Ischemic heart failure (IHF) is a life-threatening cardiac condition characterized by systolic dysfunction and reduced cardiac output. It results from an imbalance between myocardial oxygen demand and supply. If left untreated, the condition could lead to disability and death. Medical or revascularization therapy potentially restores cardiac function and improves survival, thus the selection of patients who will benefit from vascularization remains an important clinical target. However, for years, research on IHF therapies lagged behind as the focus was more on functional status of the heart than on etiology. Recently, increased research has provided greater insight into myocardial viability and contributed to new strategies for identifying patients for vascularization. In this review, we aggregate published evidence on IHF definition, etiology, pathophysiology, clinical presentation, diagnosis and clinical management of IHF. The objective is to improve clinical understanding and management of IHF.
heart failure, ischemic cardiomyopathy ischemic heart disease, ischemic heart failure, ischemic left ventricular systolic dysfunction, coronary artery disease
Ischemic heart disease (IHD), the principal component of cardiovascular diseases (CVD) [1], is the single largest cause of death in developed countries and one of the leading causes of disease burden in developing countries [2]. If the condition remains undiagnosed or untreated, it eventually leads to ischemic heart failure (IHF) – a condition characterized by a weakened myocardium and reduced cardiac output [2]. It may also lead to substantial disability, loss of productivity and increased cost of healthcare [3]. Despite these serious clinical implications, IHF lacks a definite terminology [4]. Most IHF studies refer to the condition as ischemic left ventricle (LV) systolic dysfunction or ischemic cardiomyopathy [5-7]. Many other studies refer to IHF using its sequalae – ischemic heart disease (IHD) [3,8], acute coronary syndrome (ACS) [9,10], coronary heart disease (CHD) [11,12] or coronary atherosclerotic disease [13-15].
Although studies on IHF span a period of three decades, there is a lack of strong evidence in many aspects from definition to treatment. The lack of a universal terminology complicates comparison of epidemiology, diagnosis, and treatment outcomes between studies, and impacts negatively on developing a common approach to the management of HF. Furthermore, the traditional classification systems of HF have relied on the location of cardiac dysfunction (left ventricular, right ventricular or bi-ventricular), time of onset (acute or chronic), cardiac output (high-output and low-output), or functional status (systolic or diastolic) [16]. These classification systems did not consider the etiology of IHF and contributed to the lagging behind of research into the pathophysiology of IHF with the implication of the lack of approvals for new treatment for IHF for several decades [17]. However, since 2007, new insights into pathophysiologic mechanisms of IHF began to emerge leading to the development of new anti-ischemic therapies with novel mechanisms of action [17]. In this article, which includes two meta-analysis of diagnosis methods and treatment strategies of IHF, we critically review the etiology, pathophysiology, diagnosis and clinical management of IHF.
Heart failure is a syndrome characterized by a triad of cardiac abnormality, exercise intolerance, and neuro-hormonal activation caused by an insult to the myocardium from infarction, infection, toxins, genetic abnormality, hypertension or valvular diseases [16]. The definition suggests the presence of classical HF symptoms – dyspnea, fatigue and edema – are redundant in describing HF because of the lack of established evidence to suggest the onset of symptoms represents a particular pathological event. In addition, HF therapies also appear beneficial in the asymptomatic phase to suggest HF is a continuum from asymptomatic cardiac dysfunction with neurohormonal activation to symptomatic severe cardiac dysfunction with marked neurohormonal activation [16]. Although a dominant subset of HF, IHF is not a definitive clinical condition, rather considered a syndrome representing the final pathway to a heterogeneous group of cardiac conditions that lead to decreased circulation in coronary resistance vessels and ultimately reduced myocardium oxygen supply [3].
Ischemic HF is a cardiac condition resulting from cardiomyocyte hypoxia – when oxygen supply to a part of the myocardium is insufficient to meet its metabolic needs – leading to ventricular dysfunction. The clinical phenotype accompanying cardiac ischemia can be divided into acute coronary syndrome (ACS) or chronic coronary syndrome (CCS) based on the time of onset of an ischemic episode. In ACS, a sudden drop in coronary micro-vessel circulation causes a sudden decrease in myocardial oxygen supply leading to acute ischemic cardiomyocyte injury. It may result from the obstruction of myocardial perfusion by plaque injury (rapture, erosion or hemorrhage) usually superimposed on thrombosis, endothelial dysfunction or increased smooth muscle reactivity [17]. On the other hand, in CCS, there is an abrupt increase in myocardial oxygen demand with limited cardiac ability to increase myocardial oxygen supply in the setting of disorders of coagulation, endothelial or smooth muscle cell function [18]. In IHF, the consequences of the imbalance between myocardial oxygen demand and supply is more important because it leads to a weakened myocardium and reduced cardiac ability to pump increased quantity of blood concomitant with rising metabolic demands of the body [16].
The epidemiology of IHF remains understudied as well as poorly understood. However, the epidemiology of its major sequelae (IHD) has been well established, providing valuable insights into the prevalence and incidence of IHF. The World Health Organization (WHO) estimates IHD causes 7.3 million deaths and a loss of 58 million disability-adjusted life years (DALYs: the sum of years lived with disability and years of life lost) globally [19]. About 75% of global deaths and 82% of the total DALYs due to IHD occur in low- and middle-income countries. The mortality rate across developing countries varies considerably both as a proportion of CVD deaths and as a proportion total deaths [2]. According to the WHO Global Burden of Disease 2010, IHD is the leading cause of CVD deaths globally, accounting for 43% of all CVD deaths [19]. Deaths due to CVD represent 30% of all deaths but with varying rates and patterns between high- and low- to middle-income countries. The CVD-related mortality rates in high-income countries is 38% and in low to middle-income countries is 28%. The range varies significantly from a high of 58% in Eastern Europe to 10% in Sub-Saharan Africa [19]. In all regions of the WHO except Africa, IHD the leading cause of deaths [20]. However, at the beginning of the 21 Century, IHD ranked the eight leading cause of death in Africa in both men and women [21,22]. In WHO 2005 estimates, IHD-related deaths in Africa were about 361,000 and project to double by 2030. In people aged > 60 years, it is the leading cause of death in males and the second leading cause in women [23].
Anaerobic conditions limit myocardial ability to generate sufficient energy to maintain essential cardiomyocellular processes. Thus, a sufficient constant supply of oxygen matching myocardial demand is indispensable for both cardiac viability and optimal functioning [24]. Oxygen is a major determinant of myocardial gene expression. Ischemia-induced hypoxia decreases myocardial oxygen levels significantly altering cardiac gene expression patterns [25]. Oxygen also participates in the generation of nitric oxide (NO), important for determination of vascular tone and cardiac contractility. Oxygen is also central in the generation of reactive oxygen species (ROS) that participate in cell signaling or could induce irreversible cellular damage [26,27]. Oxygen is thus both vital and deleterious to cardiac function underscoring the importance of its regulation. In IHF, vascular and non-vascular conditions cause a reduction in the myocardial oxygen supply/demand ratio in the setting of increased myocardial oxygen demand or decreased myocardial oxygen supply (Figure 1).
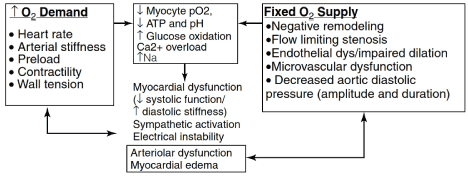
Figure 1. Pathophysiology of ischemic heart failure
The key pathophysiologic mechanisms of ischemic heart failure are increased myocardial oxygen demand and limited ability to increase myocardial oxygen supply relative to myocardial demand. Increased oxygen demand results from increased heart rate, arterial stiffness, preload, contractility and vascular wall tension. Limited ability to increase myocardial oxygen supply results from negative vascular remodeling, flow limiting stenosis, endothelial dysfunction, microvascular dysfunction, and decreased aortic diastolic pressure. Resulting myocardial oxygen demand/supply leads to myocardial dysfunction and ultimately ischemic heart failure. Adapted from Pepine & Nichols, 2007, p. I-5 [28].
Increased myocardial oxygen demand
Increased myocardial oxygen demand (the amount of oxygen required to maintain optimal cardiac function) in the absence of a concomitant increase in myocardial oxygen supply is one of the major conditions precipitating cardiac ischemia and ultimately IHF. An increase in one or more determinants of myocardial oxygen consumption leads to increased myocardial oxygen demand. The three key determinants are:
- An increase in the heart rate;
- An increase in left ventricular (LV) loading: either increased afterload due to systolic wall stress, arterial stiffness or systolic blood pressure, or increased preload due to diastolic wall stress, end diastolic pressure and volume, and wall thickness; or
- Increased contractility in the setting of heightened physical or emotional activities [28].
In addition to these three key determinants, during the actual ischemic episode, there is a secondary increase in myocardial oxygen demand but the exact mechanism underlying remains incompletely understood. This secondary increase in myocardial oxygen demand occurs in both asymptomatic and painful ischemic episodes. In painful episodes, it occurs prior to the perception of pain. Chest pain or discomfort and related symptoms may cause an increase in myocardial oxygen demand but it is not the only mechanism. The secondary increase in myocardial oxygen demand increases the magnitude or prolongs an ischemic episode and could potentially aggravate the consequences of an ischemic episode to the myocardium. Knowledge of increased myocardial oxygen demand in IHF contribute to the development of anti-ischemic therapies mainly limiting increases in myocardial oxygen demand by suppressing increases in heart rate, afterload, preload and/or contractility [28].
Reduced myocardial oxygen supply
Reduced myocardial oxygen supply refers to conditions that limit the ability of the myocardium to generate sufficient energy in the setting of a reduced coronary microvessel blood flow to the myocardium. The major conditions limiting myocardial oxygen supply and thus its ability to increase oxygen supply to match increasing cardiomyocellular demands include flow-limiting stenosis, negative vascular remodeling and coronary endothelial dysfunction [24,25].
Flow-limiting stenosis: Coronary arterial circulation consisting of conductance and resistance vessels delivers blood to the myocardium. Thus, vascular or non-vascular conditions that lead to a reduction in coronary arterial circulation contribute to the development of ischemic HF. Vascular conditions reduce myocardial oxygen supply by lowering the ceiling of myocardial blood flow through flow-limiting stenosis with insufficient collateral circulation [25]. Flow-limiting coronary stenosis mostly occurs at the epicardial vessel and at the micro-vessel level. The obstruction could be dynamic occurring as a result of altered smooth muscles negative remodeling or the rapture of platelet micro-aggregates; or fixed, occurring in the setting of atheroma, thrombus or embolus. Mostly, in chronic IHD, both dynamic and fixed obstruction are present. The extent to which flow-limiting obstruction reduces coronary circulation depends on the size of the coronary vessel lumen available for circulation, in turn determined by vascular remodeling [26].
Altered coronary reactivity: Altered coronary reactivity at the conductance and resistance vessel levels contribute to reduced myocardial oxygen supply through limiting coronary circulation. Common altered reactivity may include impaired vessel dilation in the setting of endothelial dysfunction or increased smooth muscle activation such as spasm [28]. The periodic embolization of platelet microaggreggates from roughened plaque surfaces may also contribute to flow-limiting obstruction at the micro-vessel level. Accumulating evidence supports the role of reactivity-associated flow-limiting obstruction at the coronary microcirculation level in the development of ischemic HF. It is the cause of the wide variability in effort tolerance over time; large scatter between stenosis severity and coronary flow reserve (the ratio between resting and maximal possible coronary blood flow); reduced circulation in regions perfused by non-stenotic vessels; wide variability in flow following successful stenting; and necropsy evidence for embolization in micro-vessels [26]. The role flow-limiting obstruction in ischemic heart failure supports the utility of the dilation of coronary resistance arterioles as a potential therapeutic target [29].
Coronary endothelial dysfunction: Coronary endothelium is the monolayer of cells lining the coronary vessels providing a physical barrier between the coronary vascular walls and the circulating blood. The endothelium is involved in maintaining vascular tone, regulating homeostasis and inflammation, and modulating paracellular permeability (preventing diffusion of toxic substances) [28]. Endothelial dysfunction is the inability of the endothelium to perform one or more of its functions and plays a key role in determining myocardial ischemia in all clinical manifestations of IHD [29]. Metabolic regulation of coronary circulation occurs at the resistance vessel level to match oxygen supply and demand. The secretion of free oxygen radicals mediates increased circulation in the presence of increased myocardial oxygen demand [28]. A variety of conditions such as physical, biochemical and immune-mediated injuries may damage the endothelium. Among these conditions, oxidative stress is the predominant factor producing endothelial dysfunction [29]. Conditions such as increased systolic blood pressure, low-density lipoprotein, obesity, diabetes and ageing may increase oxidant stress within the endothelium to impair the production, release or activity of nitric oxide [30]. The resulting endothelium injury negatively alters all endothelial-mediated activities such as impairs smooth muscle relaxation, stimulates smooth muscle growth, disrupts anticoagulant surface and impairs fibrinolysis, altogether termed endothelial dysfunction. The persistence of endothelial dysfunction results into negative vascular remodeling characterized by intimal thickening with atheroma formation, thus flow-limiting stenosis [31].
Disorders of the vascular smooth muscle: Disorders of the vascular smooth muscles is another mechanism proposed to cause a reduction in myocardial oxygen demand through flow-limiting obstruction in coronary vessels. Disorders such as increased vascular smooth muscle cell activity or impaired relaxation in the setting of either endothelial dysfunction or smooth muscle cell dysfunction occur in both coronary conducting and resistance vessels causing a limited flow reserve. In many patients groups such as hypertensive, diabetic, older adults, postmenopausal women and hypercholesterolemia, endothelial and smooth muscle cell dysfunction at the coronary resistance vessel level may predominate to cause ischemia or aggravate ischemic episodes in the setting of other obstructive mechanisms such as atheroma obstruction [28].
Atherothrombosis and vulnerable plaque: Stable atherothrombosis (atherosclerotic plaque disruption with superimposed thrombosis) and vulnerable plaque are other proposed IHF pathologic mechanisms leading to limited cardiac ability to increase coronary flow [32]. Atheroma formation (defined as the degeneration of arterial walls due to the accumulation of fatty deposits and scar tissues) in the presence or absence of thrombus, accounts for increases in the volumes of plaque potentially limiting coronary circulation [28]. Histologically, vulnerable plaque usually consists of a large core of extracellular lipid, a dense accumulation of macrophages, decreased numbers of vascular smooth cells and a thin fibrous cap. Plaque disruption occurs at the point where the fibrous cap is weakest and heavily infiltrated with inflammatory cells. Rapturing of the plaque exposes its highly thrombogenic, lipid-rich core with abundant tissue factor to circulating blood triggering the formation of superimposed thrombus leading to vessel occlusion and subsequent ischemic symptoms in distal areas [32]. Vulnerable plaques that are prone to rupture, erosion or inter-plaque hemorrhages with thrombus formation are pathologic mechanisms of ACS but mechanisms underlying CCS remain unclear. However, multi-vessel flow-limiting stenosis are common in CCS with a highly variable degree of lumen compromise. The coronary vessel lumen available for blood flow depends on vascular remodeling [28]. Vascular remodeling: Coronary conducting artery lumen size available for blood flow is critical to the pathophysiology of ischemic HF. Lumen size depends on vascular remodeling, a process describing the relationship between changes in atheroma volume, lumen size and external vessel size, which can be positive or negative [33-35]. The interaction of oxidative stress with vascular smooth muscles and inflammatory cells, and changes in matrix leads to vascular remodeling to determine the size of coronary vessel lumen available for circulation. Differences in vascular remodeling related to estrogen receptor alpha (ERα) expression are involved in varying smooth muscle cell phenotypes in some gender-associated differences in CAD [36]. Smooth muscle cells are responsible for extracellular matrix (ECM) synthesis and modulate the integrity of the arterial wall. Smooth muscle cells maybe decreased, apoptotic or dysfunctional in the synthesis or repair of the ECM, and in vulnerable plaque destroyed by macrophages [33].
In positive vascular remodeling, coronary vessel lumen size remains relatively the same as atheroma volume increases by compensatory remodeling – atherosclerotic mass remain external to the lumen. Atheroma volume occurs predominantly within the vascular wall accompanied with compensatory enlargement of the externa elastic membrane allowing the external vessel to enlarge and preserve the lumen size available for coronary circulation [17,35]. Compensatory remodeling is more common in ACS than in CCS [28]. Vulnerable plaque is a more common pathophysiologic mechanism in ACS while the degree of stenosis is more common in CCS, which explains the loss of compensatory remodeling as a central mechanism in CCS [28]. In negative vascular remodeling, the compensatory mechanisms become exhausted and the atheroma volume (plaque deposition) begins to compromise the coronary vessel lumen and decreases the size available for circulation. As the negative remodeling progresses, the ceiling for blood flow increases relative to increasing oxygen demand and then decreases. Because of increased atheroma volume or the exhaustion of compensatory mechanisms, or both, the coronary vascular lumen begins to narrow. Luminal narrowing of approximately 70% limits the ceiling for increasing coronary circulation in response to increased myocardial oxygen demand resulting into ischemia. Vessel shrinkage is common in CCS than in ACS suggesting a relationship between vascular distensibility and positive vascular remodeling. The absence or exhaustion of compensatory mechanisms indicates plaque stability in chronic stable ischemia [28].
Non-vascular conditions
Non-vascular conditions also contribute to the mechanisms of ischemic HF through hydraulic conditions affecting coronary blood flow and extravascular microvessel compression due myocardial hypertrophy of infiltrative diseases such as amyloid, myxedema, granuloma, and tumor cells [18]. However, the main non-vascular determinants of coronary circulation are decreased diastolic pressure-time and central arterial stiffness [28].
Decreased diastolic pressure-time: The myocardium has a very high oxygen extraction from blood perfusion it. Thus, an increase in myocardial oxygen supply can only be met by a relative increase in coronary circulation. About 80% of myocardial blood flow occurs in diastole, and thus aortic diastolic pressure amplitude and duration of diastole are the principal non-vascular determinants of myocardial perfusion. While the traditional focus for myocardial ischemia has been investigation for flow-limiting obstruction of the coronary conducting vessels with little concern for other factors limiting coronary blood flow, changes in diastolic duration exerts the same effect on coronary flow as a severe stenosis on coronary conducting vessel [37].
Central arterial stiffness: Increased central aortic stiffness in atherosclerosis is an independent predictor of adverse coronary events [38,39]. Increases in central arterial stiffness and wave reflection amplitude causes a rise in systolic aortic pressure, widening of pulse pressures and increase in myocardial systolic wall pressures and oxygen demand with decreases in diastolic (perfusion) pressure [40]. These alterations in ventricular versus vascular coupling causes an imbalance in the myocardial oxygen supply and demand ratio leading to myocardial ischemia and angina. Compared to normal (or healthy) coronary blood vessels, autoregulation preserves circulation over a wide range of perfusion pressures, for example, vasodilation during decreasing perfusion pressures [41]. In the setting of LV hypertrophy and other conditions such as tachycardia that lead to increased myocardial oxygen demand coronary circulation increases to match demand. However, when LV pumps blood into a stiff (non-compliant) aorta, systolic pressure and consequently myocardial oxygen demand increases while diastolic pressure decreases but with an increase in coronary circulation responding to increased demand with preserved contractility [42-44]. However, increased aortic stiffness reduces coronary flow reserve and during increased myocardial contractility impairs endocardial flow leading to sub-endocardial ischemia [43]. These alterations become pronounced in the setting of high-grade coronary stenosis or during reduced diastolic blood pressure [45].
Vascular and non-vascular conditions cause an imbalance between myocardial oxygen demand and supply. The imbalance maybe acute through infarct (necrosis or fibrosis) or a potentially reversible (upon resumption of adequate coronary perfusion) chronic ischemic insult on the myocardium. Both acute and chronic ischemic insult lead to a loss of ventricular function and reduced cardiac output [5-7]. The potentially reversible chronic ischemic insult to the myocardium leads to development of hibernating or stunned myocardium characterized by a transient and reversible contractile dysfunction. The concept of hibernating myocardium is usually confused with stunned myocardium. In hibernating myocardium, ischemia is ongoing but in stunned myocardium, perfusion is fully or almost restored. Both hibernating and stunned myocardium retain an inotropic reserve. However, in hibernating myocardium, the increase in contractile function occurs with deterioration in metabolic function but in stunned myocardium, there is no metabolic deterioration during inotropic stimulation [7]. Thus, inotropic stimulation in combination with metabolic imaging helps to detect viable and dysfunctional myocardium as well as distinguished hibernating from stunned myocardium [5,6]. These changes also provide insights into clinical management of transient myocardial ischemia. Therapy of hibernating myocardium is to restore coronary perfusion to the hypoperfused myocardial tissue while stunned myocardium requires no therapy since perfusion is normal and contractile function recovers spontaneously [7].
Risk factors are conditions that predispose an individual to the development of a disease. The concept of risk factors in CVD originated from the Framingham Heart Study (FHS), which published its initial findings in 1957. The study demonstrated epidemiological correlation between cigarette smoking, blood pressure and cholesterol levels, and the incidence of IHD. The findings truly revolutionized the practice of medicine by promoting minimization of risk factors as a complementary strategy to medical therapy [46]. While some risk factor may exert an independent effect on the risk of developing IHD, increased burden of risk factors significantly increases the likelihood of developing IHD [47]. The FHS divided risk factors for IHD into two: modifiable and non-modifiable risk factors. Non-modifiable risk factors such as age, gender and family history are factors that cannot be controlled while modifiable risk factors such as smoking, obesity, inactivity, excessive alcohol and stress are factors that measures can be taken to control them [47,48].
Non-modifiable risk factors
Increasing age: Increasing age is a non-modifiable and independent risk factor for the development of IHD. All clinical manifestations of atherosclerotic disease (a major sequalae to ischemic HF) increases with age suggesting an independent contribution of ageing to development of atherosclerosis [49]. Age is also the strongest predictor of IHD in patients aged ≥ 65 years who have 15 times the odds of IHD compared to patients younger than 45 years [50]. Ageing also correlates with the acquisition and increments in other major modifiable risk factors to contribute to the development of IHD. Thus, a considerable proportion of the effect of age on IHD risk could be a reflection of the intensity and duration of exposure to modifiable risk factors that accompany ageing [49].
Male gender: The male gender is an important non-modifiable risk factor for the development of IHD. The traditional consideration that IHD was a disease predominantly affecting men influenced the non-inclusion of women in earlier CVD research programs. In the 1990s, increased attention focused on female with IHD established the existence of gender difference in the utility of diagnostic and therapeutic procedures for IHD [51]. Males have 3.7 times the odds of developing IDH compared to women [50]. The lifetime risk of developing IHD at the age of 40 years is also higher for men (50%) compared to women (33%) [53]. Gender difference in the risk of IHD may emerge from differences in the intensity and prevalence of modifiable risk factors. Diabetes, high density lipoproteins and triglycerides levels have a greater impact on IHD risk in women while lipoprotein (a) has a stronger impact on men [52].
Family history: Ischemia heart disease has been known to aggregate in families. However, methodological factors such as identification and definition of a positive family history has complicated research into the contribution of family history to IHD risk especially in comparing study results [50]. In addition, the interaction between known risk factors and family history makes it difficult to quantitate its effect independent of other know factors [54]. Despite the difficulty, post hoc analysis of FHS data finds parental history of IHD or diagnosis of IHD in first-degree relative increases the probability of a premature onset of IHD [55]. The risk is higher if the father or brother has IHD before the age of 55 years or the mother or sister has the disease before the age of 65 years [56]. While a history of IHD in first-degree relatives is a risk factor for IHD, the risk is greater in women than in men [56]. Family history is important in the risk stratification of individuals who are at low risk [54].
Modifiable risk factors
Cigarette smoking: Cigarette smoking is the most preventable cause of ischemic HF. The correlation between smoking and increased risk of ischemic HF first emerged from the findings of the FHS, which demonstrated smokers had an increased risk of developing myocardial infarction (MI) or sudden cardiac death (SCD). At least 20% of MI patients go on to develop ischemic HF [24]. The risk strongly associated with the number of cigarette smoked daily and the duration of smoking [46]. Accumulating evidence continue to show that cigarette smoking doubles the risk of IHD-associated morbidity and mortality [50]. Smoking cessation significantly reduces the risk of all-cause mortality and non-fatal MI in IHD patients, [57]. The risk of morbidity and mortality associated with cigarette smoking reduces significantly after smoking cessation but may take over 20 years for a complete reversal [58]. About 20% of patients with IHD who give up smoking after acute MI achieve a 20% reduction in mortality rates and infarct recurrences [59]. Smokers < 50 years have a tenfold increase in the risk of developing IHD compared to age-matched non-smokers [50,60].
Physical inactivity: Physical inactivity or sedentary lifestyle is another potentially preventable risk factor for ischemic HF. A landmark study by Morris et al. [61] was the first demonstrate the relationship between physical inactivity and the incidence of acute MI and SCD ascribed to ischemic HF. The study reported conductors on London's double-decker buses (move up and down the bus stairs) has fewer incidence of ischemia-associated acute MI and SCD compared to their sedentary drivers. The risk of death from ischemic HF for sedentary compared with physically active individuals is 1.9 [62]. Regular physical activity has several cardiovascular benefits including reduced blood pressure, weight control, and reduced waist circumference, which are helpful in reducing the risk of developing ischemia HF. The recommended of physical exercise has become an important part of non-medical therapies as a preventive policy for all patient cohorts – older adults, children and teenagers – against cardiovascular diseases [46].
Obesity: Obesity, defined as body mass index (BMI) > 30 or overweight (BMI 25 to 30) [63], increases the likelihood of having an ischemic HF. Kannel et al. [64] in the Framingham study, was the first to described the relationship between obesity and IHD. It is an independent modifiable risk factor for all-cause mortality in patients with IHD [46]. Obesity is a metabolic disorder with associated comorbidities such as IHD, Type 2 Diabetes, hypertension and sleep apnea. It occurs as excess adipose tissue deposits, which causes alterations in metabolic profile and various adaptations on both cardiac structure and function [65]. The prevention and control of obesity and overweight had become an important strategy in the prevention of IHD and other CVD in both children and adults [66,67].
Diabetes: The involvement of diabetes in the pathogenesis of IHD was unclear until 1979 when post hoc analysis of FHS data identified diabetes as a major risk factor for ischemic HF. It increases the risk of clinical atherosclerotic disease by two to three-fold with a higher risk among females [50,68]. The American Heart Association provides statistics establishing the relationship between diabetes and IHD – at least 68% of diabetic individuals > 65 years die from heart diseases and 16% of stroke and diabetic adults have a two-fold increase to die from heart diseases compared to non-diabetic patients [69]. While diabetes is treatable with management of glucose levels, it still increases the risk of ischemic HF and stroke because of the presence of comorbidities that are also risk factors such as hypertension, smoking, high cholesterol, obesity, physical inactivity and metabolic syndrome. However, management of these risks prevents or delays the development of ischemic HF as well as improves prognosis for IHD patients [50].
Dyslipidemia: Total serum cholesterol and low-density lipoprotein (LDL) cholesterol levels have a strong relationship with the risk of developing ischemic HF and are clinical markers for predicting CVD. Dyslipidemia, unhealthy levels of one or more kinds of lipid in blood is a risk factor for IHD. The principal lipoprotein transporting cholesterol (LDL cholesterol) is directly associated with IHD and LDL cholesterol levels in young adulthood predicts development of IHD later in life [24]. LDL is a major contributor to the pathogenesis of atherosclerosis and LDL cholesterol lowering drugs reduce the risk of IHD by 50% in individuals aged 40 and 30% at the age of 60 years [70]. High-density lipoprotein (HDL) cholesterol also correlates closely and inversely with the risk of ischemic HF and are more predictive in men than in women [50]. Modest increase in HDL cholesterol levels in males with IHD and normal LDL cholesterol levels results in significant reduction in the risk of major CVD events [46].
Other risk factors: Other important risk factors include hypertension (elevated systolic pressure ≥ 160 mmHg and diastolic pressure < 90 mmHg), which increases the risk of CVD, stroke and all-cause mortality. Isolated hypertension indicates the loss of arterial elasticity and its prevalence increases with age [50]. Excessive consumption of alcohol may damage the myocardium and cause arrhythmias leading to increased risk of IHD. Alcohol can also contribute to weight gain, high triglyceride and hypertension, which increase the risk of IHD [47]. Finally, unhealthy diet high in saturated fats, cholesterol, salt and sugar may intensify other risk factors such as hypercholesterolemia, obesity and diabetes [47,48].
Ischemic HF occurs in the setting of an insult to the inner lining of the coronary conducting and resistance vessels causing atherosclerosis – a buildup of excess accumulation of fatty plaque consisting of cholesterol and other cellular waste products at the site of insult. The process leads to flow-limiting obstruction in one or more coronary conducting or resistance vessels and a consequential decrease in the amount myocardial oxygen supply [24,25]. Ischemic HF can be acute, occur suddenly quickly precipitated by a sudden decrease in coronary circulation, or can be chronic, in the setting of increased myocardial oxygen demand or progressive decrease in coronary circulation [17]. Several conditions could cause an imbalance in myocardial oxygen supply and demand contributing to ischemic episodes and ischemic HF. These conditions may include IHD, thrombosis, and coronary artery spasm.
Ischemic heart disease
Ischemic heart disease (IHD), the main sequalae to ischemic HF, encompasses a broad spectrum of clinical entities including asymptomatic subclinical atherosclerotic disease and its clinical complications such as angina pectoris, MI and SCD. IHD leads to flow-limiting obstruction to circulation in the coronary artery vessels causing myocardial ischemia [25]. IHD may present as advanced IHD, bifurcation blockage, myocardial infarction or silent ischemia [71]. Advanced IHD is common among patients with aggressive heart conditions who are at an elevated risk of developing ischemic HF and arrhythmias (irregular heartbeat or dysrhythmia). These patients may have previously undergone coronary bypass surgery, multiple stents or angioplasty procedures, and may experience chest pain despite optimal treatment [72]. Bifurcation blockage refers to a condition where the Y-junction of coronary artery has narrowed due to increased fatty acid deposition causing reduced coronary circulation. Myocardial infarction or heart attack is a fatal condition characterized by cardiomyocyte necrosis secondary to a complete loss of blood supply to an area of the myocardium leading to permanent cardiac injury [71,72]. A transient shortage of blood supply to the myocardium usually results into symptoms such as angina pectoris. However, in silent ischemia, there is no pain but it frequently disturbs cardiac rhythm leading to arrhythmias such as ventricular tachycardia or ventricular fibrillation, which potentially interferes with cardiac ability to pump and symptoms such as fainting or SCD [72].
Coronary thrombosis
Coronary artery thrombosis is another important etiology of IHD and ischemic HF. The rapture of vulnerable atherosclerotic plaque in coronary vessels in the presence or absence of severe stenosis activates thrombotic mechanisms leading to local thrombosis formation [73]. However, severe stenosis is as independent predictor of increased risk of ischemic HF. Thrombosis could cause a partial or complete occlusion of coronary conducting or resistance vessels leading to acute and severe myocardial ischemia resulting into ischemic HF [71]. Partial occlusion of coronary vessel leads to unstable angina (or non-ST Elevation Myocardial Infarction [NSTEMI]) while complete occlusion leads to ST elevation myocardial infarction (STEMI) [73].
Coronary artery spasm
Coronary artery spasms (CAS) is a transient and intense vasoconstriction of coronary arteries leading to total or incomplete occlusion of the vessel. It is an important cause of myocardial ischemic syndromes including NSTEMI, STEMI, acute MI and ischemic HF [74]. Whereas IHD has been well recognized as the key cause of ischemic HF in the setting of coronary atherosclerosis, microvascular dysfunction or acute thrombosis and luminal obstruction, CAS may cause transient ischemia leading to acute MI in the absence of observable coronary pathology [75]. CAS may also cause symptoms of chest pain, infarction, LV dysfunction, promote life-threatening arrhythmias and SCD [75].
Ischemic HF usually does not cause signs and symptoms until the setting of severe coronary stenosis or total occlusion of coronary artery. The disease usually goes undiagnosed until in a medical emergency such as a heart attack or stroke [76]. However, some patients with ischemic HF may present with signs and symptoms depending on the coronary artery affected. The most common clinical manifestation of ischemic HF is angina pectoris (AP) or ischemic chest pains, which may appear as stable angina (NSTEMI) or unstable angina (STEMI) during physical exertion, stressful situations or at rest [76]. Other clinically relevant symptoms are heart attack and SCD [77].
Angina pectoris
It has long been established that IHD comprises a wide spectrum of conditions ranging from chronic stable angina to acute MI. Unstable angina lies at the middle of the spectrum and an important symptom of ischemic HF. It results from five different but mutually inclusive causes: (a) non-occlusive coronary thrombus on a pre-existing plaque; (b) dynamic flow-limiting obstruction; (c) progressive mechanism obstruction; (d) inflammation; and (e) secondary unstable angina [77]. Unstable angina could be classified into three levels (I, II and III) based on severity, presence or absence of extracardiac conditions and time of onset (Table 1).
Table 1. Classification of unstable angina
Severity |
Description |
Clinical Circumstances |
A: Develops in the presence of extracardiac condition intensifying MI (secondary UA) |
B: Develops in the absence of extracardiac condition (primary UA) |
C: Develops within 2 weeks of acute MI (post-infarction UA) |
I |
New onset of severe or accelerated angina with no rest pain |
IA |
IB |
IC |
II |
Angina at rest within past month within 48 hours (subacute angina) |
IIA |
IIB |
IIC |
III |
Angina at rest within 48 hours (acute angina) |
IIIA |
IIIB (T-positive);
IIIB (T-Negative) |
IIIC |
MI: Myocardial Infarction; T: Troponin; UA: Unstable Angina [77]
The common triggers of typical angina include exertion, emotional stress, cold, wind or fever. Exertional angina is relieved between one and five minutes by rest or nitrates, otherwise episodes may last between two to ten minutes [76,77]. Angina manifests as heaviness or pressure in the precordium, restrosternum or epigastrium with possible radiation to outer aspects of both arms, neck, jaw and shoulder or mid-abdomen [76]. The quality of pain may be described as crushing, tightness, pressure or gripping in the presence or absence of numbness. Atypical angina on the other hand occurs especially in women and diabetic patients. It may be atypical in location and provoking factors [77].
Arrhythmias
Inadequate blood supply or injury to the myocardium could potentially interfere with cardiac electrical impulses leading to abnormal heart rhythm (arrhythmias). In IHD, two common patterns initiating fatal arrhythmias are ischemia or infarction-triggered ventricular arrhythmias or anatomical substrate (scarring from previous MI) scarring [79]. Animal models demonstrated arrhythmias in the setting of acute MI and infarction following the occlusion of coronary artery might occur in distinct phases. The first phase of arrhythmias occurs between two and ten minutes resulting from alterations in cellular electrophysiology and re-entrant mechanisms, while the second phase occur ten to thirty 30 minutes, which may have an association with accumulation of catecholamine and increased automaticity (the ability of cardiac cells to spontaneously generate an electrical impulse) [80].
The Coronary Artery Surgery Study (CASS) [81] demonstrated the LV ejection fraction impacts on Ischemic HF prognosis and treatment. However, the current definition of ischemic HF linking significant coronary obstruction and systolic dysfunction disregards important pathophysiologic and etiologic mechanisms and less useful for application in clinical diagnosis [6,7]. However, ischemic heart failure is a spectrum ranging from stunned myocardium associated with myocardial fibrosis to hibernating myocardium and repetitive ischemic episodes [82,83]. Although ischemic HF lacks focused clinical guideline for diagnosis and management, the latest American Heart Association/American College of Cardiology (AHA/ACC) [84] and European Society of Cardiology (ESC) [85] HF guidelines recommend diagnosis should be based on establishing viable myocardium – which recovers after coronary reperfusion using clinical and imaging methods [6].
Clinical evaluation
The 2012 AHA/ACC guidelines on diagnosis and management fi stable IHD [86] recommends the initial diagnosis of ischemic HF is clinical evaluation of patients with angina. Although considered the most common clinical manifestation of cardiac ischemia, the presence of angina is inconclusive since it occurs in 30% of patients with non-ischemic HF [87]. Clinical evaluation should consider accompanying signs and symptoms, patient history and risk markers to strengthen suspicion as well as categorization of angina as stable or unstable [88]. Clinical evaluation should include physical examination, resting electrocardiogram (ECG), chest x-ray, blood and exercise tests.
Physical examination is non-specific but recommended to detect evidence of alternative diagnosis for valvular heart disease, musculoskeletal disorders or hypertrophic cardiomyopathy. Surrogate markers such as severe dyslipidemia indicates IHD or third heat sound and peripheral edema indicates LV dysfunction [84]. Resting ECG is recommended for patients not having extracardiac causes of angina. Pathological Q-waves suggest prior MI [87]. Chest x-ray has limited use in diagnosing ischemic HF but provides useful information on alternative causes of anginal pain as well as surrogate markers of HF such as cardiothoracic ratio and pulmonary venous congestion [84]. Blood tests are recommended for identifying risk factors for IHD such as anemia, angina, dyslipidemia and renal function. High cholesterol levels and impaired glucose tolerance suggest additional tests for CAD [87]. Finally, exercise tests exertional symptoms, heart rate and blood pressure in response to physical activity. The test helps to detect inducible anemia by provoking cardiac ischemia through increasing myocardial activity and oxygen demand. However, coronary stenosis <70% may not be detectable by exercise testing) [88].
Imaging methods
Imaging methods establish diagnosis by detecting viable myocardium. The common imaging modalities used in the diagnosis of ischemic HF are myocardial perfusion imaging (single-photon emission computed tomography [SPECT] or positron emission tomography [PET], dobutamine stress echocardiography and cardiac magnetic resonance imaging (CMRI) [6]. Table 2 shows imaging methods and parameters for assessing myocardial viability.
Table 2. Imaging and parameters methods of assessing myocardial viability
Imaging |
SPECT/PET |
Echo |
CMRI |
CT |
Cardiac Catheterization |
Metabolic Activity |
ü |
|
|
|
|
Perfusion metabolic Mismatch |
ü |
|
|
|
|
Perfusion |
ü |
ü |
ü |
ü |
ü |
Inotropic Reserve |
|
ü |
ü |
|
ü |
Cell membrane activity |
ü |
|
ü |
ü |
|
Detecting fibrosis |
|
|
ü |
ü |
|
Electrical activity |
|
|
|
|
ü |
Arai et al. 2011, p. 11 [89]
Single-photon emission computed tomography: The SPECT modality assesses technetium-99 or thallium-201 radioisotope uptake by viable cardiomyocytes, which rely on cellular and mitochondrial integrity. The two protocols have a high sensitivity to predict contractile recovery after coronary revascularization (83% and 87% respectively) but with a lower sensitivity (65% and 54% respectively). Although widespread and availability, low spatial resolution and exposure to radiation limits their utility [6].
Positron emission tomography: The PET modality utility in assessing myocardial viability is widespread in clinical practice. The modality combines a tracer to assess blood flow and fluorine-18 fluorodeoxyglucose (18F-FDG) to evaluate cellular metabolism. The modality provides three main result patterns useful for viability assessment in ischemic HF are (a) low blood flow with preserved metabolism suggesting a mismatch compatible with hibernating myocardium); (b) decreased blood flow and metabolism suggesting a match compatible with fibrosis or necrosis; and (c) blood flow and preserved metabolism suggesting normal tissue [90]. The PET modality has high sensitivity (~92%) but with moderate specificity (63%), and a higher spatial resolution with less radiation exposure relative to SPECT. However, PET is expensive, limited availability and limited utility in diabetic patients with ischemic HF depending on sensitivity of glucose transporters [90].
Dobutamine stress echocardiography: The utility of echocardiography in myocardial viability assessment relies on three parameters: (a): ventricular wall thickness; (b) contrast enhancement by cardiomyocytes; and (c) contractile reserve with inotropic stimulation [6]. A decrease in ventricular wall thickness (end-diastolic wall thickness < 6 mm) associated with loss of tissues due to cardiomyocytes fibrosis or necrosis, has a high negative predictive value for contractile recovery following revascularization [90]. In recent years, the utility of contrast echocardiography has increased to evaluate myocardial perfusion and cellular integrity. Dobutamine stress echocardiography has been used to assess contractile reserve where following inotropic stimulation, dysfunctional segment at rest presents contractile recovery. Low doses of dobutamine is sufficient to evaluate contractile reserve because of biphasic response (initial improvement followed by worsening of contractility at higher doses) suggesting viability of the myocardium. Despite dobutamine-stress echocardiography having high sensitivity (80%) and specificity (78%), the modality has a poor acoustic window and operator-dependent [91,92].
Cardiac magnetic resonance imaging: The utility of cardiac magnetic resonance imaging (CMRI) for the assessment of myocardium viability in patients with ischemic HF is gaining importance because of better spatial resolution, no exposure to radiation, and independence of acoustic window compared to echocardiography and SPECT [93]. In the assessment of myocardial viability, CMRI uses three main parameters: (a) end-diastolic wall thickness; (b) low-dose dobutamine inotropic stimulation; and (c) late gadolinium enhancement (LGE). The CMRI assessment of end-diastolic wall thickness represents a measure of maximum thickness of myocardial wall at rest. Compared to PET (FDG uptake) wall thickness ≥5.5 mm suggests viability while < 5.0 mm suggests low probability of viability [94]. LGE is the gold standard modality for myocardial viability assessment. Greater distribution of gadolinium in the extracellular spaces (areas of necrosis or fibrosis) leading to a delayed washout. Transmural extension of scars correlated with potential contractile recovery: infarcted areas < 50% suggests increased probability of functional improvement after revascularization while > 50% suggests poorer outcomes [95].
Meta-analysis of diagnosis methods
The discovery of potentially reversible hibernating myocardium in patients with IHD motivated the need to develop, test and validate several methods to evaluate and detect myocardial viability. Viability tests help to select patients who would benefit from vascularization to reperfuse the myocardium and optimize myocardial contractility [6]. At present, non-invasive cardiac imaging modalities in widespread use are SPECT with technetium-99 or thallium-20, PET with 18F-FDG, dobutamine stress echocardiography, dobutamine stress CMRI and LGE-CMRI [84,85]. These imaging modalities assess metabolic activity, perfusion-metabolic mismatch, perfusion, inotropic reserve and cell membrane integrity to determine myocardial viability [89]. Of the four methods, CMRI is gaining widespread utility because of superior spatial resolution, no exposure to radiation, and independence of acoustic window [93]. Common CMRI methods for evaluating myocardial viability are (a) end-diastolic wall thickness; (b) low-dose dobutamine inotropic stimulation; and (c) LGE-CMRI. The objective of the present meta-analysis is to investigate the accuracy of LGE-CMRI technique in the assessment of myocardial viability.
Search criteria and inclusion: We searched for relevant studies using LGE-CMRI to assess myocardial viability before and after revascularization in patients with IHD and LV systolic dysfunction in online databases PubMed, EMBASE and Cochrane Central Register of Clinical Trials. The combination of search terms used were magnetic resonance imaging, myocardial or cardiac viability, viability assessment, viability test viable myocardium, ventricular dysfunction, cardiac dysfunction or hibernating myocardium. The search was limited to human studies including adult patients. There was no restriction on publication year or language. Additional studies were retrieved from screening citation of the included studies. Trials found only in abstract form without manuscript and with insufficient data for extraction were excluded. The inclusion criteria were the studies: (a) prospective RCTs recruiting IHD patients with chronic stable LV dysfunction; (b) CMRI was performed before and after revascularization; and (c) measured sensitivity and specificity based on provided cut-off values. Data abstracted included study name and year, patient characteristics (sample size, mean age and male proportion) and study outcomes (sensitivity and specificity, cut-off values and hyper-enhancement vs. normal intensity and mean follow-up months (Table 3).
Table 3. Characteristics of studies included in this meta-analysis on diagnosis
First Author [Ref. #] |
Year |
Size (N) |
Mean Age |
Male (%) |
Hyper-enhancement (SD > Normal Intensity) |
Cut-off |
Sensitivity |
Specificity |
Follow Up (Months) |
Ramani et al. [96] |
1998 |
24 |
64 |
92 |
NR |
< 50 |
NR |
NR |
NR |
Kim et al. [97] |
2000 |
50 |
63 |
88 |
> 6 |
NR |
NR |
NR |
3 |
Sandstede et al. [98] |
2000 |
12 |
61 |
83 |
NR |
< 50 |
98 |
76 |
3 |
Klein et al. [99] |
2002 |
31 |
59 |
84 |
NR |
< 50 |
86 |
94 |
NR |
Wellnhofer et al. [100] |
2004 |
29 |
68 |
93 |
> 2 |
< 50 |
NR |
NR |
3 |
Selvanayagam et al. [101] |
2005 |
52 |
NR |
NR |
> 2 |
< 50 |
NR |
NR |
6 |
Kuhl et al. [102] |
2006 |
29 |
66 |
72 |
> 3 |
< 50 |
97 |
68 |
6 |
Bondarenko et al. [103] |
2007 |
45 |
62 |
84 |
> 5 |
< 50 |
NR |
NR |
3 |
Wu et al. [104] |
2007 |
41 |
66 |
78 |
NR |
< 50 |
92 |
44 |
6 |
Becker et al. [105] |
2008 |
53 |
59 |
83 |
> 2 |
< 50 |
72 |
92 |
9 |
Pegg et al. [94] |
2010 |
33 |
66 |
65 |
> 2 |
< 50 |
95 |
75 |
6 |
Study characteristics and outcomes: The online search and screening of bibliographies yielded 5,543 citations for review and possible inclusion. After title, abstract and full-text screening, eleven (11) studies that met the inclusion criteria were included for analysis [94,96-105]. The 11 studies recruited 399 patients (means age 63.4 years; 82.2% male). LGE-CMRI assessment of myocardial viability was done between 3 and 9 months (mean 9 months) after revascularization but the difference was not statistically significant. The description of hyper-enhancement varied between studies: 2 SD [94,100,101,105], 3 SD [102], 5 SD [103] and 6 SD [97] with a mean 0f 3.14 that was not statistically significant. Sensitivity was calculated as the number of viable segments before vascularization divided by the number of segments with improved function after revascularization while specificities was the number of non-viable segments before revascularization divided by the total number of segments without improved function after revascularization. Pooled results show that LGE-CMRI has a good sensitivity (90.0%) and specificity (74.8%) in detecting viable myocardium in patients with IHD and LV systolic dysfunction.
Discussion
Ischemic HF is a cardiac condition characterized by hibernating myocardium, which is potentially upon restoration of myocardial perfusion. Thus, the primary clinical target in these patients is selecting those who will benefit from revascularization [85]. A previous meta-analysis of myocardial viability testing and impact of revascularization on prognosis on IHD patients with LV dysfunction reports improved survival after revascularization demonstrated by reduced annual mortality (3.2%) relative to medical therapy alone (16%) [106]. Thus, the 2016 ESC and the 2013 AHA/ACC HF diagnostic and clinical management guidelines recommend non-invasive cardiac imaging for all patients with IHD and ventricular dysfunction without angina because revascularization is recommended in all patients with angina irrespective of the extent of cardiac ischemia. While SPECT, PET, echocardiography and CMRI are common non-invasive modalities used for the assessment of myocardial viability, they have comparable diagnostic accuracies and each with unique individual limitations. However, increasing evidence suggests that LGE-CMRI maybe a more accurate modality but current evidence is fragmented and somewhat inconclusive because of the lack of systematic evaluation. In the current meta-analysis, the aim was to evaluate diagnostic accuracy of LGE-CMRI modality in myocardial viability assessment of IHD patients with LV systolic dysfunction. The analysis pools together data from pertinent prospective RCTs to determine diagnostic sensitivity and specificity of LGE-CMRI myocardial viability assessment.
Our analysis reveals that LGE-CMRI is a sensitive and specific non-invasive modality for the assessment of myocardial viability in patients with ischemic HF. In delayed contrast enhanced CMRI using gadolinium, the presence or absence of hyper-enhancement correlate closely with non-viable and viable myocardium respectively in segments that are akinetic and dyskinetic under resting conditions [96]. Thus, LGE-CMRI should be considered in patients scheduled to undergo revascularization, which should include those with abnormalities in regional wall motion on echocardiography and not having unstable angina, New York Heart Association functional class IV or contraindications to CMRI. The present findings are consistent or strengthened by those of a previous related meta-analysis. Myocardial viability identified by non-invasive image testing reduced annual mortality rates by 79.6% demonstrating patients with chronic IHD and LV dysfunction with viable myocardium benefit from revascularization but the absence of viable myocardium is associated with no significance difference in outcomes despite treatment strategy [106]. In another meta-analysis comparing EDWT, low-dose dobutamine and LGE CMRI, LGE-CMRI had the highest sensitivity and negative predictive value but low-dose dobutamine had the best specificity and positive predictive value [107]. Furthermore, another study reports that the diagnostic accuracy of LGE-CMRI is improved if used in combination with EDWT [108].
Clinical management of ischemic etiologies of LV systolic dysfunction (ischemic HF) is important since it has significantly higher mortality rates compared to non-ischemic etiologies [109]. The 2016 ESC [85] and 2013 AHA/ACC [84] HF guidelines recommend clinical management of ischemic HF should include medical and interventional therapies targeted to relieve symptoms and improve prognosis.
Medical therapy
Medical therapy is the mainstay of treatment of patients with ischemic HF providing significant improvement in both survival and quality of life. It is a priority in the management of ischemic HF because it targets the disease itself and its fundamental pathophysiologic pathways [6]. The main classes of medicine include aspirins, statins, aldosterone inhibitors, beta-blockers, angiotensin-converting-enzyme inhibitors (ACEI) or angiotensin receptor blockers (ARBs) with different clinical benefits and outcomes [84,85]. Beta-blockers are recommended for the treatment and relief of angina in patients with CAD. Beta-blockers provide additional prognostic benefits in patients with ischemic HF and LV systolic dysfunction. The CIBIC-II [110] and MERIT-HF [111] trials recruiting 65% and 50% of patients with ischemic HF respectively reported bisoprolol and metoprolol therapies significantly improved survival. Therapy with ACEI (and/or with ARBs for those contra-indicated or intolerant to ACEI) is also recommended for ischemic HF patients with CAD and LV systolic dysfunction to relieve symptoms and reduce deaths and hospital admissions [112]. Aldosterone therapy targets to block aldosterone receptors by spironolactone and in addition to standard ACEI/ARB and beta-blocker therapy, aldosterone therapy significantly reduces morbidity and mortality in MI patients complicated by LV systolic dysfunction and HF [113].
Interventional therapies
Interventional therapies are non-medical or surgical procedures aimed to relieve symptoms. The major interventional include coronary artery bypass graft surgery (CABG), sometimes combined with surgical ventricular reconstruction (SVR) or surgical mitral valve repair. Other recommended interventional therapies include implantable cardioverter-defibrillators, cardiac resynchronization therapy for those with left bundle branch block (LBBB), and orthotopic cardiac transplantation and ventricular assist devices for highly selected patients with advanced disease and less often percutaneous coronary intervention (PCI) [84,85].
Surgical revascularization: Hibernating myocardium with a potentially reversible contractile function upon myocardial reperfusion is a common manifestation of ischemic HF, thus the selection of CAD patients that would benefit from revascularization is important [6]. Three early observational studies conducted in the mid-1970s reported CABG had a 10 to > 50% reduction in mortality compared to medical therapy in CAD patients with LV systolic dysfunction [114-116]. However, the three studies were conducted before the development of beta-blockers and ACE-I/ARBs nor provided sufficient data for comparison of treatment efficacy with the current optimal medical therapy. The CASS study [81] was the first randomized clinical trial to investigate CABG vs. medical therapy on 780 patients with CAD and LV systolic dysfunction (ejection fraction < 50%). CABG has a better survival (79%) vs. medical (61%). However, CASS was done in the 1970s when more than half of the patients did not use beta-blockers and the study exclude patients with LV ejection fraction < 35% and New York Heart Association Functional Classes III and IV. Recently, the Surgical Treatment of Ischemic Heart failure (STICH) trial between 2002 and 2007 recruiting 1,212 patients with LV ejection fraction < 35% and CAD, a dual therapy of CABG and medical had better 5-year survival (death from any cause) compared to medical therapy alone [117].
Percutaneous coronary intervention: The utility of PCI in CAD patients with reduced ejection fraction has limited research support. The Bypass Angioplasty Revascularization Investigation (BARI) [118] and Angina With Extremely Serious Operative Mortality Evaluation (AWESOME) [119] trials recruited CAD patients with LV dysfunction showed no difference in clinical outcomes between PCI and CABG. However, the two trials had a small patient population (< 500) and included a dual therapy of PCI and balloon angioplasty with stents. A recent observational registry based New York study [120] involving 4,616 patients reported CABG and PCI had comparable long-term survival but PCI had a higher risk of MI and revascularization but with a lower risk of stroke. However, these findings should be interpreted with caution because of the population and device used (everolimus-eluting stent).
Meta-analysis
Ischemic HF is potentially reversible condition upon coronary revascularization (perfusion). The main methods of coronary revascularization are CABG and PCI. While CABG has received strong research support, the support for effectiveness of PCI remains fragmented and inconclusive. The primary objective of this meta-analysis is to compare published evidence on therapeutic efficacy of CABG with PCI based on the occurrence of a composite of major cardiovascular events: death, MI, and repeat revascularization.
Search strategy and inclusion criteria: The search for pertinent studies examining revascularization using CABG or PCI on patients with ischemic HF was performed on electronic databases PubMed, U.S. National Institute of Health, Cochrane Central Register of Controlled Trials, proceedings of the AHA, ACC and ESC. The search terms used included coronary artery disease, coronary artery bypass graft, percutaneous coronary intervention, stents, and angioplasty. We completed the search for studies from reference list of included studies, pertinent reviews and previous meta-analyses [121-123]. The inclusion criteria were (a) prospective randomized controlled trials; (b) recruited patients with angina; (c) compared CABG with PCI coronary revascularization strategies; (d) followed patients in at least 12 months; and reported clinical endpoints – death, MI and revascularization or composite endpoints. Studies were excluded if (a) they recruited patients with onset of acute MI within 72 hours of initial enrollment; (b) they compared different types of non-stenting techniques; and (c) used different PCI types in the same arm; (d) had only abstracts available.
Data from the included studies were extracted directly into Microsoft Excel tables by two investigators. Any discrepancies were resolved by consensus. From each included article, we recorded data on publication (first author and year of publication), patient population (size, mean age and proportion of male patients), mean duration of follow up in years, and clinical endpoints in each arm of the study –CABG and PCI – (composite endpoint, death, myocardial infarction, and repeat revascularization). Summary of the extracted data is provided in Table 4. We reported dichotomous variables as percentages, continuous variables as mean or media. We combined binary outcomes from each study with a random effect model to compute risk ratio (RR) with 95% confidence interval (CI). We calculated I2 as a measure of statistical heterogeneity 25%, 50% and 75% representing mild, moderate and severe inconsistencies.
Table 4. Characteristics of studies included in this meta-analysis on revascularization
First Author [Ref #] |
Year |
Age (yrs) |
Male (%) |
CABG (n) |
PCI (n) |
FuP (yrs.) |
Composite Endpoint |
Death (n) |
MI (n) |
Revascularization (n) |
CABG |
PCI |
CABG |
PCI |
CABG |
PCI |
CABG |
PCI |
RITA Study [124] |
1993 |
57 |
81 |
490 |
493 |
2.5 |
48 |
42 |
18 |
16 |
26 |
34 |
37 |
157 |
King et al. [125] |
1994 |
62 |
74 |
194 |
198 |
3.0 |
53 |
57 |
12 |
14 |
38 |
29 |
25 |
81 |
CABRI Study [126] |
1996 |
60 |
78 |
513 |
541 |
1.0 |
NA |
NA |
14 |
21 |
18 |
27 |
33 |
182 |
Alderman et al. [117] |
1996 |
62 |
73 |
914 |
915 |
5.4 |
98 |
125 |
98 |
125 |
179 |
195 |
73 |
586 |
Morrison et al. [118] |
2001 |
67 |
NR |
232 |
222 |
3.0 |
49 |
44 |
49 |
44 |
90 |
115 |
NA |
NA |
Rodriguez et al. [127] |
2001 |
62 |
79 |
225 |
225 |
1.5 |
28 |
8 |
17 |
7 |
15 |
5 |
11 |
38 |
Serruys et al. [128] |
2001 |
61 |
77 |
605 |
600 |
1.0 |
NA |
NA |
17 |
15 |
22 |
28 |
21 |
101 |
SoS Investigators [129] |
2002 |
61 |
79 |
500 |
488 |
2.0 |
30 |
102 |
8 |
22 |
49 |
46 |
30 |
101 |
Eefting et al. [130] |
2003 |
60 |
71 |
142 |
138 |
1.0 |
12 |
20 |
4 |
0 |
7 |
6 |
6 |
21 |
Serruys et al.[131] |
2009 |
65 |
78 |
897 |
903 |
1.0 |
65 |
68 |
30 |
39 |
28 |
43 |
50 |
120 |
Thiele et al. [132] |
2009 |
66 |
70 |
65 |
65 |
3.6 |
5 |
1 |
0 |
0 |
5 |
1 |
NA |
NA |
Boudriot et al [133] |
2011 |
68 |
75 |
101 |
100 |
1.0 |
14 |
19 |
5 |
2 |
3 |
3 |
6 |
14 |
Park et al. [134] |
2011 |
62 |
76 |
300 |
300 |
1.0 |
24 |
36 |
10 |
7 |
3 |
5 |
12 |
26 |
Mohr et al. [135] |
2013 |
65 |
78 |
805 |
871 |
5.0 |
217 |
325 |
92 |
121 |
31 |
84 |
110 |
212 |
Study characteristics and outcomes: After the screening of titles, abstract and full-text of the retrieved citation against the inclusion and exclusion criteria, fourteen (14) studies were included in the final analysis [117,118,124-135]. The included studies spanned a period of three decades, from 1993 to 2013. Table 4 shows the main study features, patient characteristics and outcomes. In total, the 14 studies enrolled 12,042 patients randomized into CABG (5983) and PCI (6,059). Overall, the mean age of the patients was 63 years with a greater male proportion (76%). The mean follow-up period was 2 years (range 1.0 to 5.4 years). Compared to PCI, patients in the CABG group had lower deaths (374 [6%] vs. 433 [7%]), MI (514 [9%] vs. 621 [10%]) and repeat revascularization (414 [7%] vs. 1639 [27%]). The analysis finds that in 12 studies [117,118,124,125,127-135], patients in the CABG group that experience composite endpoint of major cardiovascular events (death, MI, revascularization and large ischemic defects) was lower (643 [11%]) than patients in the PCI group (847 [14%]).
Pooling the data together, CABG had a 13% reduction in the odds of all-cause death (OR: 0.87, 95% CI: 0.749-1.003) but the reduction was not significant (p=0.055) (Figure 2); 20% reduction for MI (OR: 0.80, 95% CI: 0.698-0.905, p=0.001) (Figure 3) and 81% reduction for repeat revascularization (OR: 0.19, 95% CI: 0.173-0.217, p=0.001) (Figure 4). For composite clinical endpoints of death, MI, repeat vascularization or large ischemic defects, CABG reduced the odds by 27% (OR: 0.73, 95% CI: 0.653-0.821, p=0.001) (Figure 5). However, there was a high variability (inconsistency) of treatment effect across the studies (I2 = 81.39%; 95% CI, 68.57 to 88.98).
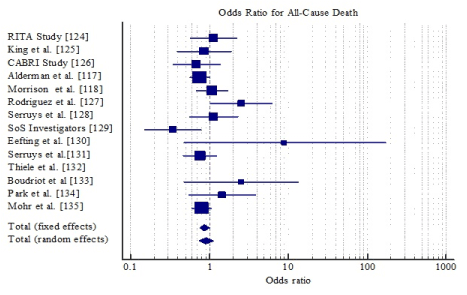
Figure 2. Odds ratio for all-cause death between CABG and PCI.
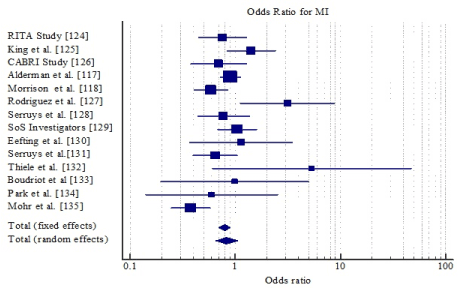
Figure 3. Odds ratio for MI between CABG and PCI.
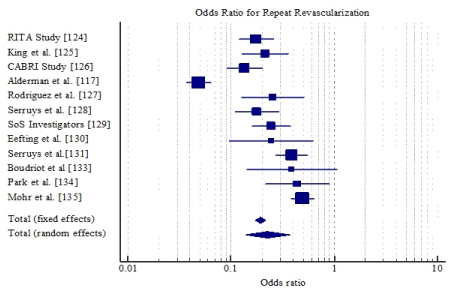
Figure 4. Odds ratio for repeat revascularization between CABG and PCI.
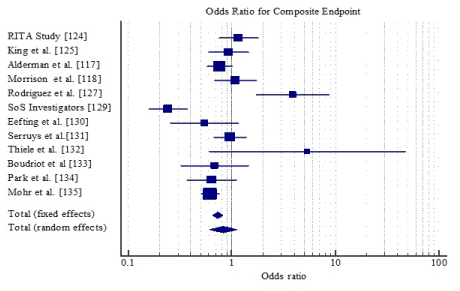
Figure 5. Odds ratio for composite clinical endpoint between CABG and PCI.
The 2014 ESC and the European Association for Cardio-Thoracic Surgery (EACTS) guidelines on myocardial revascularization recommend PCI or CABG in patients with stable CAD and with persistent symptoms despite medical treatment to reduce ischemia and its adverse clinical manifestation [136]. However, the accuracy of CABG and PCI have not been systematically compared. The present meta-analysis pooled together results from 14 prospective RCTs comparing treatment efficacy of two common revascularization procedures (CABG and PCI) in 12,042 patients with stable chronic IHD and LV systolic dysfunction (LVEF < 40%). We calculated treatment-effect size models where the former is more appropriate in the case of wide variability across trials but may inflate the importance of small RCTs. The primary analysis focused on treatment effect on death, MI and repeat revascularization, or a composite of death, MI and revascularization in a mean follow-up period of two (2) years. CABG treatment showed superior treatment efficacy reducing the odds ratio by 13%, 20% and 27% of death, MI and repeat revascularization or 11% in the composite endpoint of death, MI or repeat vascularization. The studies had a high inconsistency (I2 = 81.39%), which could be explained either by methodological quality of the inclusion criteria such as the lack of distinction between single and multi-vessel disease and wide variation in follow-up duration, or the efficacy of CABG and PCI differ based on characteristics of the patient, technique or skill of the cardiologist.
Previous meta-analyses on revascularization therapy on patients with IHD and systolic distinction provide consistent or supporting findings [121-123]. Schomig et al. [121] compared PCI and medical therapy and found PCI improved long-term survival – reduced the odds of all-cause death by 20% (OR: 0.80; 95% CI, 0.64-0.99) but with a non-significant reduction in cardiac death (OR: 0.74, 95% CI: 0.51-1.06). PCI also reduced non-fatal myocardial infarction by 10% (OR: 0.90, 95% CI: 0.66-1.23). Trikalinos et al. [122] pooled together 61 studies (25 388 patients) on PCI therapy (balloon angioplasty, bare metal stents and drug eluting stents) and medical therapy for non-acute IHD in the past twenty years. Different PCI techniques did not yield detectable improvement in deaths and MI while drug-eluting stents had a small reduction 0.04% in the risk of death and 27% in MI compared to medical therapy. Bucher et al. [123] analyzed six RCTs (953 patients) to compare PCI with medical therapy in non-acute IHD patients. PCI lead to a significant reduction in angina (10%) with non-significant reduction in non-fatal MI and death. These previous meta-analyses indicate revascularization had better treatment outcomes compared to medical therapy alone. Our analysis further suggests that, in revascularization strategies, CABG nay provide significantly better outcomes in reducing non-fatal MI and repeat revascularization compared to PCI in patients with ischemic HF.
Ischemic heart failure (HF) remains relatively understudied despite over three decades of research because of definitional and terminology variance, as well as extensive focus on non-ischemic etiologies. It is a potentially reversible type of HF caused by an imbalance between myocardial oxygen demand and supply leading to the loss of ventricular systolic function. The imbalance leads to a hibernating or stunned myocardium with a potentially reversible contractile function after coronary revascularization. The cause of increased myocardial oxygen demand include increased heart rate, abnormal loading conditions and contractility, while that of decreased myocardial oxygen supply include flow-limiting obstruction in coronary vessels caused by stenosis, endothelial dysfunction, disorders of vascular smooth cells, atherothrombosis and vascular remodeling. The major IHF risk factors include ageing, male gender, family history, smoking, physical inactivity, obesity, diabetes and dyslipidemia. The common clinical manifestations are angina (chest pains) and arrhythmias. Diagnosis is based on imaging assessment of myocardial viability and coronary flow reserve after revascularization using SPECT, PET, dobutamine stress echocardiography and CMRI (dobutamine stress or LGE). Clinical management targets to relieve symptoms and improve quality of life using medical or revascularization (CABG or PCI) therapies, or a combination of both.
- Sans S, Kesteloot H, Kromhout DO (1997) The burden of cardiovascular diseases mortality in Europe: Task Force of the European Society of Cardiology on Cardiovascular Mortality and Morbidity Statistics in Europe. Eur Heart J 18: 1231-1248. [Crossref]
- Gaziano TA, Bitton A, Anand S, Abrahams-Gessel S, Murphy A (2010) Growing epidemic of coronary heart disease in low-and middle-income countries. Current problems in cardiology 35: 72-115. [Crossref]
- Akhtar S (2006) Ischemic heart disease. Anesthesiol Clin North America 24: 461-485. [Crossref]
- Inamdar AA, Inamdar AC (2016) Heart failure: diagnosis, management and utilization. J Clin Med 5: 62. [Crossref]
- Bonow RO, Maurer G, Lee KL, Holly TA, Binkley PF, et al. (2011) Myocardial viability and survival in ischemic left ventricular dysfunction. N Engl J Med 364: 1617-1625. [Crossref]
- Lima EG, Carvalho FP, Linhares Filho JP, Pitta FG, Serrano Jr CV (2017) Ischemic left ventricle systolic dysfunction: An evidence-based approach in diagnostic tools and therapeutics. Revista da Associação Médica Brasileira 63: 793-800. [Crossref]
- Heusch G, Schulz R (1997) Characterization of hibernating and stunned myocardium. Eur Heart J 18: 102-110. [Crossref]
- Smith SW, Whitwam W (2006) Acute coronary syndromes. Emergency Medicine Clinics 24: 53-89. [Crossref]
- Hansson GK (2005) Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med 352: 1685-1695. [Crossref]
- Suwaidi JA, Hamasaki S, Higano ST, Nishimura RA, Holmes Jr DR, etal. (2000) Long-term follow-up of patients with mild coronary artery disease and endothelial dysfunction. Circ 101: 948-954. [Crossref]
- Wilson PW, D’Agostino RB, Levy D, Belanger AM, Silbershatz H, et al. (1998) Prediction of coronary heart disease using risk factor categories. Circ 97: 1837-1847. [Crossref]
- Ornish D, Brown SE, Billings JH, Scherwitz LW, Armstrong WT, et al. (1990) Can lifestyle changes reverse coronary heart disease?: The Lifestyle Heart Trial. The Lancet 336: 129-133. [Crossref]
- Kavey RE, Daniels SR, Lauer RM, Atkins DL, Hayman LL, et al. (2003) Association guidelines for primary prevention of atherosclerotic cardiovascular disease beginning in childhood. Circ 107: 1562-1566. [Crossref]
- DeFronzo RA, Ferrannini E (1991) Insulin resistance: a multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia, and atherosclerotic cardiovascular disease. Diabetes care 14: 173-194. [Crossref]
- Schmermund A, Möhlenkamp S, Stang A, Grönemeyer D, Seibel R, et al. (2002) Assessment of clinically silent atherosclerotic disease and established and novel risk factors for predicting myocardial infarction and cardiac death in healthy middle-aged subjects: rationale and design of the Heinz Nixdorf RECALL Study. Am Heart J 144: 212-218. [Crossref]
- Bolger AP, Coats AJ, Gatzoulis MA (2003) Congenital heart disease: the original heart failure syndrome. Eur Heart J 24: 970-976. [Crossref]
- Libby P, Theroux P (2005) Pathophysiology of coronary artery disease. Circ 111: 3481-8348. [Crossref]
- Naghavi M, Libby P, Falk E, Casscells SW, Litovsky S, et al. (2003) From vulnerable plaque to vulnerable patient: a call for new definitions and risk assessment strategies: Part I. Circ 108: 1664-1672. [Crossref]
- World Health Organization. (2002). The World Health Report 2002: Reducing risks, promoting healthy life. Geneva: World Health Organization. [Crossref]
- Mensah GA (2008) Ischaemic heart disease in Africa. Heart 94: 836-43. [Crossref]
- Mathers CD, Lopez AD, Murray CJ (2005) The burden of disease and mortality by condition: data, methods and results for 2001. Global burden of disease and risk factors 45:88. [Crossref]
- Rao C, Lopez A.D, Hemed Y (2006) Causes of death. In: Jamison DT, Feachem RG, Makgoba MW, et al, eds. Disease and mortality in sub-Saharan Africa. 2nd Ed. Washington, DC: The World Bank, 67–82.
- World Health Organization (2006) Department of Measurement and Health Information. Projections of mortality and burden of disease by region, 2006. [Crossref]
- Giordano FJ (2005) Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest 115: 500-508. [Crossref]
- Schoenhagen P, Ziada KM, Vince DG, Nissen SE, Tuzcu EM (2001) Arterial remodeling and coronary artery disease: the concept of “dilated” versus “obstructive” coronary atherosclerosis. J Am Coll Cardiol 38:2 97-306. [CRossref]
- Huang Y, Hickey RP, Yeh JL, Liu D, Dadak A, et al. (2004) Cardiac myocyte-specific HIF-1α deletion alters vascularization, energy availability, calcium flux, and contractility in the normoxic heart. The FASEB journal 18: 1138-1140. [Crossref]
- Davies KJ (1995, November). Oxidative stress: the paradox of aerobic life. In Biochemical Society Symposia, 61: 1-32. Portland Press, London. [Crossref]
- Pepine CJ, Nichols WW (2007) The pathophysiology of chronic ischemic heart disease. Clinical Cardiology: An International Indexed and Peer‐Reviewed Journal for Advances in the Treatment of Cardiovascular Disease 30: I-4. [Crossref]
- Pepine CJ, Kerensky RA, Lambert CR, Smith KM, von Mering GO, et al. (2006) Some thoughts on the vasculopathy of women with ischemic heart disease. J Am Coll Cardiol 47: S30-S35. [Crossref]
- Matsuzawa Y, Lerman A (2014) Endothelial dysfunction and coronary artery disease: assessment, prognosis and treatment. Coronary artery disease 25: 713-724. [Crossref]
- Esper RJ, Nordaby RA, Vilariño JO, Paragano A, Cacharrón JL, et al. (2006) Endothelial dysfunction: a comprehensive appraisal. Cardiovascular diabetology 5: 4. [Crossref]
- Viles-Gonzalez JF, Fuster V, Badimon JJ (2004) Atherothrombosis: a widespread disease with unpredictable and life-threatening consequences. Eur Heart J 25: 1197-1207. [Crossref]
- Schoenhagen P, Nissen SE, Tuzcu EM (2003) Coronary arterial remodeling: from bench to bedside. Curr Atheroscler Rep 5: 150-154. [Crossref]
- Kinlay S (2001) What has intravascular ultrasound taught us about plaque biology? Curr Atheroscler Rep 3: 260-266. [Crossref]
- Glagov S, Weisenberg E, Zarins CK, Stankunavicius R, Kolettis GJ (1987) Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med 316: 1371-1375. [Crossref]
- Pepine CJ, Nichols WW, Pauly DF (2006) Estrogen and different aspects of vascular disease in women and men. Circ Res 99: 459-461. [Crossref]
- Ferro G, Duilio C, Spinelli L, Liucci GA, Mazza F, et al. (1995) Relation between diastolic perfusion time and coronary artery stenosis during stress-induced myocardial ischemia. Circ 92: 342-347. [Crossref]
- Boutouyrie P, Tropeano AI, Asmar R, Gautier I, Benetos A, et al. (2002) Aortic stiffness is an independent predictor of primary coronary events in hypertensive patients: a longitudinal study. Hypertens 39: 10-15. [Crossref]
- Fukuda D, Yoshiyama M, Shimada K, Yamashita H, Ehara S, et al, (2006) Relation between aortic stiffness and coronary flow reserve in patients with coronary artery disease. Heart 92: 759-762. [Crossref]
- O’rourke MF, Nichols WW (2005) Aortic diameter, aortic stiffness, and wave reflection increase with age and isolated systolic hypertension. Hypertens 45: 652-658. [Crossref]
- Hoffman JI (1987) A critical view of coronary reserve. Circ 75: I6-I11. [Crossref]
- Watanabe H, Ohtsuka S, Kakihana M, Sugishita Y (1992) Decreased aortic compliance aggravates subendocardial ischaemia in dogs with stenosed coronary artery. Cardiovascular research 26: 1212-1218. [Crossref]
- Saeki A, Recchia F, Kass DA (1995) Systolic flow augmentation in hearts ejecting into a model of stiff aging vasculature: influence on myocardial perfusion-demand balance. Circ Res 76: 132-141. [Crossref]
- Ohtsuka S, Kakihana M, Watanabe H, Sugishita Y (1994) Chronically decreased aortic distensibility causes deterioration of coronary perfusion during increased left ventricular contraction. J Am Coll Cardiol 24: 1406-1414. [Crossref]
- Isoyama S, Maruyama Y, Ashikawa K, Sato S, Suzuki H, et al. (1983) Effects of afterload reduction on global left ventricular and regional myocardial functions in the isolated canine heart with stenosis of a coronary arterial branch. Circ 67: 139-147. [Crossref]
- Hajar R (2017) Risk factors for coronary artery disease: Historical perspectives. Heart views: the official journal of the Gulf Heart Association 18: 109. [Crossref]
- Lloyd-Jones DM, Leip EP, Larson MG, d’Agostino RB, Beiser A, et al. (2006) Prediction of lifetime risk for cardiovascular disease by risk factor burden at 50 years of age. Circ 113: 791-798. [Crossref]
- Nadeem M, Ahmed SS, Mansoor S, Farooq S (2013) Risk factors for coronary heart disease in patients below 45 years of age. Pakistan journal of medical sciences 29: 91-96. [Crossref]
- Kannel WB, Vasan RS (2009) Is age really a non-modifiable cardiovascular risk factor? Am J Cardiol 104: 1307-1310. [Crossref]
- Al-Shudifat AE, Johannessen A, Azab M, Al-Shdaifat A, AbuMweis SS, et al. (2017) Risk factors for coronary artery disease in patients undergoing elective coronary angiography in Jordan. BMC cardiovascular disorders 17: 183. [Crossref]
- Roeters van Lennep JE, Westerveld HT, Erkelens DW, van der Wall EE (2002) Risk factors for coronary heart disease: implications of gender. Cardiovascular research 53: 538-549. [Crossref]
- Rafique R (2015) Ischemic Heart Disease in Pakistani Women: A Case Control Study. Journal of Asian Health. [Crossref]
- Lloyd-Jones DM, Larson MG, Beiser A, Levy D (1999) Lifetime risk of developing coronary heart disease. The Lancet 353: 89-92. [Crossref]
- Shea S, Ottman R, Gabrieli C, Stein Z, Nichols A (1984) Family history as an independent risk factor for coronary artery disease. J Am Coll Cardiol 4: 793-801. [Crossref]
- Snowden CB, Mcnamara PM, Garrison RJ, Feinleib M, Kannel WB, et al. (1982) Predicting coronary heart disease in siblings—a multivariate assessment: the Framingham Heart Study. Am J Epidemiol 115: 217-222. [Crossref]
- Pohjola-Sintonen S, Rissanen A, Liskola P, Luomanmäki K (1998) Family history as a risk factor of coronary heart disease in patients under 60 years of age. Eur Heart J 19: 235-239. [Crossref]
- Critchley JA, Capewell S (2003) Mortality risk reduction associated with smoking cessation in patients with coronary heart disease: a systematic review. Jama 290: 86-97. [Crossref]
- Wald NJ (1997) Prospective study of effect of switching from cigarettes to pipes or cigars on mortality from three smoking related diseases. BMJ 314: 1860. [Crossref]
- Jolly K, Bradley F, Sharp S, Smith H, Thompson S, et al. (1999) Randomised controlled trial of follow up care in general practice of patients with myocardial infarction and angina: final results of the Southampton heart integrated care project (SHIP). Bmj 318: 706-711. [Crossref]
- Wilhelmsen L (1988) Coronary heart disease: epidemiology of smoking and intervention studies of smoking. Am Heart J 115: 242-249. [Crossref]
- Morris JN, Heady JA, Raffle PA, Roberts CG, Parks JW (1953). Coronary heart-disease and physical activity of work. The Lancet 262: 1111-1120. [Crossref]
- Berlin JA, Colditz GA (1990) A meta-analysis of physical activity in the prevention of coronary heart disease. Am J Epidemiol 132: 612-628. [Crossref]
- Center for Disease Control and Prevention. (2017). Defining Adult Overweight and Obesity. [Crossref]
- Kannel WB, Lebauer EJ, Dawber TR, Mcnamara PM (1967) Relation of body weight to development of coronary heart disease: The Framingham Study. Circ 35: 734-744. [Crossref]
- Poirier P, Giles TD, Bray GA, Hong Y, Stern JS, Pi-Sunyer FX, et al. (2006) Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss: an update of the 1997 American Heart Association Scientific Statement on Obesity and Heart Disease from the Obesity Committee of the Council on Nutrition, Physical Activity, and Metabolism. Circ 113: 898-918. [Crossref]
- Lama RM, Alonso AF, Gil-Campos M, Leis RT, Martínez VS, et al. (2006) Recommendations of the nutrition committee of the Spanish association of pediatrics. Part I. Prevention. Early detection. Role of the pediatrician. InAnales de pediatria 65: 607-615. [Crossref]
- McTigue KM, Harris R, Hemphill B, Lux L, Sutton S, et al. (2003) Screening and interventions for obesity in adults: summary of the evidence for the US Preventive Services Task Force. Annals of internal medicine 139: 933-949. [Crossref]
- Kannel WB, McGee DL (1979) Diabetes and cardiovascular disease: the Framingham study. Jama 241: 2035-2038. [Crossref]
- Loud and Clear that there is a Strong Correlation between Cardiovascular Disease (CVD) and Diabetes. Heart Org. [Crossref]
- Law MR, Wald NJ, Thompson SG (1994) By how much and how quickly does reduction in serum cholesterol concentration lower risk of ischaemic heart disease?. Bmj 308: 367-372. [Crossref]
- Dai X, Wiernek S, Evans JP, Runge MS (2016) Genetics of coronary artery disease and myocardial infarction. World journal of cardiology 8: 1-23. [Crossref]
- Thiriet,M (2015) Coronary artery disease and myocardial infarction. In Diseases of the Cardiac Pump (pp. 379-471). Springer, Cham. [Crossref]
- Srikanth S, A Ambrose J (2012) Pathophysiology of coronary thrombus formation and adverse consequences of thrombus during PCI. Current cardiology reviews 8: 168-176. [Crossref]
- Hung MJ, Hu P, Hung MY (2014) Coronary artery spasm: review and update. Int. J. Med. Sci. 11: 1161-1171. [Crossref]
- Slavich M, Patel RS (2016) Coronary artery spasm: Current knowledge and residual uncertainties. IJC Heart & Vasculature 10: 47-53. [Crossref]
- Hamm CW, Braunwald E (2000) A classification of unstable angina revisited. Circ 102: 118-122. [Crossref]
- Braunwald E (2018) Unstable angina: an etiologic approach to management Circ 98: 2219-2222 [Crossref]
- Wee Y, Burns K, Bett N (2015) Medical management of chronic stable angina. Australian Prescriber 38: 131-136. [Crossref]
- Meissner MD, Akhtar M, Lehmann MH (1991) Nonischemic sudden tachyarrhythmic death in atherosclerotic heart disease. Circ 84: 905-912. [Crossref]
- Ghuran AV, Camm AJ (2001) Ischaemic heart disease presenting as arrhythmias. British medical bulletin 59: 193-210. [Crossref]
- Alderman EL, Bourassa MG, Cohen LS, Davis KB, Kaiser GG, et al. (1990) Ten-year follow-up of survival and myocardial infarction in the randomized Coronary Artery Surgery Study. Circ 82: 1629-1646. [Crossref]
- Rahimtoola SH, Dilsizian V, Kramer CM, Marwick TH, Vanoverschelde JL (2008) Chronic ischemic left ventricular dysfunction: from pathophysiology to imaging and its integration into clinical practice. JACC: Cardiovascular Imaging 1: 536-555. [Crossref]
- Braunwald E, Rutherford JD. Reversible ischemic left ventricular dysfunction: evidence for the hibernating myocardium. J Am Coll Cardiol 8: 1467-1470. [Crossref]
- Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, et al. (2013) 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 62: e147-e239. [Crossref]
- Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, et al. (2016) 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J 37: 2129-2200. [Crossref]
- Fihn SD, Gardin JM, Abrams J, Berra K, Blankenship JC, et al. (2012) 2012 ACCF/AHA/ACP/AATS/PCNA/SCAI/STS guideline for the diagnosis and management of patients with stable ischemic heart disease: a report of the American College of Cardiology Foundation/American Heart Association task force on practice guidelines, and the American College of Physicians, American Association for Thoracic Surgery, Preventive Cardiovascular Nurses Association, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol 60: e44-164. [Crossref]
- Felker GM, Thompson RE, Hare JM, Hruban RH, Clemetson DE, et al. (2000) Underlying causes and long-term survival in patients with initially unexplained cardiomyopathy. N Engl J Med 342: 1077-1084. [Crossref]
- Davies SW (2001) Clinical presentation and diagnosis of coronary artery disease: stable angina. British medical bulletin 59: 17-27. [Crossref]
- Arai AE (2011) The cardiac magnetic resonance (CMR) approach to assessing myocardial viability. J Nucl Cardiol 18: 1095-1102. [Crossref]
- Cwajg JM, Cwajg E, Nagueh SF, He ZX, Qureshi U, et al. (2000) End-diastolic wall thickness as a predictor of recovery of function in myocardial hibernation: relation to rest-redistribution Tl-201 tomography and dobutamine stress echocardiography. J Am Coll Cardiol 35: 1152-1161. [Crossref]
- Senior R, Lahiri A (1995) Enhanced detection of myocardial ischemia by stress dobutamine echocardiography utilizing the “biphasic” response of wall thickening during low and high dose dobutamine infusion. J Am Coll Cardiol 26: 26-32. [Crossref]
- Schinkel AF, Bax JJ, Poldermans D, Elhendy A, Ferrari R, et al. (2007) Hibernating myocardium: diagnosis and patient outcomes. Current problems in cardiology 32: 375-410. [Crossref]
- Morgan RB, Kwong R (2015) Role of cardiac MRI in the assessment of cardiomyopathy. Current treatment options in cardiovascular medicine 17: 53. [Crossref]
- Baer FM, Voth E, Schneider CA, Theissen P, Schicha H, et al. (1995) Comparison of low-dose dobutamine–gradient-echo magnetic resonance imaging and positron emission tomography with [18F] fluorodeoxyglucose in patients with chronic coronary artery disease: a functional and morphological approach to the detection of residual myocardial viability. Circ 91: 1006-1015. [Crossref]
- Pegg TJ, Selvanayagam JB, Jennifer J, Francis JM, Karamitsos TD, et al. (2010) Prediction of global left ventricular functional recovery in patients with heart failure undergoing surgical revascularisation, based on late gadolinium enhancement cardiovascular magnetic resonance. J Cardiovasc Magn Reson 12: 56. [Crossref]
- Ramani K, Judd RM, Holly TA, Parrish TB, Rigolin VH, et al. (1998) Contrast magnetic resonance imaging in the assessment of myocardial viability in patients with stable coronary artery disease and left ventricular dysfunction. Circ 98: 2687-2694. [Crossref]
- Kim RJ, Wu E, Rafael A, Chen EL, Parker MA, et al. (2000) The use of contrast-enhanced magnetic resonance imaging to identify reversible myocardial dysfunction. N Engl J Med 343: 1445-1453. [Crossref]
- Sandstede JJ, Lipke C, Beer M, Harre K, Pabst T, et al. (2000) Analysis of first-pass and delayed contrast-enhancement patterns of dysfunctional myocardium on MR imaging: use in the prediction of myocardial viability. Am J Roentgenol 174: 1737-1740. [Crossref]
- Klein C, Nekolla SG, Bengel FM, Momose M, Sammer A, et al. (2002) Assessment of myocardial viability with contrast-enhanced magnetic resonance imaging: comparison with positron emission tomography. Circ 105: 162-167. [Crossref]
- Wellnhofer E, Olariu A, Klein C, Gräfe M, Wahl A, et al. (2004) Magnetic resonance low-dose dobutamine test is superior to SCAR quantification for the prediction of functional recovery. Circ 109: 2172-2174. [Crossref]
- Selvanayagam JB, Kardos A, Francis JM (2005) Value of delayed-enhancement cardiovascular magnetic resonance imaging in predicting myocardial viability after surgical revascularization. ACC Current Journal Review 1: 28. [Crossref]
- Kühl HP, Lipke CS, Krombach GA, Katoh M, Battenberg TF, et al. (2006) Assessment of reversible myocardial dysfunction in chronic ischaemic heart disease: comparison of contrast-enhanced cardiovascular magnetic resonance and a combined positron emission tomography–single photon emission computed tomography imaging protocol. Eur Heart J 27: 846-853. [Crossref]
- Bondarenko O, Beek AM, Nijveldt R, McCann GP, van Dockum WG, et al. (2007) Functional outcome after revascularization in patients with chronic ischemic heart disease: a quantitative late gadolinium enhancement CMR study evaluating transmural scar extent, wall thickness and periprocedural necrosis. J Cardiovasc Magn Reson 9: 815-821. [Crossref]
- Wu YW, Tadamura E, Yamamuro M, Kanao S, Marui A, et al. (2007) Comparison of contrast-enhanced MRI with 18F-FDG PET/201Tl SPECT in dysfunctional myocardium: relation to early functional outcome after surgical revascularization in chronic ischemic heart disease. Journal of Nuclear Medicine 48: 1096-1103. [Crossref]
- Becker M, Lenzen A, Ocklenburg C, Stempel K, Kühl H, et al. (2008) Myocardial deformation imaging based on ultrasonic pixel tracking to identify reversible myocardial dysfunction. J Am Coll Cardio 51: 1473-1481. [Crossref]
- Allman KC, Shaw LJ, Hachamovitch R, Udelson JE (2002) Myocardial viability testing and impact of revascularization on prognosis in patients with coronary artery disease and left ventricular dysfunction: a meta-analysis. J Am Coll Cardio 39: 1151-1158. [Crossref]
- Romero J, Xue X, Gonzalez W, Garcia MJ (2012) CMR imaging assessing viability in patients with chronic ventricular dysfunction due to coronary artery disease: a meta-analysis of prospective trials. JACC: Cardiovascular Imaging 5: 494-508. [Crossref]
- Krittayaphong R, Laksanabunsong P, Maneesai A, Saiviroonporn P, Udompunturak S, et al. (2008) Comparison of cardiovascular magnetic resonance of late gadolinium enhancement and diastolic wall thickness to predict recovery of left ventricular function after coronary artery bypass surgery. J Cardiovasc Magn Reson 10: 41. [Crossref]
- Felker GM, Shaw LK, O’Connor CM (2002) A standardized definition of ischemic cardiomyopathy for use in clinical research. J Am Coll Cardio 39: 210-218. [Crosref]
- Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): A randomised trial (1999). Lancet 353: 9-13. [Crossref]
- Merit-HF Study Group (1999) Effect of metoprolol CR/XL in chronic heart failure: metoprolol CR/XL randomised intervention trial in-congestive heart failure (MERIT-HF). The Lancet 353: 2001-2017. [Crossref]
- Pfeffer MA, Swedberg K, Granger CB, Held P, McMurray JJ, et al. (2003) Effects of candesartan on mortality and morbidity in patients with chronic heart failure: the CHARM-Overall programme. The Lancet 362: 759-766. [Crossref]
- Pitt B, Remme W, Zannad F, Neaton J, Martinez F, Roniker B, et al. (2003) Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 348: 1309-1321. [Crossref]
- Manley JC, King JF, Zeft HJ, Johnson WD (1976) The" bad" left ventricle. Results of coronary surgery and effect on late survival. J Thorac Cardiovasc Surg 72: 841-848. [Crossref]
- Vlietstra RE, Assad-Morell JL, Frye RL, Elveback LR, Connolly DC, et al. (1977) Survival predictors in coronary artery disease. Medical and surgical comparisons. Mayo Clinic Proceedings 52: 85-90. [Crossref]
- Faulkner SL, Stoney WS, Alford WC, Thomas CS, Burrus GR, et al. (1977) Ischemic cardiomyopathy: medical versus surgical treatment. J Thorac Cardiovasc Surg 74: 77-82. [Crossref]
- Velazquez EJ, Lee KL, Deja MA, Jain A, Sopko G, et al. (2011) Coronary-artery bypass surgery in patients with left ventricular dysfunction. N Engl J Med 364: 1607-1616. [Crossref]
- Alderman EL, Andrews K, Bost J, Bourassa M, Chaitman BR, et al. (1996) The Bypass Angioplasty Revascularization Investigation (BARI) Investigators Comparison of coronary bypass surgery with angioplasty in patients with multivessel disease. N Engl J Med 335: 217-225. [Crossref]
- Morrison DA, Sethi G, Sacks J, Henderson W, Grover F, et al. (2001) Angina With Extremely Serious Operative Mortality Evaluation (AWESOME). Percutaneous coronary intervention versus coronary artery bypass graft surgery for patients with medically refractory myocardial ischemia and risk factors for adverse outcomes with bypass: a multicenter, randomized trial. Investigators of the Department of Veterans Affairs Cooperative Study# 385, the Angina With Extremely Serious Operative Mortality Evaluation (AWESOME). J Am Coll Cardiol 38: 143-149. [Crossref]
- Bangalore S, Guo Y, Samadashvili Z, Blecker S, Hannan EL (2016) Revascularization in patients with multivessel coronary artery disease and severe left ventricular systolic dysfunction: everolimus eluting stents vs. coronary artery bypass graft surgery. Circ 133: 2132-2140. [Crossref]
- Schomig A, Mehilli J, de Waha A, Seyfarth M, Pache J, et al. (2008) A meta-analysis of 17 randomized trials of a percutaneous coronary intervention-based strategy in patients with stable coronary artery disease. J Am Coll Cardiol 52: 894-904. [Crossref]
- Trikalinos TA, Alsheikh-Ali AA, Tatsioni A, Nallamothu BK, Kent DM (2009) Percutaneous coronary interventions for non-acute coronary artery disease: a quantitative 20-year synopsis and a network meta-analysis. The Lancet 373: 911-918. [Crossref]
- Bucher HC, Hengstler P, Schindler C, Guyatt GH (2000) Percutaneous transluminal coronary angioplasty versus medical treatment for non-acute coronary heart disease: meta-analysis of randomised controlled trials. Bmj 321: 73-77. [Crossref]
- Participants RT (1993) Coronary angioplasty versus coronary artery bypass surgery: the Randomised Intervention Treatment of Angina (RITA) trial. The Lancet 341: 573-580. [Crossref]
- King SB, Lembo NJ, Weintraub WS, Kosinski AS, Barnhart HX, et al. (1994) A randomized trial comparing coronary angioplasty with coronary bypass surgery. N Engl J Med 331: 1044-1050. [Crossref]
- CABRI Trial Participants (1995) First-year results of CABRI (coronary angioplasty versus bypass revascularisation investigation). Lancet 346: 1179-1184. [Crossref]
- Rodriguez A, Bernardi V, Navia J, Baldi J, Grinfeld L, et al. (2001) Argentine randomized study: coronary angioplasty with stenting versus coronary bypass surgery in patients with multiple-vessel disease (ERACI II): 30-day and one-year follow-up results. J Am Coll Cardiol 37: 51-58. [Crossref]
- Serruys PW, Unger F, Sousa JE, Jatene A, Bonnier HJ, et al. (2001) Comparison of coronary-artery bypass surgery and stenting for the treatment of multivessel disease. N Engl J Med 344: 1117-1124. [Crossref]
- SoS Investigators (2002) Coronary artery bypass surgery versus percutaneous coronary intervention with stent implantation in patients with multivessel coronary artery disease (the Stent or Surgery trial): a randomized controlled trial. The Lancet 360: 965-970. [Crossref]
- Eefting F, Nathoe H, van Dijk D, Jansen E, Lahpor J, etal. (2003) Randomized comparison between stenting and off-pump bypass surgery in patients referred for angioplasty. Circ 108: 2870-2876. [Crossref]
- Serruys PW, Morice MC, Kappetein AP, Colombo A, Holmes DR, et al. (2009) Percutaneous coronary intervention versus coronary-artery bypass grafting for severe coronary artery disease. N Engl J Med 360: 961-972. [Crossref]
- Thiele H, Neumann-Schniedewind P, Jacobs S, Boudriot E, Walther T, et al. (2009) Randomized comparison of minimally invasive direct coronary artery bypass surgery versus sirolimus-eluting stenting in isolated proximal left anterior descending coronary artery stenosis. J Am Coll Cardiol 53: 2324-2331. [Crossref]
- Boudriot E, Thiele H, Walther T, Liebetrau C, Boeckstegers P, et al. (2011) Randomized comparison of percutaneous coronary intervention with sirolimus-eluting stents versus coronary artery bypass grafting in unprotected left main stem stenosis. J Am Coll Cardiol 57: 538-545. [Crossref]
- Park SJ, Kim YH, Park DW, Yun SC, Ahn JM, et al. (2011) Randomized trial of stents versus bypass surgery for left main coronary artery disease. N Engl J Med 364: 1718-1127. [Crossref]
- Mohr FW, Morice MC, Kappetein AP, Feldman TE, Ståhle E, et al. (2013) Coronary artery bypass graft surgery versus percutaneous coronary intervention in patients with three-vessel disease and left main coronary disease: 5-year follow-up of the randomised, clinical SYNTAX trial. The Lancet 381: 629-638. [Crossref]
- Windecker S, Kolh P, Alfonso F, Collet JP, Cremer J, et al. (2014) 2014 ESC/EACTS guidelines on myocardial revascularization: the Task Force on Myocardial Revascularization of the European Society of Cardiology (ESC) and the European Association for Cardio-Thoracic Surgery (EACTS) developed with the special contribution of the European Association of Percutaneous Cardiovascular Interventions (EAPCI). Eur Heart J 35: 2541-2619. [Crossref]