Lesch–Nyhan syndrome (LNS) is a rare X - linked recessive disorder, that occurs almost exclusively in males. The disorder is caused by a deficiency of the enzyme hypoxanthine-guanine phosphoribosyltransferase (HGPRT). It is characterized by neurological and behavioral abnormalities and the overproduction of uric acid. LNS affects about 1 in 380,000 live births. The HGPRT deficiency causes a build-up of uric acid in all body fluids. Hypoxanthine degraded into xanthine and uric acid in the liver by the enzyme xanthine oxidase. The combination of increased synthesis and decreased utilization of purines leads to high levels of uric acid production. LNS is characterized by three major signs: uric acid overproduction (hyperuricemia), neurologic dysfunction, and cognitive and behavioral disturbances including self-mutilation. We present a case of a 27-day-old full term male infant, that was admitted to our department due to failure to thrive, he was not eating and growing as he should, laboratory results revealed high levels of uric acid, hypocalcaemia, renal failure with metabolic acidosis. On admission, his weight 3300-gram, low height for his age, Irritability, poor sucking, and muscle atrophy. After full investigation, diagnosis of LNS was established through biochemical analysis and molecular examinations.
Lesch–Nyhan syndrome, hypoxanthine, xanthine, xanthine oxidase, renal failure
The Lesch-Nyhan syndrome (LNS) is a rare x-linked excessive disorder of purine metabolism the gene is carried by the mother and passed on to her son, female carriers are at an increased risk for gouty arthritis but are usually otherwise unaffected. The disorder is caused by the congenital absence of hypoxanthine guanine phosphoribosyl transferase (HGPRT) [1]. The lack of HPRT causes a build-up of uric acid in all body fluids, and leads to symptoms such as delayed motor development and nervous system impairment, the most common presenting features are abnormally decreased muscle tone hypotonia and developmental delay, which are evident by three to six months of age [2,3].
Self-injuring behavior, it is rarely to be the presenting feature, but eventually develops in nearly all cases. patients affected are cognitively impaired and have behavioral disturbances that emerge between two and three years of age [4]. Overproduction of uric acid, "orange sand" in the diapers - uric acid crystalluria, it is due to HGPRT deficiency that causes a build-up of uric acid in all body fluids. Increased synthesis and decreased utilization of purines leads to high levels of uric acid production. This results in both high levels of uric acid in the blood and urine, associated with severe gout and kidney problems such as renal failure or frank hematuria-nephrolithiasis [5,6]. Clinical manifestations according to age: At birth- no apparent neurologic dysfunction. After several months-developmental, retardation and neurologic signs become apparent. Before 4 months- hypotonic, recurrent vomiting, By age of 8-12 months - extrapyramidal signs. The age at onset of self-injury may be as early as 1 year and occasionally as late as the teens [7,8].
physical examination may show: growth retardation, cognitive dysfunction and average IQ=60, all patients are wheelchair bound, self-mutilation like partial amputations of the fingers, lips, tongue, or oral mucosa, scarring from repetitive self-abrasion or hitting, in addition due to high levels of uric acid gouty arthritis and arthritic tophi may be seen. Lab Studies can show hyperuricemia, hyperuricosuria, macrocytic anemia. The diagnosis of Lesch-Nyhan syndrome may be confirmed by a thorough clinical evaluation, including a detailed patient history and specialized blood tests. Children with this disorder have abnormally high concentrations of uric acid in the blood. The absence of the HPRT enzyme in cells from any tissue confirms the diagnosis. Molecular genetic testing for the HPRT1 gene is available to determine the specific disease-causing mutation. Carrier testing for Lesch-Nyhan syndrome is possible using molecular genetic testing [7,8]. Prenatal diagnosis and preimplantation genetic diagnosis are possible if the disease-causing HPRT1 gene mutation has been identified in an affected family member. Prenatal diagnosis can also be done by enzyme analysis. Few reports about LNS patients are available. We report a case of LNS in an 27-day-old male infant, who presented with characteristic with FTT.
27-day-old infant was brought by his parents, the infant was admitted due to FTT, Weight 3300 grams, the infant had muscle atrophy and suffered from severe dehydration. The history was obtained from the mother and it revealed that during his first three weeks his nutrition was through breastfeeding only, and due to weight loss switched to a milk-based diet. After switching to formula- weight stabilized without increasing. The infant did not have fever, vomiting, or diarrhea. Pregnancy course- gestational diabetes, cesarean delivery due to breech presentation. Birth weight- 3590 grams Family History showed a 3-year-old sister suffered from low height and weight, and a brother who died at the age of two from optic glioma. Laboratory studies showed kidney failure, and metabolic acidosis, Creatinine=3.2, BUN=35, PH=7.26, PCO2=26.4, HCO3=11.7, BE=-13.4, HG=12.2, MCV=98.3. During hospitalization: Uric acid>40mg%, Phosphor>10 mg%, Calcium=6.5 mg%, albumin=3.3g%. Abdominal ultrasound - Hyperacogenic kidney with crown-shaped sediment around the kidney pelvis, suitable for uric-acid sediment (Figure 1). Abdominal x-ray: no stones in the kidney have been seen (Figure 2).
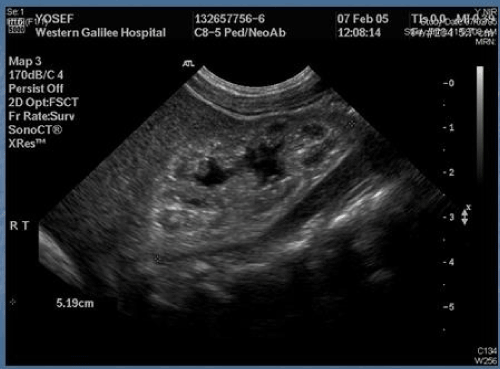
Figure 1. Abdominal ultrasound
Figure 2. Abdominal X-ray
The initial treatment aimed to reduce uric acid, and according to this hydration, Allopurinol, Sodium Bicarbonate were given. For electrolyte repair - Calcium Gluconate, Potassium Absorber. In addition, for nutrition - Similac 80 cc X 8 /D was used.
After initial treatment - CREAT=2.3, BUN=12. PH=7.39, PCO2=55, HCO3 = 32, BE=5.1, HG=8.2, MCV=98.2, Uric acid=16.5, Phosphorus=5.3, Calcium=7.9. Following two seizure events (presumably against low blood calcium), the need for a central vein line, was transferred to pediatric intensive care.
The infant was in Intensive Care for 2 days: -
A central vein was inserted, treatment continued similar to the treatment received at the department. Laboratory tests showed CREAT=2.1, BUN=10, PH=7.38, PCO2=52.5, HCO3=30, BE=4.1, HG=7.4, MCV=97.9, Uric acid=10.4, Phosphorus=4.5, Calcium=10.5.
Diagnosis of LNS is based on HPRT enzyme activity, preferably measured in live cells such as cultured fibroblasts, and on molecular genetic techniques demonstrating the gene mutation. Results might provide predictive clues about ultimate disease severity. In addition clinical and biochemical (hyperuricemia and hyperuricosuria), together with psychomotor are significant findings [8]. HGPRT deficiency leads to characteristic triad of features: hyperuricemia, neurological dysfunctions and cognitive and behavioral disturbances. Hyperuricemia may lead to formation of crystals of uric acid in the renal system which may be passed in the urine. An orange crystal in the diapers of a newborn is one of the early clues of the disease that may be the observed. Overproduction of uric acid may lead to the development of uric acid crystals or stones in the kidneys, ureters, or bladder. Such crystals deposited in joints later in the disease may produce gout-like arthritis, with swelling and tenderness.
Persons affected are cognitively impaired and have behavioral disturbances that emerge between two and three years of age. The self-injury begins with biting of the lips and tongue; as the disease progresses, affected individuals frequently develop finger biting and head banging. Hypotonia is a common feature which may render the child non-ambulatory. Within the first few years of life, extrapyramidal involvement causes abnormal involuntary muscle contractions such as loss of motor control (dystonia), writhing motions (choreoathetosis), and arching of the spine (opisthotonus) [3,4]. The definitive diagnosis of LNS is primarily based on activity of HGPRT enzyme which is decreased in the cells from all the tissues of the body. Prenatal diagnosis for LNS can be performed with amniotic cells obtained by amniocentesis at about 15-18 weeks' gestation, or chorionic villus biopsy obtained at about 10-12 weeks gestation. Treatment for LNS is symptomatic. Allopurinol treatment reduces the over production levels of uric acid. Self-injurious behavior can be managed with a various combination of physical restraints, behavioral and pharmacological treatment [9,10].
LNS is a rare disorder but it can easily be diagnosed. Patients may die from aspiration pneumonia or complications from chronic nephrolithiasis and renal failure. With optimal care, few patients live beyond 40 years and most are confined to a wheelchair. Diagnosis is suspected when psychomotor delay occurs in a patient with elevated UA in blood and urine. Undetectable HPRT enzyme activity in peripheral blood or in intact cells (erythrocyte, fibroblast) and molecular genetic testing confirm the diagnosis.
- Seegmiller JE, Rosenbloom FM, Kelly WN (1967) Enzyme defect associated with a sex linked human neurological disorder and excessive purine synthesis. Science 155: 1682-1684.
- Jinnah HA, Friedmann T (2001) Lesch-Nyhan disease and its variants. In: Scriver CR, Beaudet AL, Sly WS, et al., eds. The metabolic and molecular bases of inherited disease. New York, NY: McGraw-Hill; pp: 2537-2570.
- García Puig J, Torres Jiménez R, Mateos F, Ramos T, Arcas J, et al. (2001) The spectrum of Hypoxanthine-Guanine Phosphoribosyltransferase (HPRT) deficiency. Clinical experience based on 22 patients from 18 Spanish families. Medicine 80: 102-112.
- Anderson LT, Ernst M (1994) Self-injury in Lesch-Nyhan disease. J Autism Dev Disord 24: 67-81.
- Nyhan WL, O'Neill JL, Jinnah HA, Harris JC (2014) Lesch-Nyhan Syndrome. Gene Reviews.
- Augoustides-Savvopoulou P, Papachristou F, Fairbanks LD, Dimitrakopoulos K, Marinaki AM, et al. (2002) Partial hypoxanthine-Guanine phosphoribosyl transferase deficiency as the unsuspected cause of renal disease spanning three generations: a cautionary tale. Pediatrics 109: E17.
- Lesch M, Nyhan WL (1964) A familial disorder of uric acid metabolism and central nervous system function. Am J Med 36: 561-570.
- Jinnah HA, Ceballos-Picot I, Torres RJ, Visser JE, Schretlen DJ, et al. (2010) Lesch-Nyhan Disease international study group. Attenuated variants of Lesch-Nyhan disease. Brain 133: 671-689.
- Zilli EA, Hasselmo ME (2008) A model of behavioural treatments for self-mutilation behaviour in Lesch-Nyhan syndrome. Neuroreport 19: 459–462.
- Olson L, Houlihan D (2000) A Review of Behavioural Treatments used for Lesch-Nyhan Syndrome. Behav Modif 24: 202-222.