboembolism
Venous thromboembolism (VTE) is a disease of the vasculature and represents a major threat to the health and wellbeing, and occasionally, the lives of a large number of patients worldwide [1]. Its clinical forms are thrombus formation in the deep vein predominantly within the lower limbs and pelvis referred to as deep vein thrombosis (DVT) and embolization of the thrombus leading to occlusion of part of the pulmonary arterial vasculature referred to as pulmonary embolism (PE) [2]. PE is the most dangerous and life-threatening clinical manifestation of VTE. It is the third leading cause of mortality from cardiovascular diseases (CVD) after coronary artery disease (CAD) and cerebrovascular stroke [3]. Massive PE (MPE) is the most lethal form of PE characterized by a significant obstruction of part of the pulmonary vasculature resulting into sustained arterial hypotension and cardiogenic shock [4]. Mainly, MPE has non-specific signs and symptoms making diagnosis challenging and delaying the institution of definitive treatment. In addition, despite the availability of evidence-based and guideline directed treatment, mortality remains high. Optimal use of advanced therapies such as thrombolysis and catheter-based therapies also remains uncertain [5]. The present paper reviews recent data on MPE, including two meta-analyses of diagnostic and therapeutic methods, to advance the understanding of its etiology, pathophysiology, diagnosis and clinical management strategies.
Historically, the classification and definition of MPE relied on angiographic evidence of anatomic burden of the thrombus in the pulmonary vasculature based on the Miller’s Index [6]. However, this definition had limited clinical use. Registry data have since established that clinical outcomes of PE patients are more dependent on the extent of PE-associated hemodynamic injury such as circulatory arrest, hypotension or RV dysfunction. The International Cooperative Pulmonary Embolism Registry (ICOPER) involving 2,454 patients from seven countries reported the 90-day mortality rate for patients with acute PE and systolic BP < 90 mm Hg at presentation was 52% against 15% for the remainder of the cohort [7]. The Germany-based Management Strategy and Prognosis of Pulmonary Embolism Registry (MAPPET) analyzed in-hospital mortality for 1,001 patients with acute PE and reported significantly higher rates in patients requiring cardiopulmonary resuscitation (65%) and those with cardiogenic shock (25%) compared to hemodynamically stable patients (8%) [8]. The Geneva and Pulmonary Embolism Severity Index (PESI) clinical scores also identify hypotension (BP < 100 mm Hg) as a significant predictor of unfavorable prognosis [9,10]. The presence of RV dysfunction has also been associated with a two-fold increase in 90-day mortality rate [11].
The 2011 scientific statement from the American Heart Association (AHA) on management of massive and submassive PE, iliofemoral DVTs, and chronic thromboembolic pulmonary hypertension (PH) [12] and the 2008 European Society of Cardiology (ESC) Guidelines on the diagnosis and management of acute PE [13] reclassified PE based on hemodynamic compromise (hypotension and cardiogenic shock). They categorized PE as massive (high-risk), submassive (intermediate risk) and non-massive (low risk). Consequently, they defined MPE as an acute onset of PE with sustained hypotension (systolic blood pressure [BP] < 90 mm Hg for at least 15 minutes or requiring inotropic support not due to any other cause such as arrhythmias, hypovolemia, sepsis or left ventricular (LV) dysfunction, pulselessness or persistence profound bradycardia (heart rate < 40 beats per minute (bpm) with signs or symptoms of shock [12,13]. Table 1 provides a brief summary of the current definitions of all the three clinical forms of PE.
Table 1. Classification and clinical definitions of categories of pulmonary embolism
Category |
Clinically defined as PE in the presence of … |
Massive |
- Arterial hypotension (systolic BP < 90 mm Hg or a decrease of > 40 mm Hg) for > 15 minutes or requiring inotropic support not caused by a new onset of arrhythmia.
- Cardiogenic shock (oliguria, lactic acidosis, cool extremities or altered levels of consciousness).
- Circulatory collapse in patients with syncope or undergoing cardiopulmonary resuscitation.
|
Submassive |
Systolic BP > 90 mm Hg but with RV dysfunction or hypokinesis confirmed by echocardiography, or PH or the presence of increased biomarkers of myocardial injury. |
Nonmassive |
Systolic BP > 90 mm Hg and no evidence of RV dysfunction, PH or increased markers of myocardial injury |
Adapted from the AHA [12] and ESC [13]
VTE, encompassing DVT and PE, is the third most frequent CVD with an overall annual incidence of 100 to 200 per 100,000 inhabitants [14,15]. Although VTE may be life threatening in the acute phase (MPE) or lead to chronic disease and disability [16-19], it is often preventable [20]. MPE is the most serious clinical manifestation of VTE. Since PE is mostly the consequence of DVT, most of the existing epidemiology data are derived from studies examining VTE as a whole. The epidemiology of PE is difficult to determine because it may remain asymptomatic, diagnosis may be incidental or the initial presentation may be sudden cardiac death (SCD) [12]. In Europe, PE is a major cause of mortality, morbidity and hospitalization. Analysis of six countries of the European Union with a population of 454.4 million reported 317,000 VTE-related deaths [15]. These deaths comprised of sudden fatal PE (34%) and PE that remained undiagnosed (59%) and diagnosed PE (7%). The ICOPER reported 4.2% of 2,454 patients from seven countries had MPE. In the United States, MPE diagnosis per year are ~150,000 patients but many more additional deaths may go undiagnosed possibly mistaken for myocardial infarction or ventricular arrhythmias [21].
Identification of risk factors have important clinical implications for MPE patients. They aid in clinical diagnosis, guide decisions about prophylactic measures and repeat testing in borderline cases [22]. Since PE is usually preceded by VTE, risk factors for the two conditions are the same and broadly fit the Virchow’s triad of venous stasis, local injury to the vascular wall and increased coagulability of blood [23]. Table 2 provides an extensive list of environmental and genetic risk factors for thromboembolic disease.
Table 2. Risk factors for massive pulmonary embolism heart failure
Risk Factors |
Specific Risk Items |
Venous stasis/injury,
secondary hypercoagulable states |
Immobilization or other causes of venous stasis such as stroke |
Major trauma or surgery within 4 weeks |
Active cancer treatment within 6 months/palliative therapy |
History of thromboembolism |
Reduced cardiac output (congestive HF) |
Obesity, advanced age |
Pregnancy, early puerperium, contraceptive pill (high estrogen content) |
Acquired thrombotic disorders |
Primary hypercoagulable states
(thrombophilia) |
Deficient antithrombin III, protein C or S |
Resistance to activated protein C |
Elevated plasminogem activator inhibitor |
Hyperhomocysteinaemia |
High plasma concentration of factor VIII |
Prothrombin gene mutation |
Adapted from Riedel, 2001, p. 229 [22]
Patient-related factors
Risk factors for MPE may be categorized into patient-related and setting-related. Patient-related risk factors are non-modifiable and often genetic. They increase the risk of MPE through inducing primary hypercoagulability state (or thrombophilia), which increases the risk of thrombosis. Common patient-related factors include deficient antithrombin III and proteins C or S, resistance to activated protein C, elevated plasminogem activator inhibitor, hyperhomocysteinaemia, high plasma concentration of Factor VIII, or prothrombin gene mutation. Other patient-related factors such as polycythemia (increased production of red blood cells), pregnancy and age [3,22] may increase the risk of MPE due to genetic factors. Patient age in particular is a non-modifiable factor and a secondary cause of hypercoagulable state. The incidence of MPE has been shown to increase exponentially with age, with the mean age of presentation of 62 years affecting both men and women equally [23-25].
Setting-related factors
Setting-related factors refer to modifiable or reversible factors occurring in the clinical setting that may precipitate or aggravate thrombosis. They increase the risk of MPE by causing venous stasis, causing local injury to the venous wall or inducing secondary hypercoagulable state. Often, setting-related risk factors include surgery, trauma, prolonged immobilization or hormone replacement therapy, which could provoke MPE within the last six weeks to three months before diagnosis [26]. Major trauma, surgery, lower limb fractures and joint replacement, and spinal cord injury are strong provoking factors for MPE [27,28]. Medical conditions such as cancer predisposes patient to MPE with the risk varying with different types [29,30]. Hematological malignancies, lung cancer, gastrointestinal cancer, pancreatic cancer and brain cancer have the highest risk for MPE [31,32]. In addition, cancer also is a strong predictor of all-cause mortality following an episode of MPE [33].
In women of childbearing age, oral contraceptives is the most common risk factor for MPE [34,35]. Pregnancy is also a risk factor and VTE is a major cause of maternal mortality [36]. The risk is greatest during the third trimester and over six weeks of the postpartum period, about 60 times higher compared to non-pregnant women [36]. In-vitro fertilization has a 6-fold increase in the risk of pregnancy-associated VTE in the first trimester [37]. In post-menopausal women, the risk of using hormone replacement therapy on MPE varies depending on the formulation used [38]. Blood transfusion and erythropoiesis stimulating agents have also been associated with an elevated risk of MPE [5,22]. Thromboembolism may be considered part of the CVD disease continuum and common risk factors such as cigarette smoking, obesity, hypercholesterolemia, hypertension and diabetes mellitus are shared with arterial disease such as atherosclerosis but mediated by effects of CAD and cancer in the case of smoking [28,39-44]. Heart diseases such as myocardial infarction and HF increases the risk of PE as well as MPE may increase the risk of myocardial infarction and stroke [45,46]. The presence of persistent risk factors rather than major temporary risk factors, may guide the decision on the duration of anticoagulation therapy following the first MPE episode [5].
Mechanism of circulatory failure
Heart failure in the setting of MPE results from a combination of increased vascular wall stress and cardiac ischemia leading to RV dysfunction and decreased LV output (Figure 1).
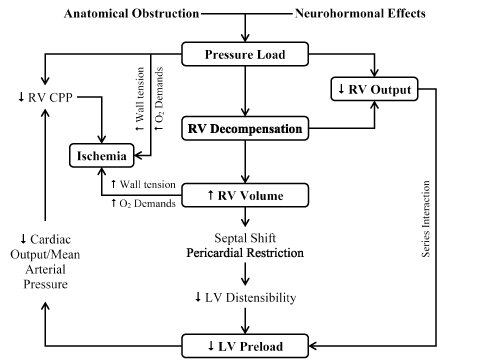
Figure 1.Pathophysiologic cycle in massive pulmonary embolism.
Anatomical obstruction and neurohormonal effect conspire to cause a series of pathophysiological RV and LV changes in MPE patients. They cause an increase in RV pressure afterload leading to RV decompensation, in turn causing a reduction in RV output and an increase in RV volume. Leftward shift in the interventricular septum, together with reduced RV output causes an increase in LV distensibility and LV preload in turn reducing cardiac output, coronary perfusion pressure leading to ischemia. Increase in RV volume also causes an increase in wall tension and oxygen demand, and may lead to ischemia. CPP: Coronary Perfusion Pressure. Reproduced from Wood, 2002, p. 882 [47].
The degree of RV overload is associated with the interaction between mechanical pulmonary vascular obstruction and the underlying cardiopulmonary status. Additional factors associated with increased RV afterload include pulmonary vasoconstriction induced by neural reflexes, the release of humoral factors from platelets (serotonin and platelet-activating factor), plasma (thrombin and vasoactive peptides), tissue (histamine) and systemic arterial hypoxia [47,48]. Increasing RV pressure afterload induces multiple effects on both the RV and LV function. RV stroke volume diminishes because of an inverse relationship with increasing vascular load [49]. The resulting RV dilatation and increased cavitary pressure leads to increased RV wall stress. Since RV wall stress is the primary determinant of RV oxygen consumption, it creates the potential for RV ischemia [50]. Increasing RV load and wall stress depresses RV systolic function and diminishes cardiac output. Increases in RV load sufficient to decrease cardiac output by 20% causes a higher increase in end-systolic volume relative to end-diastolic volume [47].
The afterload mismatch causes the LV preload to decrease because the ventricles are aligned in series. The LV preload is additional decreased by a reduced LV distensibility resulting from a leftward shift of the inter-ventricular septum and pericardial restraint, both of which are related to the degree of RV dilatation [51,52]. Additional decrease in LV flow leads to systemic hypotension. Decreases in mean arterial pressure (MAP) associated with increases in RV end-diastolic pressure impairs the sub-endocardial perfusion and oxygen supply [53]. Increased oxygen demand due to increased wall stress together with decreased oxygen supply precipitate RV ischemia suggested as the cause of RV failure [47]. Clinical evidence of RV infarction due to preceding condition has been demonstrated in patients with and without obstructive coronary disease. In individuals without cardiopulmonary disease, the RV generates 40 mm Hg mean pulmonary arterial pressure [54]. Thus, pulmonary arterial pressure > 40 mm Hg in MPE suggests recurrent PE or chronic thromboembolic PH [47,48].
Mechanism of respiratory failure
The effect of PE on the respiratory system is complex. It is associated with the size and characteristics of the embolus, the degree of occlusion, the underlying cardiopulmonary status and the duration of embolization [47]. Hypoxia (deficiency in the amounts of oxygen reaching tissues) has been linked to increased alveolar dead space, right-to-left shunting, ventilation-perfusion mismatch and a low mixed venous oxygen level [5,47,55]. The two latter mechanisms may explain for a majority of cases of observed hypoxia and hypocapnia (reduced carbon dioxide in blood) pre- and post-treatment [3,48]. Areas of reduced flow in obstructed vessels combined with areas of overflow in the capillary bed in unobstructed vessels leads to ventilation-perfusion mismatch, contributing to hypoxia. In addition, diminished cardiac output leads to low-mixed venous oxygen level [55].
Presentation
The clinical presentation of MPE is neither very sensitive nor specific. The absence or presence of any individual clinical symptom or sign is insufficient to confirm or exclude the diagnosis of MPE. However, in about a third of patients, clinical suspicion of MPE is because of pleuritic chest pain in the presence or absence of dyspnea [56,57]. Isolated dyspnea, often acute but sometimes gradually progressive in the absence of demonstrated alternative cause, suggests MPE in 20% of the patients [X]. Syncope and cardiogenic shock are rare in MPE patients (< 10%) and may manifest despite hemodynamic instability [5,58]. Finally, MPE may be discovered in the absence of clinical suspicion (incidental diagnosis) on CT scans performed for other indications such as cancer staging.
Syndromes
Symptomatic MPE may manifest as three distinct syndrome of different pathology and variable severity: alveolar hemorrhage, isolated dyspnea and syncope or shock [59].
Alveolar hemorrhage: Alveolar hemorrhage is a clinical syndrome of MPE due to peripheral emboli. Its clinical hallmark is pleuritic pain caused by the irritation of the visceral pleura and more rarely by hemoptysis. Sometimes alveolar hemorrhage may be incorrectly referred to as pulmonary infarction, its histopathological correlate is alveolar hemorrhage provoked by an influx of blood from elevated pressure in the bronchial circulation in the embolus-occluded segment [60]. The classical radiological image of alveolar hemorrhage syndrome is a wedge-shaped plural-based infiltrate affecting about 20% of the patients [61]. Other typical chest anomalies include plate-like atelectasis and pleural effusion. Tachycardia and dyspnea are less common, often reflecting peripheral character and lesser hemodynamic repercussion of pulmonary emboli [59].
Isolated dyspnea: Isolated dyspnea is another syndrome of MPE. This syndrome lacks systematic pleuritic pain because of a more proximal embolization level in the pulmonary vasculature. Patients may present with oppressive retrosternal chest pain suggesting differential diagnosis of angina pectoris [57]. Such pain possible reflects true myocardial ischemia due to increased RV wall tension and diminished right coronary artery flow. Tachycardia is present in slightly half of these patients (45%) [59]. Electrocardiogram is rarely normal and abnormalities are non-specific. Dyspnea usually has a rapid onset but in some patients it may progress gradually over several days or even weeks [57].
Syncope or shock: Syncope or cardiogenic shock are the clinical manifestation of MPE patients implicated as the underlying cause of acute severe pulmonary hypertension and ventricular failure [5]. It is usually due to a large central embolus. Although suggestive of MPE in patients with demonstrated risk factors such as recent surgery, syncope maybe a misleading presentation [62,63]. Suspected MPE with cardiogenic shock is a distinct situation that usually requires a specific diagnosis. However, since all these signs and symptoms are non-specific and may be encountered in many patients with potentially lethal cardio=pulmonary conditions, diagnostic tests are usually by exclusion of PE [57].
Definitive diagnosis of MPE is challenging because of the lack of specific symptoms and a reliable non-invasive cardiac imaging test. In the 2014 ESC guidelines on PE, diagnosis of MPE is based on therapeutic decision. Positive diagnosis indicates the need for PE-specific treatment and the exclusion of PE indicates withholding PE-specific treatment. Diagnosis is usually achieved using a validated scoring system (prediction rules) and/or D-dimer tests to raise clinical suspicion, and for suspected patients, non-invasive imaging tests usually using CT angiography is recommended to assess ventricular function and to confirm diagnosis [5].
Scoring system
Over the past two decades, there has been a trend to test more patients for MPE, which has resulted in a decrease in the proportion of confirmed cases, as few as 5% of patients with suspected PE are diagnosed with MPE [64]. The result is the question of when and in whom, a physician should suspect MPE. In most studies on the diagnosis of MPE, sudden onset or worsening of dyspnea or chest pain without any other demonstrated cause was a common criterion for inclusion of patients [65,66]. The criteria was difficult to standardize leading to a trade-off between over-suspecting and over-testing, and missing diagnosis. Testing MPE in all patients presenting with dyspnea would increase cost and tests complications without a meaningful improvement in health. The challenge is to selected patients suspected with MPE for testing while excluding others. Thus, a combination of clinical features and risk factors (including findings from common tests such as chest X-ray and electrocardiogram for differential diagnosis) have been incorporated into a clinical scoring system to select patients suspected with MPE for additional diagnostic tests. Scoring systems have been shown to improve physicians’ ability to classify patients with suspected MPE into distinct categories of clinical probability that correspond to actual prevalence of confirmed MPE [5]. The value of prediction rules on risk stratification and on raising clinical suspicion of MPE have been already demonstrated in several large series [58, 67-70]. The most frequently used clinical scoring systems for MPE in clinical settings are the Wells score [71] and the revised Geneva score [58] (Tables 3 and 4).
Table 3. Wells scores for predicting pulmonary embolism
Variables |
Items |
Scores |
Risk factors |
Previous PE or DVT |
1.5 |
Surgery or immobilization (within 4 weeks) |
1.5 |
Active cancer |
1 |
Symptoms |
Hemoptysis |
1 |
Clinical signs |
Heart Rate ≥ 100 bpm |
1.5 |
Clinical signs of DVT |
3 |
Alternative diagnosis less likely than PE |
3 |
Clinical probability |
Three level: Low |
0-1 |
Intermediate |
2-6 |
High |
≥ 7 |
Two level: PE unlikely |
0-4 |
PE likely |
≥ 5 |
bpm: beats per minute; DVT: Deep Vein Thrombosis; PE: Pulmonary Embolism.
Table 4. Revised geneva scores for predicting pulmonary embolism
Variables |
Items |
Score |
Risk factors |
Previous PT or DVT |
3 |
Age > 65 years |
1 |
Surgery or fracture within the past month |
2 |
Active cancer |
2 |
Symptoms |
Unilateral lower limb pain |
3 |
Hemoptysis |
2 |
Clinical signs |
Pain on lower limb deep venous palpation and unilateral edema |
4 |
Heart rate 74-94 bpm |
3 |
Heart rate ≥ 95 bpm |
5 |
Clinical probability |
Three level score: low |
0-3 |
Intermediate |
4-10 |
high |
≥ 11 |
Two level score: PE unlikely |
0-5 |
PE likely |
≥ 6 |
bpm: beats per minute; DVT: Deep Vein Thrombosis; PE: Pulmonary Embolism
The Wells score is simple and based on information that is easy to obtain. However, the weight of one subjective item (alternative diagnosis less likely than PE) may potentially reduce inter-observe variability [5]. The Wells score raises clinical suspicion of PE using three-level (low, moderate and high clinical probability) or two levels (likely and unlikely) prediction rules. Both three and two level prediction rules have been extensively validated [66,72-75]. The revised Geneva prediction scores for clinical suspicion of MPE is also simple and standardized. Both the Wells score and revised Geneva scores have been adequately validated [76-78]. The Wells and the Geneva scores were recently revised and simplified, and externally validated to improve their adoption in clinical settings [5]. Whichever scores used (Wells or revised Geneva) the proportion of patients with confirmed PE is approximately 10% in the low-probability category and 30% in the moderate probability category and 65% in the high probability category when using the three-level classification. In the case of the two-level classification, the proportion of patients confirmed with PE in the unlikely category is approximately 12% [78]. The 2014 ESC Guidelines on the diagnosis and management of acute PE recommends the use of Wells score or revised Geneva scores to improve clinical identification of patients suspected with MPE as well as improve risk stratification of individuals suspected with MPE [5].
D-Dimer tests
For patients with a clinical suspicion of MPE, the 2014 ESC on acute PE guidelines recommends d-dimer tests to rule out the presence of inappropriate thrombus in MPE patients or to determine the need for further tests. Plasma D-dimer is a degradation product of cross-linked fibrin and acts as a surrogate marker for coagulation and subsequent fibrosis [2]. Thus, acute thrombosis due to simultaneous activation of coagulation and fibrinolysis cause an elevation of plasma D-dimer levels. The negative predictive value of D-dimer tests is higher for MPE diagnosis while normal plasma D-dimer levels indicate diagnosis of MPE is very unlikely. Conversely, since fibrin production also occurs in a variety of other conditions such as cancer, inflammation, bleeding, trauma, surgery, pregnancy and necrosis, the positive predictive value of D-dimer test (elevated D-dimer levels) is low and the test is not useful in the confirmation of MPE [79,80].
A good number of D-dimer assays are available and validated for clinical use: (a) the quantitative enzyme-linked immunosorbent assay (ELISA); (b) the quantitative latex-derived assays; and (c) a whole-blood agglutination assay [5]. ELISA-derived assays have a diagnostic sensitivity of ≥ 95% and useful in ruling out MPE in patients with low or moderate pre-test probability. In the emergency department, the use of negative ELISA in combination with clinical probability can exclude MPE without further testing in ~30% of patients suspected with MPE [66,81,82]. Quantitative latex-derived assays and whole-blood agglutination assays have a diagnostic sensitivity of < 95%, which is considered as moderately sensitive. They are useful in excluding MPE in PE-unlikely patients and in patients with low clinical probability based on Wells or revised Geneva scores [66,75,77]. However, the safety of excluding MPE in the intermediate and clinical probability category have not been established [5]. Since the specificity of D-dimer tests in patients suspected with MPE decreases with age to almost 10% in patients > 80 years, utility of age-adjusted cut-offs would improve the performance of D-dimer tests in the elderly [55,83-86].
Imaging methods
The prevalence of definitive diagnosis of MPE based on clinical scoring system has been low in large clinical series, ranging between 10% and 35% [66,81,83,87]. To improve diagnosis, the 2014 ESC guidelines on acute PE proposed a straightforward diagnostic workout for patients suspected with MPE for non-invasive imaging (Figure 2).
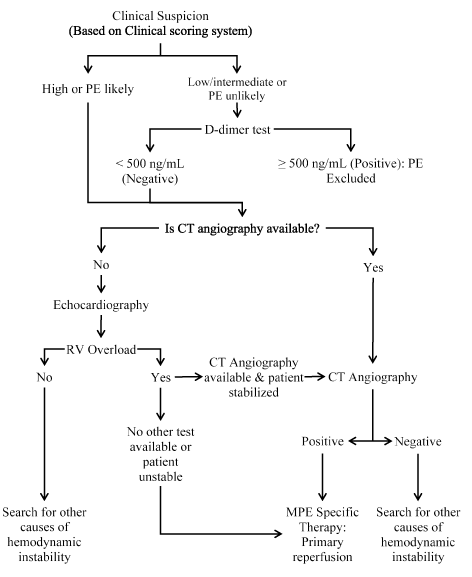
Figure 2.Proposed diagnostic workout for patients with suspected MPE.
Recommended diagnosis for patients suspected with MPE (clinical scoring and D-dimer test) is CT angiography (CTA) or echocardiography if CTA unavailable. Positive echocardiography findings of RV overload require further tests using CT angiography but if negative, test for other causes of hemodynamic instability should be considered. Negative findings of CTA should prompt for additional tests for the cause of hemodynamic instability while positive findings should inform MPE specific therapy. CT: Computed Tomography; MPE: Massive Pulmonary Embolism; RV: Right Ventricular. Reproduced and modified from 2014 ESC Guidelines on Acute PE, 2014, p. 3 [5]
CT angiography: Since the introduction of multi-detector computed tomography (MDCT) angiography with high spatial and temporal resolution and quality of arterial opacification, computed tomography (CT) pulmonary angiography has emerged as the method of choice for imaging the pulmonary vasculature in patients suspected with MPE. CT angiography (CTA) allows adequate visualization of the pulmonary arteries down to the segmented level [87-88], has a high sensitivity of 83% and specificity of 96%, and is consistent with clinical scoring system for PE [67]. CTA has a high negative predictive value for patients with low (96%) or intermediate (89%) clinical probability of PE by Wells score and 60% for those with high-pre-test probability. CTA has a high positive predictive value (92% to 96%) in patients with intermediate or high clinical probability but lower in patients with low-probability (58%) [67]. Negative CTA excludes PE in patients with non-high clinical probability of PE. CAT revealing PE at the segmental level or more proximal level is adequate evidence of PE in patients with non-low clinical probability. On the other hand, the positive predictive value of CTA is low in patients with low clinical probability of PE and may require additional tests is the thrombus are limited to segmental arteries. If CTA is positive, PE-specific therapy should be initiated, otherwise search for other causes of hemodynamic instability [5].
Echocardiography: Echocardiography is the recommended alternative non-invasive imaging method when CTA is not immediately available. The method is able to detect RV pressure overload and RV dysfunction that are associated with MPE. However, due to the peculiar geometry of the RV, there is no individual echocardiographic parameter to provide fast and reliable information on RV size and function. For this reason, echocardiographic criteria has differed greatly between studies. Echocardiography also has a low negative predictive value of between 40% and 50% and thus a negative result cannot rule out MPE [90-92]. Echocardiography signs of RV overload and RV dysfunction are non-specific to MPE. They also appear in the absence of MPE and may result from co-occurring cardiac or respiratory disease. At least 25% of patients with MPE exhibit RV dilation, and its detection, by echocardiography is useful for risk stratification of MPE patient [5].
Echocardiographic findings based on impaired RV ejection pattern or decreased contractility of the RV free wall compared with the RV apex are associated with a high positive predictive value for MPE patients even in the presence of cardiorespiratory disease. Additional echocardiographic evidence of pressure overload may be required to avoid false diagnosis of MPE in patients with RV free wall hypokinesia or akinesia secondary to RV infarction [93]. New echocardiographic parameters of RV function such as Doppler Tissue Imaging and wall strain are non-specific and are affected by the presence of acute PE [94]. RV overload or dysfunction excludes PE as the cause of hemodynamic instability and echocardiography may be useful in differential diagnosis of the cause of shock by detecting pericardial tamponade, acute valvular dysfunction, severe global or regional LV dysfunction, aortic dissection, or hypovolemia [95]. In highly unstable patients, echocardiography evidence of RV dysfunction with or without visualization of right heart thrombi is sufficient to prompt reperfusion without further testing [96,97].
Other imaging tests: For nearly three decades, ventilation-perfusion lung scintigraphy was a safe and favored non-invasive method allowing indirect diagnosis of MPE based on mismatch defect (perfusion defect with normal ventilation) usually representing PE. The basis of lung scintigraphy is an intravenous injection of technetium (Tc)-99m-labelled macroaggregated albumin particles that block a small fraction of capillaries to allow scintigraphy assessment of lung perfusion. In hypoperfused segments, ventilation is expected to be normal [5]. However, its main limitation were high frequency of intermediate or inconclusive findings, low-probability studies do not exclude MPE, prolonged time required to obtain scans, frequency of inaccessibility of the technology and ambiguous reporting method [68]. Pulmonary angiography was the reference standard for the diagnosis or exclusion of MPE but its use is limited since the less-invasive CTA provides similar diagnostic accuracy [98]. Magnetic resonance angiography although showing promising results, is not yet ready for clinical practice because of low availability in emergency settings, low sensitivity, and high proportion of inconclusive MRA scans [5].
Meta-analysis of clinical scoring and d-dimer for pe exclusion
The diagnosis of MPE remains a common challenge confronting physicians in daily clinical practice. MPE is considered in the differential diagnosis of many clinical presentations and in a wide variety of clinical settings. In particular, non-specificity and non-sensitivity of clinical signs and symptoms has led to a low threshold for suspicion and increased referral for CT angiography [99]. However, only a small proportion (~10-15%) of all suspected cases are confirmed during CTA diagnostic investigation [64]. To improve the selection of patients for additional diagnosis, the utility of clinical scoring systems has been recommended and are widely used [57]. The present meta-analysis seeks to evaluate the accuracy of Wells score or revised Geneva score combined with D-dimer tests in the selection of patients for diagnostic CTA investigation.
Study search and inclusion criteria: Search for studies investigating the value of clinical scoring systems combined with D-dimer tests to exclude PE in suspected patients was conducted on online databases: PubMed and EMBASE. A combination of the following subject headings “Wells score” OR “Geneva scores”, AND “D-dimer tests”, AND “pulmonary embolism” OR “thromboembolism” OR “thrombosis”. Also searched were references lists of the included studies and review articles. The following inclusion criteria was applied to select eligible studies. Studies that (a) were prospective or retrospective clinical trials; (b) investigated Wells score or revised Geneva scores in combination with D-dimer tests to exclude PE in suspected patients; (c) used CT angiography, ultrasonography or ventriculography, as the reference standard; and (d) reported quantitative outcomes including number of patients excluded using a combination of clinical rules and D-dimer tests, false positive detected using reference standard and of thromboembolic events of the excluded patients during follow-up. Case series or reports, editorial and conference papers were excluded. There was no restriction on publication language.
The following data was extracted from the individual studies included in this meta-analysis: study details (first author, publication year, and country the study was conducted), patient characteristics (sample size, mean age and proportion of male patients), clinical characteristics (type of scoring system and D-dimer test) and clinical outcomes (prevalence of in PE population, clinical probability, patients excluded, positive diagnosis (false positive) and thromboembolic events at 3 to 6 months follow-up) (Table 5). Two investigators reviewed all data and resolved any disagreement by consensus. The primary outcome was negative prediction (exclusion) of PE in patients, secondary outcomes was positive prediction (selection) of patients for additional CTA diagnostic investigation and thromboembolic events within three to six months. Summary estimates were calculated for all studies as well as weighted outcomes, their 95% clearance intervals and heterogeneity test (I2).
Table 5. Summary of studies on MPE diagnosis
1st Author |
Year |
Country |
Sample Size (N) |
Female (n) |
Mean Age (yrs.) |
Reference Standard |
Scoring & D-dimer Test |
Prevalence of PE n(%) |
Clinical Probability |
Patients Excluded |
Positive Diagnosis |
T-Events @Follow-up |
Wells et al. [75] |
2001 |
Canada |
930 |
583 |
50.5 |
Pulmonary angiography |
Wells & SimpliRED |
86 (9.5) |
Low probability |
437 |
1 |
1 |
Kruip et al. [100] |
2001 |
Netherlands |
234 |
150 |
51.0 |
Ultrasonography, Pulmonary angiography |
Wells & VIDAS |
52 (22.0) |
Low probability |
147 |
25 |
0 |
Perrier et al. [81] |
2004 |
Switzerland/ France |
965 |
562 |
61.0 |
CT pulmonary angiography |
Geneva & ELISA or VIDAS |
222 (23) |
Low probability |
280 |
NR |
0 |
Guo et al. [101] |
2005 |
China ≥ 65 |
196 |
96 |
76.1 |
CT pulmonary angiography |
Wells & turbidimetric |
56 (28.6) |
Unlikely |
58 |
0 |
NR |
|
|
< 65 |
140 |
59 |
48.0 |
CT pulmonary angiography |
Wells & turbidimetric |
40 (28.6) |
Unlikely |
42 |
0 |
NR |
Ten Wolde et al. [102] |
2004 |
Netherlands |
631 |
378 |
53.0 |
Perfusion lung scintigraphy or ultrasonography |
Wells & Tinaquant |
NR |
Low probability (< 20%) |
95 |
0 |
0 |
Kearon et al [73] |
2006 |
Canada |
1126 |
357 |
57.0 |
Pulmonary angiography |
Wells & SimpliRED |
171 (15.2) |
Low probability |
186 |
4 |
1 |
Page [66] |
2006 |
Netherlands |
3306 |
1897 |
53.0 |
CT Pulmonary angiography |
Wells & VIDAS or Tinaquant |
674 (20.4) |
Unlikely |
1057 |
4 |
5 |
Lucassen et al. [103] |
2010 |
Netherlands |
2701 |
NR |
NR |
Pulmonary angiography |
Wells & SimpliRED |
545(20.2) |
Unlikely |
1876 |
50 |
NR |
Geersing et al. [104] |
2012 |
Netherlands |
598 |
425 |
48 |
Spiral CT, pulmonary angiography |
Wells & SimpliRED |
73 (12.2) |
Unlikely |
272 |
4 |
NR |
Study characteristics and outcomes: Our search for pertinent studies on clinical scoring system and D-dimer tests for the exclusion of PE in suspected patients yielded 594 studies. Of these studies, 573 were excluded because they failed to meet the inclusion criteria. Of the 21 eligible studies, full text screening excluded a further 12 studies because they did not investigate combined utility of clinical scoring and D-dimer tests. Thus, nine (9) studies published between 2001 and 2012 were finally included in this meta-analysis [66,73,75,81,100-104]. A majority of studies were conducted in Netherlands and others in Canada, Switzerland/France and China. One study [101] divided patients into two (2) cohorts: the elderly (≥ 65%) and non-elderly (< 65 years). The two groups were analyzed separately. The main reference standard used were CT pulmonary angiography and perfusion lung/leg ultrasonography. All the studies used Wells score except one study [81] that used Geneva score. The studies used either PE likely or unlikely [66,101,103,104], or low, moderate (intermediate) or high scoring levels [73,75,81,100-102] to exclude patients. The most common D-dimer test used was simpliRED [73,75,103,104], VIDAS [66,81,100] and tinaquant [66,102] while one study used turbidimetric immunoassay [101].
The total patient population enrolled in the nine studies was 10,827. In eight studies that provided patient characteristics [66,73,75,81,100-102,104] enrolling 8,126 patients, there was an almost equal gender representation, with 4,507 female patients (55%), and the mean age was 55.3 years. In eight studies [66,73,75,81,100,101,103,104] that reported prevalence of PE, the overall prevalence was 18.62% (95% CI: 17.87-19.39). The efficiency of the combined utility of Wells or Geneva score and D-dimer tests based on the number of patients excluded (4,178) was 40.55% (95% CI: 39.620 to 41.478). Of these 4,178 patients, 88 cases of PE was confirmed by CT pulmonary angiography: false negative rate of 4.56% (95% CI: 3.95 to 5.24). In six studies [66,7375,81,100,102] reporting thromboembolic events in the excluded patients within three to six months post randomization, 7 patients had either fatal or non-fatal thromboembolic events, translating into 0.39% (95% CI: 0.17 to 0.69) (Table 6). Our findings reveal that Wells or Geneva scoring system used in combination with a negative D-dimer test results can safely exclude PE from the need of additional diagnostic imaging tests.
Table 6. Weighted mean of prevalence, excluded patients and false negative rate
Variables (Weighted Mean) |
Weighted Mean |
95% CI |
Prevalence of PE in the enrolled population |
18.62 |
17.87 to 19.39 |
Efficiency (No. of PE patients excluded) |
40.55 |
39.62 to 41.48 |
False negative rate (Cases diagnosed) |
4.56 |
3.95 to 5.24 |
Thromboembolic events within 3-6 months |
0.39 |
0.17 to 0.69 |
Discussion: The number of patients with suspected PE referred for hospital emergency unit for CT angiographic diagnostic test is on the increase but the proportion of whom diagnosis is confirmed is decreasing. The main challenge in the diagnosis workout of clinically suspected PE is accurate and rapid identification of ~25% of patients with PE and requires PE-specific reperfusion or anticoagulation therapy from ~75% who would not benefit from PE-specific treatment [66]. Thus, a more safe and efficient diagnostic strategy is needed. While clinical scoring systems such as Wells and Geneva, and point of care D-dimer tests have been independently shown to exclude patients from further diagnostic tests or the need for PE-specific treatment, the accuracy and efficiency of their combined use has not been well demonstrated.
The present meta-analysis investigated the combined effect of scoring system with D-dime tests in the exclusion of PE patients from additions angiographic diagnostic tests. Our findings reveal that Wells score of < 4 combined with point of care negative D-dimer (< 500 ng/ml) results is efficient – is able to classify about 40% of patients suspected with PE at low risk or unlikely to have the condition. In addition, the failure rate of the method is low, about 4.6% are diagnosed with PE on CT angiography. The exclusion method is also safe, since only about 0.39% of patients will develop thromboembolic within three to six months after exclusion.
The present results are in accordance with studies done in primary and secondary care as documented in two previous systematic reviews on the performance of several clinical scoring systems including the Wells and Geneva [105,106]. Analysis of five models of clinical scoring (original Wells, modified Wells, simplified Wells, revised Geneva, and simplified revised Geneva) are accurate and applicable in primary care settings. They have a high sensitivity (88% to 96%) and specificity (48% to 53%); efficiency (43% to 48%) and low failure rates (1.2% to 3.1%) [105]. The low values of specificity means they are unsuitable for diagnosing PE. In a systematic analysis of 11 studies with 6,837 VTE patients, the combination of clinical scoring system and D-dimer is safe. The overall rate of thromboembolic events in the excluded patients (2,056) was nine (0.44%). The findings demonstrate that withholding PE-specific treatment in patients suspected with PE based on Wells score < 4 and negative D-dimer test results is safe [106]. Other studies have also reported the safety of low clinical probability of PE in combination with normal D-dimer test results for the exclusion of PE [107,108].
The present findings have important clinical implications. An increasing number of patients are referred to emergency units with a clinical suspicion of PE but the proportion confirmed remains low. In addition, if left untreated, PE could be fatal or cause considerable morbidity [5]. While thrombolysis or anticoagulant reduces morbidity and mortality, unnecessary treatment should be avoided due to an elevated risk of bleeding [106]. Based on the present findings, primary care physician can safely exclude PE in a large proportion of patients thereby reducing their need for PE-specific treatment (thrombolysis or anticoagulation). In addition, it allows primary care facilities to reduce the cost and burden to the patient associated with unnecessary referral to secondary care facilities. Although the present findings only identify low-risk patients, additional studies are required to investigate if moderate or intermediate risk patients could also benefit from exclusion from treatment based on clinical scoring system and negative or normal D-dimer test results.
Low cardiac output is the primary cause of death in MPE patients. Short-term mortality increases based on the extent of hemodynamic insult due to the obstruction to RV outflow. The choice of initial therapy should depend on the severity of hemodynamic insult [2].
Treatment strategies
Patients with MPE are at an elevated risk of in-hospital death, in particular during the first few hours post-admission. Initial treatment strategies is hemodynamic and respiratory support with concomitant anticoagulation. Thrombolytic therapy is the recommended choice for high-risk MPE patients. In patients contraindicated to thrombolytic therapy or failure to improve hemodynamic status, surgical embolectomy is recommended if expertise and resources are available. Alternative to surgical embolectomy, percutaneous catheter-directed therapy should be considered if expertise and resources are available. In such cases, an inter-disciplinary team involving thoracic surgeons or interventional cardiologists should make treatment decision [12].
Hemodynamic and respiratory support: Low cardiac output secondary to acute RV failure is the leading cause of death in high-risk MPE patients. Thus, the primary therapeutic target is hemodynamic and respiratory support through reperfusion. In MPE patients with low cardiac output and normal BP, a fluid bolus therapy (500mL) is beneficial to increase cardiac output [109].
Vasopressors as either monotherapy or dual therapy with pharmacological, surgical or interventional reperfusion treatment are necessary in MPE patients. Vasopressors such as norepinephrine improves RV function through its direct positive inotropic effect to improve RV function, improves RV coronary perfusion through peripheral vascular alpha-receptor stimulation and increased systemic BP [109]. However, its use is limited to hypotensive patients. Dobutamine and/or dopamine may be considered for MPE patients with low cardiac index and normal BP. However, cardiac index should not be increased above physiological values because of the risk of aggravating the ventilation-perfusion mismatch by redistributing flow from obstructed to unobstructed vessels [110]. Epinephrine has been found to combine the beneficial effects of norepinephrine and dobutamine without the systemic vasodilatory effect of dobutamine and thus recommended in MPE patients [2].
Vasodilators are recommended for MPE patients to decrease pulmonary arterial pressure and vascular resistance but lack specificity for pulmonary vasculature after intravenous administration. Vasodilators such as inhaled nitric oxide has been shown to improve hemodynamic status and respiration in MPE patients [111,112] while levosimendan restores RV-pulmonary arterial coupling in MPE via combining vasodilation with increased RV contractility [113,114]. Hypoxemia and hypocapnia are common in MPE patients but mostly of moderate severity. Oxygen administration reverses hypoxemia. However, mechanism ventilation induces positive intrathoracic pressure and may decrease venous return to worsen RV failure in MPE patients. Thus, positive end-expiratory pressure should be applied with caution to limit its adverse hemodynamic effects [5].
Anticoagulation therapy: The 2011 AHA guidelines on PE recommends that unless there is a strong contraindication, anticoagulation should be recommended in all patients with MPE. The objective is to prevent early death and recurrent symptomatic VTE [12]. Anticoagulant options for acute phase include low molecular weight heparin (LMWH), intravenous unfractionated heparin (UFH) or subcutaneous fondaparinux for the first 5 to 10 days [12]. The 2014 ESC PE guidelines [5] also recommends parenteral heparin should overlap with the initiation of vitamin K antagonist (VKA) or followed by the administration of one of the novel anticoagulants such as dabigatran or edoxaban. However, if oral rivaroxaban or apixaban is administered, they should be started directly or after 1 to 3 days of administration of UFH, LWMH or fondaparinux with increased doses over three weeks (rivaroxaban) or 7 days (apixaban).
Thrombolytic therapy: Compared to passive reduction of thrombus size by heparin, thrombolytic agents actively promote the hydrolysis of fibrin molecules to achieve a more rapid restoration of pulmonary perfusion [115,116]. Early reduction of pulmonary obstruction results in prompt reduction in pulmonary artery pressure and resistance in turn leading to improvement in RV function [117]. Other important benefits include rapid resolution of symptoms – dyspnea, chest pain, and psychological distress – respiratory stabilization and cardiovascular function without the need for mechanical ventilation or vasopressor support, decreased RV dysfunction, improved exercise tolerance, prevention of PE recurrence and increased probability of survival [12]. However, the benefits of thrombolytic therapy is limited to the first few days and the does not appear after seven days after initiation of treatment. Echocardiographic evidence of improvement of RV function within 36 hours appears in almost all patients (90%) [118]. Prompt initiation of therapy, within 48 hours of symptom onset, conveys the greatest benefit to MPE patients but it also useful for MPE patients who have had symptoms for between 6 and 14 days [119]. Thrombolytic therapy also provides a protective effect against cardiac death, in-hospital mortality or recurrent PE in MPE patients [120,121]. On the other hand, thrombolytic therapy has been associated with an elevated risk of major bleeding including intracranial in 1.9 to 2.2% of the MPE patients [122,123], which worsens with increasing age and in the presence of comorbidities [124]. The current evidence strongly suggest the need for improving the safety of thrombolytic therapy in patients at an elevated risk of life-threatening bleeding.
Pulmonary embolectomy: Pulmonary embolectomy refers to surgical removal of obstructive emboli on pulmonary arterial vasculature and the institution of cardiopulmonary bypass (CPB) usually with bicaval venous cannulation. To minimize myocardial ischemia, the procedure is performed using normothermic CPB on a beating heart without cross clamping the aorta. Surgical embolectomy is indicated for high-risk MPE patients with contraindication to thrombolytic therapy due to potential hemorrhagic complications or patients with persistence hemodynamic compromise or RV dysfunction despite thrombolytic therapy [2,125]. Surgical embolectomy is also indicated in patients with free-floating thrombus within the right atrium, RV or with impending paradoxic embolism through a patent foramen ovale [126,127]. Prior to surgical embolectomy, it is important to radiologically identify a centrally accessible PE within the main pulmonary trunk or left/right main pulmonary artery since patients with most thrombus burden located peripherally do not benefit from surgery [2].
Catheter-directed thrombectomy: Catheter-directed thrombectomy is an alternative therapeutic strategy that could be used for treatment of MPE patients with hemodynamic instability. It is recommended in patients with MPE contraindicated for thrombolytic therapy or has failed to improve RV function and surgical intervention is not readily available or contraindicated [13]. The therapeutic target of catheter-directed thrombectomy is rapid resolution of pulmonary and systemic perfusion by lysing large central occlusive thrombus to reduce RV afterload and strain. The fragmentation process redistributes thrombus into multiple smaller branches further downstream in a larger volume of the peripheral arterial tree [128] and in the process increases the surface area for the exposure of the fibrinolytic agent (intrinsic thrombolytic enzymes) causing thrombus dissolution [129]. Catheter-directed thrombectomy is performed through the femoral vein to disrupt thrombus using rheolytic or rotational techniques in combination with aspiration of the thrombus [2]. Complications of catheter-directed thrombectomy include thrombus embolization, perforation or dissection of the pulmonary artery, injury to the RG, arrhythmias, pulmonary hemorrhage, pericardial tamponade and femora vein injury. To minimize the risk of perforation, only pulmonary artery branches > 6 mm should be treated and the procedure stopped upon improvement of hemodynamic status [130].
Meta-analysis of thrombolysis and anticoagulant therapies
The primary treatment target for MPE patients is pulmonary reperfusion. Thrombolytic treatment has been shown to result in a more rapid resolution of emboli in the pulmonary vasculature than anticoagulation treatment [118]. However, whether this rapid resolution translates into improved clinical outcomes compared to anticoagulation alone remains unclear. This meta-analysis seeks to compare the efficacy and safety of thrombolytic therapy and anticoagulation in patients diagnosed with MPE.
Study search and inclusion criteria: Randomized clinical trials investigating treatment outcomes of thrombolytic and anticoagulation therapies on patients with PE were identified through computer-aided search on online databases MEDLINE, EMBASE, the Cochrane Register of Controlled Trials and CINAHL databases, until September 2018. Additional studies were identified through scrutiny of bibliographies of the included studies and review articles. The key search terms used were pulmonary embolism, thrombolytic therapy, thrombolysis and anticoagulation (heparin, vitamin K antagonist, dabigatran, edoxaban, rivaroxaban or apixaban).
Trials were eligible if they met the following criteria: (a) recruited patients diagnosed with PE; (b) compared thrombolytic and anticoagulation therapies; (c) were followed up for a minimum of 30 days (1 month); and (d) provided data on major clinical endpoints: major bleeding, death or recurrence of PE. There was no limitation to publication language date, patient age or gender. Two investigators independently reviewed individual trials for inclusion and extracted data from the included studies. Any discrepancy between the results obtained by the two investigators regarding inclusion and data extraction was resolved through consensus. Data extracted from the individual studies were author, publication year, patient population, mean age, proportion of female patients, and incidence of death, PE recurrence and major bleeding events (Table 7).
Table 7. Summary of data on studies comparing thrombolysis and anticoagulation therapy
1st Author |
Year |
PE Status |
No. of Patients |
Mean Age |
Men (%) |
Thrombolytic Agent |
Anticoagulant |
FUP (Months) |
Thrombolytic Therapy (n) |
Anticoagulation |
|
|
|
TP |
AC |
|
|
|
|
|
Death |
RPE |
MB |
Death |
RPE |
MB |
Konstantinides et al. [131] |
2017 |
Stable |
359 |
350 |
66 |
46 |
Tenecteplase |
UFH |
38 |
73 |
0 |
1 |
63 |
2 |
0 |
Kline et al. [132] |
2014 |
Stable |
40 |
34 |
57 |
59 |
Tenecteplase |
LWMH, enoxaparin or dalteparin |
3 |
1 |
1 |
NR |
1 |
1 |
NR |
Meyer et al. [133] |
2014 |
Stable |
506 |
499 |
66 |
70 |
Tenecteplase |
Heparin |
1 |
12 |
1 |
58 |
16 |
5 |
12 |
Sharifi et al. [134] |
2013 |
Stable |
61 |
60 |
58 |
45 |
Low dose TPA |
UFH or enoxaparin |
28 |
1 |
0 |
0 |
3 |
3 |
0 |
Fasullo et al. [135] |
2011 |
Stable |
37 |
35 |
57 |
57 |
Alteplase |
UFH |
6 |
0 |
2 |
0 |
6 |
1 |
1 |
Becattini et al.[116] |
2010 |
Stable |
28 |
30 |
64 |
40 |
Tenecteplase |
UFH |
1 |
0 |
1 |
2 |
0 |
1 |
1 |
Konstantinides et al. [136] |
2002 |
Stable |
118 |
138 |
61 |
48 |
Alteplase |
UFH |
1 |
4 |
4 |
1 |
3 |
4 |
5 |
Jerjes-Sanchez et al. [137] |
1995 |
Unstable |
4 |
4 |
51 |
63 |
Streptokinase |
Heparin |
24 |
NR |
NR |
0 |
NR |
NR |
4 |
Dalla-Volta et al. [114] |
1992 |
Stable |
20 |
16 |
33 |
65 |
Alteplase |
Heparin |
1 |
2 |
NR |
3 |
1 |
NR |
2 |
AC: Anticoagulant; FUP: Follow-up Period; MB: Major Bleeding; NR: Not Reported PE: Pulmonary Embolism; RPE: Recurrent PE; TP: Thrombolytic Therapy; TPA: Tissue Plasminogen Activator; UFH: Unfractionated Heparin
The primary clinical outcomes investigated in this meta-analysis was the incidence of major clinical outcomes events: death, major bleeding events and/or recurrence of PE. Major bleeding events were reviewed and the International Society of Hemostasis and Thrombosis (ISTH) criteria for major bleeding were applied if sufficient information was available. Otherwise, major bleeding events were defined according to the original study we calculated odds ratio (OR) and associated 95% CI using the fixed-effect or random-effect model based on outcome of heterogeneity (I2) test.
Study characteristics and outcomes: The search for pertinent studies identified 656 potentially eligible studies. After scanning titles and abstracts, 24 studies were retained for full-text evaluation against the inclusion criteria. Studied that used animal models, non-randomization, and/or compared thrombolytic regimes or old versus new anticoagulants were excluded. Finally, nine (9) studied published between 1992 and 2017 were included in this meta-analysis [114,116,131-137].
While the intention of this meta-analysis was evaluating MPE patients, only one study specifically compared the safety and efficacy of thrombolysis and anticoagulation [137] in patients diagnosed with MPE while the rest of the studies recruited PE patients with an acute onset of symptoms but hemodynamically stable. The combined patient population in the nine studies was 2,339 PE patients randomized into thrombolytic therapy (1,173) and anticoagulation therapy (1,166). Mean age at presentation was 57 years. There was almost equal gender representation (male patients = 55%). The mean follow- up period was 11 months (range 1 month [114,116,132,136] to 38 months [131].
Pooled analysis of data from the individual studies reveals thrombolytic therapy has comparable efficacy with anticoagulation measured using death (OR, 0.987; 95% CI, 0.675-1.443, p = 0.944, 12 = 5.98%) (Figure 3) and recurrent PE (OR, 0.655; 95% CI, 0.278-1.543, p = 0.333, 12 = 0.0%) (Figure 4). However, thrombolytic therapy had significantly reduced patient safety indicated by major bleeding events (OR, 2.635; 95% CI, 1.647-4.214, p < 0.001, 12 = 67.16%) (Figure 5). Measure for heterogeneity indicated significant variation between individual studies possible due to larder differences in follow-up period, duration of hospitalization, dosage and types of anticoagulants used in combination with thrombolytic agents.
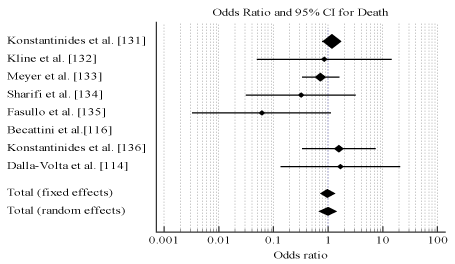
Figure 3.Odds ratio for death between thrombolysis and anticoagulation therapies.
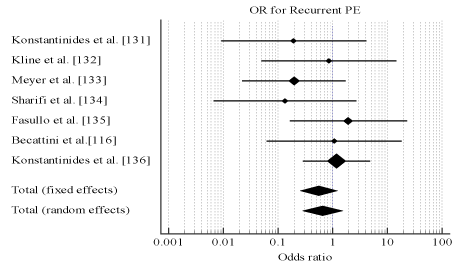
Figure 4.Odds ratio for recurrent pe between thrombolysis and anticoagulation therapies.
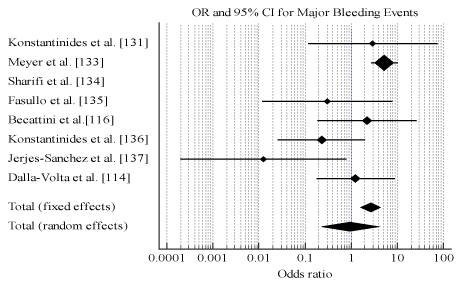
Figure 5.Odds ratio for major bleeding between thrombolysis and anticoagulation.
Convincing evidence of thrombolytic agents achieving a much faster lysis of emboli and improved hemodynamics compared to a monotherapy of heparin informed the approval of tenecteplase streptokinase or alteplase in combination with anticoagulation in the treatment of patients with acute PE with hemodynamic instability. However, evidence regarding the effect of thrombolytic therapy on mortality and major bleeding events in the long-term has remained inconclusive [138-141]. The present meta-analysis of randomized clinical trials provide no significant evidence on the efficacy of thrombolytic therapy relative to heparin for the initial treatment of selected patients with acute PE. Thrombolytic therapy has comparable efficacy to anticoagulation in the prevention of death or recurrence of PE. However, thrombolytic therapy causes safety issues – significantly increased risk of major bleeding events – compared to anticoagulation therapy alone. Two previous meta-analysis have provided similar findings. Thrombolytic therapy reduces mortality and recurrence of PE but the reduction is non-significant. On the other hand, thrombolytic therapy increases the risk of both major and non-major bleeding events [142,143].
However, the present findings should be interpreted with caution when applied to MPE patients. Only one study [137] recruited MPE patients with cardiogenic shock. The study was terminated after enrolling eight patients, four in each arm of the study, and reported a significant difference in mortality. All the four MPE patients in the thrombolytic arm survived (/0% mortality) while all the four in the heparin group died (100% mortality). Thus, thrombolytic therapy is considered the standard care for MPE patients with hemodynamic instability and cardiogenic shock. Studies on the safety of thrombolysis in MPE patients are not forthcoming due to ethical consideration in MPE cohort. Thrombolytic agents and regimen used in the individual studies differ with regard to choice, dose and length of treatment, while some agents used in earlier studies are no longer in use presently. Finally, most of the studies provided information on short-term clinical outcomes and data on the long-term effect was not sufficient to determine the effect on death, PE recurrence and bleeding events in the long-term.
The present findings have important clinical implications. Systemic use of thrombolytic therapy should not be recommended as the initial therapy for PE patients with RV dysfunction but hemodynamically stable because of increased risk of major bleeding events and non-superiority in the short-term prevention of recurrent PE or death. However, for, MPE patients who are hemodynamically unstable they may benefit from thrombolytic therapy although available evidence is insufficient for a definitive determination. In the absence of high quality data, leading international heart associations such as the AHA [12] and the ESC [13] have published in their guidelines common absolute contraindication to systemic thrombolytic therapy based on major risks to bleeding including hemorrhagic or ischemia stroke, neoplasms, surgery within three weeks, gastrointestinal hemorrhage within four weeks, and other known bleeding disorders. These contraindications focus on MPE patients with high-risk of bleeding.
Heart failure secondary to massive pulmonary embolism (MPE) is a consequence of sustained hypotension and cardiogenic shock in the absence of other potential causes such as arrhythmias, hypovolemia, sepsis or left ventricular (LV) dysfunction, pulselessness or persistence profound bradycardia. Individuals with the Virchow’s triad of venous stasis, local injury to the vascular wall and hypercoagulability state are at an elevated risk of MPE. The cardinal pathophysiological mechanisms for MPE are circulatory and respiratory failure. In circulatory failure, anatomical occlusion of the pulmonary vasculature and neurohormonal effects causes an increase in RV vascular wall stress and oxygen consumption leading to cardiac ischemic, RV dysfunction and ultimately decreased LV output. In respiratory failure, occlusion of pulmonary vasculature leads to increased alveolar dead space, right-to-left shunting, ventilation-perfusion mismatch and a low mixed venous oxygen level causing hypoxia and hypocapnia. The most frequently encountered clinical symptoms are severe dyspnea at rest, syncope or cardiac arrest. Non-specific signs and symptoms, and the lack of a reliable non-invasive imaging test complicate diagnosis. Diagnosis is based on the need for PE-specific treatment. Clinical scoring system such as the Wells score or revised Geneva score use a combination of clinical features and risk factors to predict patients suspected to have MPE. In addition to scoring system, CT pulmonary angiography usually confirms the diagnosis of MPE. Clinical management targets improving pulmonary perfusion through hemodynamic and respiratory support, and resolution of thrombi using thrombolytic or anticoagulation therapy.
- Konstantinides SV, Barco S, Lankeit M, Meyer G (2016) Management of pulmonary embolism: an update. J Am Coll Cardiol 67: 976-990. [Crossref]
- Moorjani N, Price S (2013) Massive pulmonary embolism. Cardiol Clin 31: 503-518. [Crossref]
- Kostadima E, Zakynthinos E (2004) Pulmonary embolism: pathophysiology, diagnosis, treatment. Hellenic J Cardiol 48: 94-107. [Crossref]
- Sekhri V, Mehta N, Rawat N, Lehrman SG, Aronow WS (2012) Management of massive and nonmassive pulmonary embolism. Arch Med Sci 8: 957-969. [Crossref]
- Konstantinides SV, Torbicki A, Agnelli G, Danchin N, Fitzmaurice D, et al. (2014) 2014 ESC Guidelines on the diagnosis and management of acute pulmonary embolism: The Task Force for the Diagnosis and Management of Acute Pulmonary Embolism of the European Society of Cardiology (ESC) Endorsed by the European Respiratory Society (ERS). Eur Heart J 35: 3033-3073. [Crossref]
- Miller GA, Sutton GC, Kerr IH, Gibson RV, Honey M (1971) Comparison of streptokinase and heparin in treatment of isolated acute massive pulmonary embolism. Br Med J 2: 681-684. [Crossref]
- Goldhaber SZ, Visani L, De Rosa M (1999) Acute pulmonary embolism: clinical outcomes in the International Cooperative Pulmonary Embolism Registry (ICOPER). Lancet 353: 1386–1389 [Crossref]
- Kasper W, Konstantinides S, Geibel A, Olschewski M, Heinrich F, et al. (1997) Management strategies and determinants of outcome in acute major pulmonary embolism: results of a multicenter registry. J Am Coll Cardiol. 30: 1165-1171. [Crossref]
- Aujesky D, Obrosky DS, Stone RA, Auble TE, Perrier A, et al. (2005) Derivation and validation of a prognostic model for pulmonary embolism. Am J Respir Crit Care Med 172: 1041-1046. [Crossref]
- Wicki J, Perrier A, Perneger TV, Bounameaux H, Junod AF (2000) Predicting adverse outcome in patients with acute pulmonary embolism: a risk score. Thromb Haemost. 84:548-552. [Crossref]
- Ten Wolde M, Söhne M, Quak E, Mac Gillavry MR, Büller HR (2004) Prognostic value of echocardiographically assessed right ventricular dysfunction in patients with pulmonary embolism. Arch Intern 164: 1685-1689. [Crossref]
- Jaff MR, McMurtry MS, Archer SL, Cushman M, Goldenberg N, et al. (2011) Management of massive and submassive pulmonary embolism, iliofemoral deep vein thrombosis, and chronic thromboembolic pulmonary hypertension: a scientific statement from the American Heart Association. Circ 123: 1788-1830. [Crossref]
- Torbicki A, Perrier A, Konstantinides S, Agnelli G, Galiè N, et al. (2008) Guidelines on the diagnosis and management of acute pulmonary embolism: the Task Force for the Diagnosis and Management of Acute Pulmonary Embolism of the European Society of Cardiology (ESC). Eur Heart J 29: 2276–2315 [Crossref]
- Heit JA (2008) The epidemiology of venous thromboembolism in the community. Arterioscler Thromb Vasc Biol 28: 370-372. [Crossref]
- Cohen AT, Agnelli G, Anderson FA, Arcelus JI, Bergqvist D, et al. (2007) Venous thromboembolism (VTE) in Europe. The number of VTE events and associated morbidity and mortality. Thromb Haemost 98: 756-764. [Crossref]
- Fanikos J, Piazza G, Zayaruzny M, Goldhaber SZ (2009) Long-term complications of medical patients with hospital-acquired venous thromboembolism. Thromb Haemost 102: 688-693. [Crossref]
- Bonderman D, Wilkens H, Wakounig S, Schafers HJ, Jansa ,, et al. (2009) Risk factors for chronic thromboembolic pulmonary hypertension. Eur Respir J 33: 325-331. [Crossref]
- Condliffe R, Kiely DG, Gibbs JS, Corris PA, Peacock AJ, et al. (2009) Prognostic and aetiological factors in chronic thromboembolic pulmonary hypertension. Eur Respir J 33: 332-338. [Crossref]
- Klok FA, van Kralingen KW, van Dijk AP, Heyning FH, Vliegen HW, et al. (2010) Quality of life in long-term survivors of acute pulmonary embolism. Chest 138: 1432-1440. [Crossref]
- Kearon C, Akl EA, Comerota AJ, Prandoni P, Bounameaux H, Goldhaber SZ, et al. (2012) Antithrombotic therapy for VTE disease: antithrombotic therapy and prevention of thrombosis: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 141: e419S-96S. [Crossref]
- Kucher N, Goldhaber SZ (2005) Management of massive pulmonary embolism. Circ 112: e28-32. [Crossref]
- Riedel M (2001) Acute pulmonary embolism 1: pathophysiology, clinical presentation, and diagnosis. Heart 85: 229-240. [Crossref]
- Nordstrom M, Lindblad B (1998) Autopsy-verified venous thromboembolism within a defined urban population--the city of Malmo, Sweden. APMIS: acta pathologica, microbiologica, et immunologica Scandinavica 106: 378-384. [Crossref]
- Oger E, EPI-GETBO study group (2000) Incidence of venous thromboembolism: a community-based study in Western France. Thromb Haemost 84: 657-660. [Crossref]
- Silverstein MD, Heit JA, Mohr DN, Petterson TM, O'fallon WM, Melton LJ (2001) Trends in the incidence of deep vein thrombosis and pulmonary embolism: a 25-year population-based study. Arch Intern Med 158: 585-593. [Crossref]
- Kearon C, Akl EA (2014) Duration of anticoagulant therapy for deep vein thrombosis and pulmonary embolism. Blood 1: blood-2013. [Crossref]
- Anderson FA Jr, Spencer FA (2003) Risk factors for venous thromboembolism. Circ 107: I9-I16. [Crossref]
- Rogers MA, Levine DA, Blumberg N, Flanders SA, Chopra V,, et al. (2012) Triggers of hospitalization for venous thromboembolism. Circ 125: 2092–2099. [Crossref]
- Ku GH, White RH, Chew HK, Harvey DJ, Zhou H,, et al. (2009) Venous thromboembolism in patients with acute leukemia: incidence, risk factors, and effect on survival. Blood 113: 3911-3917. [Crossref]
- Chew HK, Wun T, Harvey D, Zhou H, White RH (2006) Incidence of venous thromboembolism and its effect on survival among patients with common cancers. Arch Intern Med 166: 458-464. [Crossref]
- Blom JW, Doggen CJ, Osanto S, Rosendaal FR (2005) Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA 293: 715-722. [Crossref]
- Timp JF, Braekkan SK, Versteeg HH, Cannegieter SC (2013) Epidemiology of cancer-associated venous thrombosis. Blood 122: 1712-1723. [Crossref]
- Gussoni G, Frasson S, La Regina M, Di Micco P, Monreal M (2013) Three-month mortality rate and clinical predictors in patients with venous thromboembolism and cancer. Findings from the RIETE registry. Thromb Res 131: 24-30. [Crossref]
- Blanco-Molina A, Rota LL, Di Micco P, Brenner B, Trujillo-Santos J, et al. (2010) Venous thromboembolism during pregnancy, postpartum or during contraceptive use. Thromb Haemost 103: 306-311. [Crossref]
- Blanco-Molina A, Trujillo-Santos J, Tirado R, Canas I, Riera A,, et al. (2009) Venous thromboembolism in women using hormonal contraceptives. Findings from the RIETE Registry. Thromb Haemost 101: 478-482. [Crossref]
- Pomp ER, Lenselink AM, Rosendaal FR, Doggen CJ (2008) Pregnancy, the postpartum period and prothrombotic defects: risk of venous thrombosis in the MEGA study. J Thromb Haemost 6: 632-637. [Crossref]
- Henriksson P, Westerlund E, Wallen H, Brandt L, Hovatta O,, et al. (2013) Incidence of pulmonary and venous thromboembolism in pregnancies after in vitro fertilization: cross sectional study. BMJ 346: e8632. [Crossref]
- Sweetland S, Beral V, Balkwill A, Liu B, Benson VS,, et al. (2012) Venous thromboembolism risk in relation to use of different types of post-menopausal hormone therapy in a large prospective study. J Thromb Haemost 10(11):2277-2286. [Crossref]
- Piazza G, Goldhaber SZ (2010. Venous thromboembolism and atherothrombosis: an integrated approach. Circ 121: 2146-2150. [Crossref]
- Severinsen MT, Kristensen SR, Johnsen SP, Dethlefsen C, Tjonneland A,, et al. (2009) Smoking and venous thromboembolism: a Danish follow-up study. J Thromb Haemost 7: 1297-1303. [Crossref]
- Steffen LM, Cushman M, Peacock JM, Heckbert SR, Jacobs DR Jr,, et al. (2009) Metabolic syndrome and risk of venous thromboembolism: Longitudinal Investigation of Thromboembolism Etiology. J Thromb Haemost 7: 746-751. [Crossref]
- Ageno W, Becattini C, Brighton T, Selby R, Kamphuisen PW (2008) Cardiovascular risk factors and venous thromboembolism: a meta-analysis. Circ 117: 93-102. [Crossref]
- Montecucco F, Mach F (2001) Should we focus on “venous vulnerability” instead of “plaque vulnerability” in symptomatic atherosclerotic patients? Thromb Haemost 106: 995-996. [Crossref]
- Piazza G, Goldhaber SZ, Lessard DM, Goldberg RJ, Emery C,, et al. (2011) Venous thromboembolism in patients with symptomatic atherosclerosis. Thromb Haemost 106: 1095-1102. [Crossref]
- Prandoni P, Pesavento R, Sorensen HT, Gennaro N, Dalla VF,, et al. (2009) Prevalence of heart diseases in patients with pulmonary embolism with and without peripheral venous thrombosis: findings from a cross-sectional survey. Eur J Intern Med 20: 470-473. [Crossref]
- Sorensen HT, Horvath-Puho E, Pedersen L, Baron JA, Prandoni P (2007) Venous thromboembolism and subsequent hospitalization due to acute arterial cardiovascular events: a 20-year cohort study. Lancet 370: 1773-1779. [Crossref]
- Wood KE (2002) Major pulmonary embolism: review of a pathophysiologic approach to the golden hour of hemodynamically significant pulmonary embolism. Chest 121: 877-905. [Crossref]
- Yamamoto T (2008) Management of patients with high-risk pulmonary embolism: a narrative review. J Crit Care 6: 16. [Crossref]
- Calvin Jr JE, Baer RW, Glantz SA (1985) Pulmonary artery constriction produces a greater right ventricular dynamic afterload than lung microvascular injury in the open chest dog. Circ Res 56: 40-56. [Crossref]
- Stein PD, Sabbah HN, Anbe DT, Marzilli M (1979) Performance of the failing and nonfailing right ventricle of patients with pulmonary hypertension. Am J Cardiol 44: 1050-1055. [Crossref]
- Jardin F, Dubourg O, Guéret P, Delorme G, Bourdarias JP (1987) Quantitative two-dimensional echocardiography in massive pulmonary embolism: emphasis on ventricular interdependence and leftward septal displacement. J Am Coll Cardiol 10: 1201-1206. [Crossref]
- Belenkie I, Dani R, Smith ER, Tyberg JV (1989) Effects of volume loading during experimental acute pulmonary embolism. Circ 80: 178-188. [Crossref]
- Vlahakes GJ, Turley K, Hoffman JI (1981) The pathophysiology of failure in acute right ventricular hypertension: hemodynamic and biochemical correlations. Circ 63: 87-95. [Crossref]
- Sharma GV, McIntyre KM, Sharma S, Sasahara AA (1984) Clinical and hemodynamic correlates in pulmonary embolism. Clin Chest Med 5: 421-437. [Crossref]
- Burrowes KS, Clark AR, Tawhai MH (2011) Blood flow redistribution and ventilation-perfusion mismatch during embolic pulmonary arterial occlusion. Pulmonary circulation. 1: 365-376. [Crossref]
- Pollack CV, Schreiber D, Goldhaber SZ, Slattery D, Fanikos J et al, (2011) Clinical characteristics, management, and outcomes of patients diagnosed with acute pulmonary embolism in the emergency department: initial report of EMPEROR (Multicenter Emergency Medicine Pulmonary Embolism in the Real World Registry). J Am Coll Cardiol. 57: 700-706. [Crossref]
- Righini M, Robert-Ebadi H, Le Gal G (2017) Diagnosis of acute pulmonary embolism. J. Thromb. Haemost 15: 1251-1261. [Crossref]
- White RH, Zhou H, Romano PS (2003) Incidence of symptomatic venous thromboembolism after different elective or urgent surgical procedures. Thromb Haemost 89: 446-455. [Crossref]
- Stein PD, Henry JW (1997) Clinical characteristics of patients with acute pulmonary embolism stratified according to their presenting syndromes. Chest 112: 974-979. [Crossref]
- Dalen JE, Haffajee CI, Alpert JS, Howe III JP, Ockene IS, et al. (1977) Pulmonary embolism, pulmonary hemorrhage and pulmonary infarction. N Engl J Med 296: 1431-1435. [Crossref]
- Elliott CG, Goldhaber SZ, Visani L, DeRosa M (2000) Chest radiographs in acute pulmonary embolism: results from the International Cooperative Pulmonary Embolism Registry. Chest 118: 33-38. [Crossref]
- Prandoni P, Lensing AW, Prins MH, Ciammaichella M, Perlati M, et al. (2016) Prevalence of pulmonary embolism among patients hospitalized for syncope. N Engl J Med 375: 1524-1531. [Crossref]
- Thames MD, Alpert JS, Dalen JE (1977) Syncope in patients with pulmonary embolism. JAMA 238: 2509–2511. [Crossref]
- Le Gal G, Bounameaux H (2004) Diagnosing pulmonary embolism: running after the decreasing prevalence of cases among suspected patients. J Thromb Haemost 2: 1244-1246. [Crossref]
- Righini M, Le Gal G, Aujesky D, Roy PM, Sanchez O, et al. (2008) Diagnosis of pulmonary embolism by multidetector CT alone or combined with venous ultrasonography of the leg: a randomised non-inferiority trial. The Lancet 371: 1343-1352. [Crossref]
- Page P (2006) Effectiveness of managing suspected pulmonary embolism using an algorithm combining clinical probability, D-dimer testing, and computed tomography. Jama 295: 172-179. [Crossref]
- Le Gal G, Righini M, Roy PM, Sanchez O, Aujesky D, et al. (2006) Prediction of pulmonary embolism in the emergency department: the revised Geneva score. Ann Intern. 2006 Feb 7;144(3):165-71. [Crossref]
- Pioped Investigators (1990) Value of the ventilation/perfusion scan in acute pulmonary embolism. Results of the prospective investigation of pulmonary embolism diagnosis (PIOPED). Jama 263: 2753-2759. [Crossref]
- Miniati M, Pistolesi M, Marini C, Di Ricco G, Formichi B, et al. (1996) Value of perfusion lung scan in the diagnosis of pulmonary embolism: results of the Prospective Investigative Study of Acute Pulmonary Embolism Diagnosis (PISA-PED). Am J Respir Crit Care Med 154: 1387-1393. [Crossref]
- Musset D, Parent F, Meyer G, Maître S, Girard P, et al. (1992) Diagnostic strategy for patients with suspected pulmonary embolism: a prospective multicentre outcome study. The Lancet 360: 1914-1920. [Crossref]
- Wells PS, Anderson DR, Rodger M, Ginsberg JS, Kearon C, et al. (2000) Derivation of a simple clinical model to categorize patients probability of pulmonary embolism: increasing the models utility with the SimpliRED D-dimer. Thromb Haemost 83: 416-420. [Crossref]
- Anderson DR, Kovacs MJ, Dennie C, Kovacs G, Stiell I, et al. (2005) , Dreyer J, McCarron B, Pleasance S, Burton E, Cartier Y, Wells PS. Use of spiral computed tomography contrast angiography and ultrasonography to exclude the diagnosis of pulmonary embolism in the emergency department. J Emerg Med 29: 399-404. [Crossref]
- Kearon C, Ginsberg JS, Douketis J, Turpie AG, Bates SM, et al. (2006) An evaluation of D-dimer in the diagnosis of pulmonary embolism: a randomized trial. Ann Intern Med 144: 812-821. [Crossref]
- Sohne M, Kamphuisen PW, Van Mierlo PJ, Buller HR (2005) Diagnostic strategy using a modified clinical decision rule and D-dimer test to rule out pulmonary embolism in elderly in-and outpatients. Thromb Haemost 94: 206-210. [Crossref]
- Wells PS, Anderson DR, Rodger M, Stiell I, Dreyer JF, et al. (2001) Excluding pulmonary embolism at the bedside without diagnostic imaging: management of patients with suspected pulmonary embolism presenting to the emergency department by using a simple clinical model and d-dimer. Ann Intern Med 135: 98-107. [Crossref]
- Lucassen W, Geersing GJ, Erkens PM, Reitsma JB, Moons KG, et al. (2011) Clinical decision rules for excluding pulmonary embolism: a meta-analysis Ann Intern Med 155: 448-460. [Crossref]
- Douma RA, Mos IC, Erkens PM, Nizet TA, Durian MF, et al. (2011) Performance of 4 clinical decision rules in the diagnostic management of acute pulmonary embolism: a prospective cohort study. Ann Intern Med 154: 709-718. [Crossref]
- Ceriani E, Combescure C, Le Gal G, Nendaz M, Perneger T, et al. (2010) Clinical prediction rules for pulmonary embolism: a systematic review and meta‐analysis. J Thromb Haemost 8: 957-970. [Crossref]
- Di Nisio M, Squizzato A, Rutjes AW, Buller HR, Zwinderman AH, et al. (2007) Diagnostic accuracy of D‐dimer test for exclusion of venous thromboembolism: a systematic review. J Thromb Haemost 5: 296-304. [Crossref]
- Stein PD, Hull RD, Patel KC, Olson RE, Ghali WA, et al. (2004) D-dimer for the exclusion of acute venous thrombosis and pulmonary embolism: a systematic review Ann Intern Med 140: 589-602. [Crossref]
- Perrier A, Roy PM, Aujesky D, Chagnon I, Howarth N, et al. (2004) Diagnosing pulmonary embolism in outpatients with clinical assessment, D-dimer measurement, venous ultrasound, and helical computed tomography: a multicenter management study. Am J Med 116: 291-299. [Crossref]
- Perrier A, Roy PM, Sanchez O, Le Gal G, Meyer G, et al. (2005) Multidetector-row computed tomography in suspected pulmonary embolism. N Engl J Med 352: 1760-1768. [Crossref]
- Righini M, Goehring C, Bounameaux H, Perrier A (2000) Effects of age on the performance of common diagnostic tests for pulmonary embolism. Am J Med 109: 357-361. [Crossref]
- Douma RA, Le Gal G, Sohne M, Righini M, Kamphuisen PW, et al. (2010) Potential of an age adjusted D-dimer cut-off value to improve the exclusion of pulmonary embolism in older patients: a retrospective analysis of three large cohorts. Bmj 340: c1475. [Crossref]
- Penaloza A, Roy PM, Kline J, Verschuren F, Le Gal G, et al. (2012) Performance of age‐adjusted D‐dimer cut‐off to rule out pulmonary embolism. J Thromb Haemost 10: 1291-1296. [Crossref]
- Kline JA, Webb WB, Jones AE, Hernandez-Nino J (2004) Impact of a rapid rule-out protocol for pulmonary embolism on the rate of screening, missed cases, and pulmonary vascular imaging in an urban US emergency department. Ann Emerg Med 44: 490-502. [Crossref]
- Ghaye B, Szapiro D, Mastora I, Delannoy V, Duhamel A, et al. (2001) Peripheral pulmonary arteries: how far in the lung does multi-detector row spiral CT allow analysis? Radiology 219: 629-636. [Crossref]
- Patel S, Kazerooni EA, Cascade PN (2003) Pulmonary embolism: optimization of small pulmonary artery visualization at multi-detector row CT. Radiology 227: 455-460. [Crossref]
- Remy-Jardin M, Remy J, Wattinne L, Giraud F (1992) Central pulmonary thromboembolism: diagnosis with spiral volumetric CT with the single-breath-hold technique: comparison with pulmonary angiography. Radiology 185: 381-387. [Crossref]
- Roy PM, Colombet I, Durieux P, Chatellier G, Sors H, et al. (2005) Systematic review and meta-analysis of strategies for the diagnosis of suspected pulmonary embolism. Bmj 331: 259. [Crossref]
- Grifoni S, Olivotto I, Cecchini P, Pieralli F, Camaiti A et al, (2000) Short-term clinical outcome of patients with acute pulmonary embolism, normal blood pressure, and echocardiographic right ventricular dysfunction. Circ 101: 2817-2822. [Crossref]
- Torbicki A, Kurzyna M, Ciurzynski M, Pruszczyk P, Pacho R, et al. (1999) Proximal pulmonary emboli modify right ventricular ejection patternEur Respir J 13: 616-621. [Crossref]
- Casazza F, Bongarzoni A, Capozi A, Agostoni O (2005) Regional right ventricular dysfunction in acute pulmonary embolism and right ventricular infarction. Eur J Echocardiogr 6: 11-14. [Crossref]
- Rudski LG, Lai WW, Afilalo J, Hua L, Handschumacher MD, et al. (2010) Guidelines for the echocardiographic assessment of the right heart in adults: a report from the American Society of Echocardiography: endorsed by the European Association of Echocardiography, a registered branch of the European Society of Cardiology, and the Canadian Society of Echocardiography. J Am Soc Echocardiogr 23: 685-713. [Crossref]
- Hsiao SH, Chang SM, Lee CY, Yang SH, Lin SK, et al. (2006) Usefulness of tissue Doppler parameters for identifying pulmonary embolism in patients with signs of pulmonary hypertension. Am J Cardiol 98: 685-690. [Crossref]
- Krivec B, Voga G, Zuran I, Skale R, Pareznik R et al, (1997) Diagnosis and treatment of shock due to massive pulmonary embolism: approach with Transesophageal echocardiography and intrapulmonary thrombolysis. Chest 112): 1310-1316. [Crossref]
- Pruszczyk P, Torbicki A, Kuch-Wocial A, Szulc M, Pacho R (2001) Diagnostic value of transoesophageal echocardiography in suspected haemodynamically significant pulmonary embolism. Heart 85: 628-634. [Crossref]
- Van Beek EJ, Reekers JA, Batchelor DA, Brandjes DP, Buller HR (1996) Feasibility, safety and clinical utility of angiography in patients with suspected pulmonary embolism. Eur Radiol 6: 415-419. [Crossref]
- Raji H, JavadMoosavi SA, Dastoorpoor M, Mohamadipour Z, Ghanavati SP (2018) Overuse and underuse of pulmonary CT angiography in patients with suspected pulmonary embolism. "Med J Islam Repub Iran 32: 3. [Crossref]
- Kruip MJ, Slob MJ, Schijen JH, van der Heul C, Büller HR (2002) Use of a clinical decision rule in combination with D-dimer concentration in diagnostic workup of patients with suspected pulmonary embolism: a prospective management study. Arch Intern Med 162: 1631-1635. [Crossref]
- Guo DJ, Zhao C, Zou YD, Huang XH, Hu JM, et al. (2005) Values of the Wells and revised Geneva scores combined with D-dimer in diagnosing elderly pulmonary embolism patients. Chin Med J 128: 1052-1057. [Crossref]
- Ten Wolde M, Hagen PJ, Macgillavry MR, Pollen IJ, Mairuhu AT, et al. (2004) Non‐invasive diagnostic work‐up of patients with clinically suspected pulmonary embolism; results of a management study. J Thromb Haemos 2: 1110-1117. [Crossref]
- Lucassen WA, Douma RA, Toll DB, Büller HR, van Weert HC (2010) Excluding pulmonary embolism in primary care using the Wells-rule in combination with a point-of care D-dimer test: a scenario analysis. BMC family practice 11: 64. [Crossref]
- Geersing GJ, Erkens PM, Lucassen WA, Büller HR, ten Cate H, et al. (2012) Safe exclusion of pulmonary embolism using the Wells rule and qualitative D-dimer testing in primary care: prospective cohort study. Bmj. 345: e6564. [Crossref]
- Hendriksen JM, Geersing GJ, Lucassen WA, Erkens PM, Stoffers HE, et al. (2015) Diagnostic prediction models for suspected pulmonary embolism: systematic review and independent external validation in primary care. BMJ 351: h4438. [Crossref]
- Ten Cate‐Hoek AJ, Prins MH (2005) Management studies using a combination of D‐dimer test result and clinical probability to rule out venous thromboembolism: a systematic review. J Thromb Haemos 3: 2465-2470. [Crossref]
- Wolf SJ, McCubbin TR, Feldhaus KM, Faragher JP, Adcock DM (2004) Prospective validation of Wells Criteria in the evaluation of patients with suspected pulmonary embolism. Ann Emerg Med 44: 503-510. [Crossref]
- Wells PS, Anderson DR, Rodger M, Forgie M, Kearon C, et al. (2003) Evaluation of D-dimer in the diagnosis of suspected deep-vein thrombosis. N Engl J Med 349: 1227-1235. [Crossref]
- Mercat A, Diehl JL, Meyer G, Teboul JL, Sors H (1999) Hemodynamic effects of fluid loading in acute massive pulmonary embolism. Crit Care Med 27) :540-544. [Crossref]
- Manier G, Castaing Y (1992) Influence of cardiac output on oxygen exchange in acute pulmonary embolism. Am Rev Respir Dis 145: 130-136. [Crossref]
- Capellier G, Jacques T, Balvay P, Blasco G, Belle E, et al. (1997) Inhaled nitric oxide in patients with pulmonary embolism. Intensive Care Med 23: 1089-1092. [Crossref]
- Szold O, Khoury W, Biderman P, Klausner JM, Halpern P, et al. (2006) Inhaled nitric oxide improves pulmonary functions following massive pulmonary embolism: a report of four patients and review of the literature. Lung 184: 1-5. [Crossref]
- Kerbaul F, Gariboldi V, Giorgi R, Mekkaoui C, Guieu R, et al. (2007) Effects of levosimendan on acute pulmonary embolism-induced right ventricular failure. Crit Care Med 35: 1948-1954. [Crossref]
- Dalla-Volta S, Palla A, Santolicandro A, Giuntini C, Pengo V, et al. (1992) PAIMS 2: alteplase combined with heparin versus heparin in the treatment of acute pulmonary embolism. Plasminogen activator Italian multicenter study 2. J Am Coll Cardiol 20: 520-526. [Crossref]
- Goldhaber SZ, Haire WD, Feldstein ML, Miller M, Toltzis R et al, (1993) Alteplase versus heparin in acute pulmonary embolism: randomized trial assessing right-ventricular function and pulmonary perfusion. Lancet 341: 507-511. [Crossref]
- Becattini C, Agnelli G, Salvi A, Grifoni S, Pancaldi LG, et al. (2010) Bolus tenecteplase for right ventricle dysfunction in hemodynamically stable patients with pulmonary embolism. Thromb Res 125: e82– e86. [Crossref]
- Konstantinides S, Tiede N, Geibel A, Olschewski M, Just H, et al. (1998) Comparison of alteplase versus heparin for resolution of major pulmonary embolism. Am J Cardiol 82: 966-970. [Crossref]
- Meneveau N, Seronde MF, Blonde MC, Legalery P, Didier-Petit K, et al. (2006) Management of unsuccessful thrombolysis in acute massive pulmonary embolism. Chest 129: 1043-1050. [Crossref]
- Daniels LB, Parker JA, Patel SR, Grodstein F, Goldhaber SZ (1997) Relation of duration of symptoms with response to thrombolytic therapy in pulmonary embolism. Am J Cardiol 80: 184-188. [Crossref]
- Stein PD, Matta F (2012) Thrombolytic therapy in unstable patients with acute pulmonary embolism: saves lives but underused. Am J Med 125: 465-470. [Crossref]
- Wan S, Quinlan DJ, Agnelli G, Eikelboom JW (2004) Thrombolysis compared with heparin for the initial treatment of pulmonary embolism: a meta-analysis of the randomized controlled trials. Circ 110: 744-749. [Crossref]
- Kanter DS, Mikkola KM, Patel SR, Parker JA, Goldhaber SZ (1997) Thrombolytic therapy for pulmonary embolism. Frequency of intracranial hemorrhage and associated risk factors. Chest 111: 1241-1245. [Crossref]
- Levine MN, Goldhaber SZ, Gore JM, Hirsh J, Califf RM (1995) Hemorrhagic complication of thrombolytic therapy in the treatment of myocardial infarction and venous thromboembolism. Chest 108: 291S-301S. [Crossref]
- Mikkola KM, Patel SR, Parker JA, Grodstein F, Goldhaber SZ (1997) Increasing age is a major risk factor for hemorrhagic complications after pulmonary embolism thrombolysis. Am Heart J 134: 69-72. [Crossref]
- Fukuda I, Daitoku K (2017) Surgical Embolectomy for Acute Pulmonary Thromboembolism. Ann Vasc Dis 10: 107-114. [Crossref]
- Kasper W, Geibel A, Tiede N, Just H (1992) Patent foramen ovale in patients with hemodynamically significant pulmonary embolism. The Lancet 340: 561-564. [Crossref]
- Torbicki A, Galie N, Covezzoli A, Rossi E, De Rosa M, et al. (2003) Right heart thrombi in pulmonary embolism: results from the International Cooperative Pulmonary Embolism Registry. J Am Coll Cardiol 41: 2245-2251. [Crossref]
- Girard P, Simonneau G (1999) Catheter fragmentation of pulmonary emboli. Chest 115: 1759. [Crossref]
- Kuo WT (2002) Endovascular therapy for acute pulmonary embolism. J Vasc Interv Radiol 23: 167-179. [Crossref]
- Biederer J, Charalambous N, Paulsen F, Heller M, Müller-Hülsbeck S (2006) Treatment of acute pulmonary embolism: local effects of three hydrodynamic thrombectomy devices in an ex vivo porcine model. J Endovasc Ther 13: 549-560 [Crossref]
- Konstantinides SV, Vicaut E, Danays T, Becattini C, Bertoletti L, et al. (2017). Impact of thrombolytic therapy on the long-term outcome of intermediate-risk pulmonary embolism. J Am Coll Cardiol 69: 1536-1544. [Crossref]
- Kline JA, Nordenholz KE, Courtney DM, Kabrhel C, Jones AE, et al. (2014) Treatment of submassive pulmonary embolism with tenecteplase or placebo: cardiopulmonary outcomes at 3 months: multicenter double‐blind, placebo‐controlled randomized trial. J Thromb Haemost 12: 459-468. [Crossref]
- Meyer G, Vicaut E, Danays T, Agnelli G, Becattini C, et al. (2014) Fibrinolysis for patients with intermediate-risk pulmonary embolism. N Engl J Med 370: 1402-1411. [Crossref]
- Sharifi M, Bay C, Skrocki L, Rahimi F, Mehdipour M (2013) Moderate pulmonary embolism treated with thrombolysis (from the “MOPETT” Trial). Am J Cardiol 111: 273-277. [Crossref]
- Fasullo S, Scalzo S, Maringhini G, Ganci F, Cannizzaro S, et al. (2011) Six-month echocardiographic study in patients with submassive pulmonary embolism and right ventricle dysfunction: comparison of thrombolysis with heparin. Am. J. Med. Sci 341: 33-39. [Crossref]
- Konstantinides S, Geibel A, Heusel G, Heinrich F, Kasper W (2002) Heparin plus alteplase compared with heparin alone in patients with submassive pulmonary embolism. N Engl J Med 347: 1143-1150. [Crossref]
- Jerjes-Sanchez C, Ramírez-Rivera A, de Lourdes García M, Arriaga-Nava R, Valencia S, et al. (1995) Streptokinase and heparin versus heparin alone in massive pulmonary embolism: a randomized controlled trial. J Thromb 2: 227-229. [Crossref]
- Levine M, Hirsh J, Weitz J, Cruickshank M, Neemeh J, et al. (1990) A randomized trial of a single bolus dosage regimen of recombinant tissue plasminogen activator in patients with acute pulmonary embolism. Chest 98: 1473-1479. [Crossref]
- Tibbutt DA, Davies JA, Anderson JA, Fletcher EW, Hamill J, et al. (1974) Comparison by controlled clinical trial of streptokinase and heparin in treatment of life-threatening pulmonary embolism. Br Med J 1: 343-347. [Crossref]
- Katchan BM (2000) Thrombolytic therapy for pulmonary embolism. Can J Surg 43: 411. [Crossref]
- Meneveau N, Bassand JP, Schiele F, Bouras Y, Anguenot T, et al. (1993) Safety of thrombolytic therapy in elderly patients with massive pulmonary embolism: a comparison with nonelderly patients. J Am Coll Cardiol 22: 1075-1079. [Crossref]
- Wan S, Quinlan DJ, Agnelli G, Eikelboom JW (2004) Thrombolysis compared with heparin for the initial treatment of pulmonary embolism: a meta-analysis of the randomized controlled trials. Circ 110: 744-749. [Crossref]
- Agnelli G, Becattini C, Kirschstein T (2002) Thrombolysis vs heparin in the treatment of pulmonary embolism: a clinical outcome–based meta-analysis. Arch Intern Med 162: 2537-2541. [Crossref]