The generation of reactive oxygen species (ROS) results in oxidative stress, leading to damage of tissue via many cellular molecular pathways. ROS can result in damage of principal cellular components of the cell such as lipids, proteins, and nucleic acids which then cause cell death via different modes of necrosis or apoptosis. Therefore, it is very important to maintain the redox status in our body. This is maintained by balance in the production of free radicals and antioxidants. Oxidative stress is the main phenomena which occur in progression of many diseases such as diabetes, neurodegenerative diseases, cancers etc. Alzheimer disease (AD) is one of the neurodegenerative disease and is common form of dementia in elderly people. The etiology of this disease is multifactorial; pathologically it is accompanied with accumulation of amyloid beta and neurofibrillary tangles. Accumulation of amyloid beta and mitochondrial dysfunction leads to oxidative stress. There is natural antioxidant defence system in our body which helps us to prevent us from various diseases via different mechanisms. Antioxidants are believed to act against the detrimental effects of ROS and thereby preventing or treating oxidative stress-related diseases. The nuclear factor erythroid 2-related factor 2 (Nrf2) is an emerging regulator of cellular resistance to oxidants. It controls the basal and induced expression of antioxidant response element-dependent genes. The current review examined the extensive role of oxidative stress in AD. Further investigation into the role that oxidative stress mechanisms seem to play in the pathogenesis of Alzheimer disease may lead to novel clinical interventions.
oxidative stress, antioxidant machinery, alzhemier disease, ros, nrf2
Oxidative stress is generated by pertubance of balance between reactive oxygen species (ROS) and antioxidants. Damage of biomolecules- lipids, proteins and nucleic acids in response to increased ROS levels leads to oxidative stress. It plays a very crucial role in the pathogenesis of many diseases like cardiovascular diseases, cancers, neurodegeneration, cancers, immune disorders, diabetes, aging, etc [1]. There arises a shift in the balance between oxidants and antioxidants in favour of oxidants in oxidative stress. Regulation of redox state is very crucial for cell viability, activation, proliferation, and cellular integrity. Therefore, redox homeostasis plays a paramount role in disease prevention [2]. Aerobic organisms have evolved integrated antioxidant systems, which include many enzyme systems such as superoxide dismutase, catalase, and glutathione system [3]. Furthermore, various transcriptional factors get activated by oxidative stress. These systems tend to repair oxidative damage as they act as oxidative sensors in signal transduction pathway [4]. Recent interest has focused on the intricate ways by which redox signalling integrates these converse properties. Redox balance is maintained by prevention, interception, and repair, and concomitantly the regulatory potential of molecular thiol-driven master switches such as Nrf2/Keap1 or NF-κB/IκB is used for system-wide oxidative stress response [5]. Organisms come across various types of oxidants from internal metabolism and external environmental toxic exposure. The reactive oxygen and nitrogen species result in oxidative stress and are considered as harmful [6,7]. On the other hand, regularized generation of oxidants in normal cells serve to regulate various signalling pathways. Our intricate antioxidant defence systems which is regulated by web of various pathways is adequate to counterbalance the reactive oxidants [8]. The nuclear factor erythroid 2-related factor 2 (Nrf2) emerges as a regulator of cellular resistance to oxidants. It regulates and controls the basal expression of an array of ARE antioxidant response element-dependent genes to regulate various physiological outcomes of oxidant exposure [9].
The vulnerability of the nervous system
The neuronal system is vulnerable to ROS mediated injury as compared to other biological systems due to different physiological and biochemical properties of the brain [10]. The nervous system - including the brain, spinal cord, and peripheral nerves - is very prone to oxidative damage [11]. It is due to various reasons such as; high oxygen consumption of the brain as it demands higher energy, which in turn, results in excessive ROS produced; the neuronal membranes possess higher polyunsaturated fatty acids (PUFA), PUFA being susceptible to free radical damage; high aerobic metabolic activity; elongated axonal morphology which is vulnerable to peripheral injury [12] the excitotoxic glutamate is the major effector that causes oxidative stress; the high Ca2+ traffic across neuronal membranes and interference of ion transport increase intracellular Ca2+, often leading to oxidative stress [13]. Moreover, the elevated levels of iron which can be essential during brain development, facilitates oxidative stress via iron-catalysed formation of ROS [14]. Furthermore, the regions of the brain which possess substantial amount of catecholamines, adrenaline, noradrenaline and dopamine are exceptionally very prone to free radical generation. Catecholamines can generate free radicals via spontaneous breakdown or by being metabolized by endogenous enzymes such as monoamine oxidase [15]. Antioxidant defines mechanisms are modest, in particular, low levels of catalase, glutathione peroxidase, and vitamin E; activated microglia produce ROS and cytokines in a perpetual process; neuronal mitochondria generate O2 [16]. The interaction of NO with superoxide can be implicated also in neuronal degeneration [17] neuronal cells are nonreplicating and thus are sensitive to ROS (Figure 1).
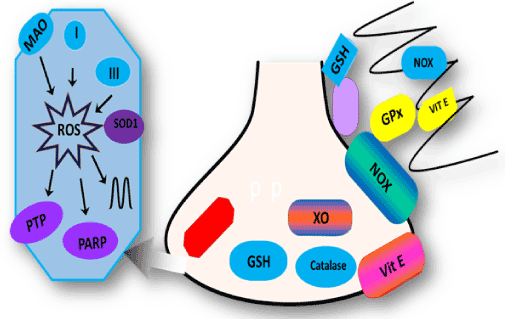
Figure 1. Schematic diagram of main producers of ROS and antioxidant system in neurons and glia. Main ROS producers are monoamine oxidase (MAO), complex I and within mitochondria. ROS generated in mitochondria target the permeability transition pore (PTP), PARP, and mitochondrial DNA. In the cytosol, NADPH oxidase (NOX) and xanthine oxidase (XO) are the main producers of ROS. The major antioxidant systems are shown in green and include superoxide dismutase (SOD) in the mitochondria, glutathione (GSH), catalase (C), and glutathione peroxidase (GPx)
Oxidative stress in Alzhemier disease
Alois Alzhemier a German physician for the first time described AD when he was practicing in Asylum state in Frankfurt. He investigated on patient August D, 51 year old lady in 1901, who suffered from symptoms of cognition, aggressive behaviour and hallucinations. AD is the common form of dementia in elderly people. According to World Health Organization (WHO) estimation, 71% of 81.1 million dementia cases will be reported by 2040 [18,19]. The majority of people which suffer from AD are older individuals usually aged 65 or more. They have sporadic or late onset form of AD, there can be rare case of early onset or we can say familial AD which occur in individuals before age 65 [20]. These people possess autosomal dominant mutation on either one of the presenilin genes located on chromosomes 1 and 14 or in the amyloid precursor protein (APP) gene located on chromosome 21. In addition, there is increased risk of developing early onset AD in individuals suffering from Down’s syndrome [21]. The genetics of sporadic AD is less understood and very complex. It is well known that the epsilon four allele of the apolipoprotein E (APOE) gene located on chromosome 19 is a risk factor for the development of sporadic AD [22].
The etiology of this disease is multifactorial. Pathologically it is characterized by neuronal death, intracellular neurofibrillary tangles and extracellular amyloidal protein deposits contributing to senile plaques [23]. Many different hypotheses have been given from time to time to elucidate the causative factors of this disease in order to explain the multifactorial nature of disease such as cholinergic hypothesis, Aβ hypothesis, tau hypothesis, oxidative stress hypothesis and inflammation hypothesis [24]. Aging has been associated with oxidative damage and is extensive in the brain in Alzheimer disease [25,26]. There are ample evidences supporting the exposure of brain tissue in patients with AD to oxidative stress (e.g., protein oxidation, lipid oxidation, DNA oxidation and glycoxidation) during the course of the disease [27]. The neurons are exposed to higher levels of ROS as compared to other systems of our body [28], and brain accounts for approximately 20% of body’s total oxygen. Moreover, AGEs (Advanced glycation endproducts) found in amyloid plaques and its extracellular accumulation in AD is believed to be caused by an enhanced oxidation of glycated proteins [29]. AGEs cause neuronal cell death directly (chemical) and indirectly (cellular) by increasing oxidative stress due to generation of free radicals. AGEs, a diverse class of posttranslational modifications, are generated by the non-enzymatic reaction of a sugar ketone or aldehyde group with the free amino groups of a protein or amino-acid specifically lysine, arginine and possibly histidine. Accumulation of AGEs in the brain is a feature of aging and it also is also implicated in the development of pathophysiology in age-related diseases such as diabetes mellitus, atherosclerosis, and AD [30].
The most notable feature of degenerative change in AD brain is increased lipid peroxidation as the membrane phospholipids of brains which mainly consists of PUFA is highly vulnerable to free radical attack [31]. Furthermore, the protein oxidation by free radicals might be significant in AD, as the oxidation of proteins of the brain, affect enzymes which are necessary to neuron and glial functions [32]. Two enzymes are very sensitive to oxidative modification namely glutamine synthetase and creatine kinase, which are remarkably low in AD brains, depicting the change of glutamate concentrations and increase of excitotoxicity. While as oxidative impairment of creatine kinase might cause decreased energy metabolism in AD [33]. Moreover neurofibrillary tangles results from aggregation and hyperphosphorylation of tau protein [34]. Phosphorylation is connected to oxidation via microtubule associated protein kinase pathway and through activation of the transcription factor nuclear factor-κB, thus potentially linking oxidation to the hyperphosphorylation of tau (τ) proteins [35]. Oxidation of proteins can also induce advanced glycation end products (AGEs) [36]. Furthermore, oxidation of the proteins can affect DNA, producing strand breaks, sister chromatid exchange, DNA-protein crosslinking, and base modification [37]. As aging process is connected with increase in production of ROS, together with the decrease in the defence system against them, not surprisingly, studies on Alzheimer’s disease over the past ten years have established that oxidative stress and damage are not only in the lesions of AD but also in the neurons at risk of death.
Various lines of evidence have shown that oxidative stress plays a crucial role in the initiation of the AD via various cell signalling pathways [38]. Among them, mitochondrial and metal abnormalities also contribute to oxidative stress. Mitochondria act as source of ROS production [26]. Damaged mitochondria have been observed in AD and the common defect in mitochondria is deficiency in many important enzymes which are responsible for oxidative metabolism including a-ketoglutarate dehydrogenase complex (KGDHC) and pyruvate dehydrogenase complex (PDHC) [39]. These enzymes are involved in rate limiting step of tricarboxylic acid cycle, and another enzyme being the terminal enzyme cytochrome oxidase responsible for reducing molecular oxygen [40]. The production of ROS is the result of these functional abnormalities in mitochondria. Furthermore, damaged mitochondria and formation of mitochondria derived lysosomes and lipofuscin was evident in almost all of AD neurons [26]. Neurons in AD show significantly higher number of completely damaged mitochondria compared to an aged-matched control group [41]. Abnormality in mitochondrial morphology, membrane potential and ROS production was evident from the studies on cybrid cell lines with mitochondria DNA from AD patients [42]. Increased sporadic mutations in the mtDNA control region, with some being unique to AD, were found in AD patients compared to controls [43]. Another study in Tg2567 mice model demonstrated that at mRNA level, gene expression related with mitochondrial metabolism and apoptosis were changed, suggesting mitochondrial energy metabolism is impaired by the expression of APP/Aβ [44].
Iron (Fe) is another cause of oxidative stress because of its excess concentration in brain. It has been found that it accumulates in other parts like hippocampus, cerebral cortex and basal nucleus of Meynert, and colocalizes with AD lesions, senile plaques and neurofibrillary tangles (NFT) [45].
Copper (Cu) is another metal ion that is important for many enzymes in brain metabolism and that has been implicated in disease pathogenesis [46]. The homeostasis of copper is disrupted in AD. There are two pathways connected with copper related oxidative stress. One being the changes in ceruloplasmin and second being the copper interaction with amyloid-β protein precursor (AβPP). The ceruloplasmin is a copper binding protein which is responsible for entry of copper in brain. It plays a pivotal role in protection of cell against oxidative stress. It is a key protein involved in interconversion of redox state of iron by converting the ROS catalytic-Fe (II) to a less reactive Fe (III). It is increased in brain and cerebrospinal fluid in AD, whereas neuronal levels of ceruloplasmin remain unchanged. Thus, higher levels of ceruloplasmin might indicate a compensatory response to oxidative stress, if it fails, so it plays a crucial role in metal catalyzed damage [46].
Copper has also shown to play a role in producing ROS through its binding to Aβ. As with iron, copper concentrations are highly concentrated within Aβ plaques; Aβ binds copper in AD tissue, and Aβ: Cu complexes form a catalytic centre of H2O2, reducing Cu (II) to Cu (I) involving an electron-transfer reaction that can enhance the production of radical •OH. A recent study also reported that tau protein can also bind to Cu, and inappropriately binding with tau protein may trigger oxidative stress [47].
Various studies have shown that Aβ exerts its toxic effects by generating oxidative stress. It induces the oxidation of biomolecules which include peroxidation of lipids and lipoproteins, generates H2O2 and hydroxynonenal (HNE) in neurons, damages DNA and inactivates transport enzymes [27,48]. But there are three conditions which are required by Aβ to induce oxidation. Fibrillation; presence of transition metals and methionine (met) 35. If the peptide is aged the aggregation occurs. Also, the presence of transition metals is a requisite for Aβ aggregation and its pro-oxidant activity [49]. The toxicity of Aβ is likely to be mediated by a direct interaction between this peptide and transition metals with subsequent generation of ROS. It has been demonstrated that the substitution of this met residue by another amino acid abrogates or diminishes significantly the prooxidant action of Aβ [50,51]. Furthermore, AGEs, in the presence of transition metals can undergo redox cycling with consequent ROS production. Additionally, AGEs and amyloid-β activate specific receptors such as the receptor for advanced glycation end products (RAGE) and the class A scavenger-receptor to increase ROS production [29] and modulate gene transcription of various factors involved in inflammation through NF κB activation [52]. Similar to situations in the periphery where damaged tissue and the chronic presence of inert abnormal materials cause inflammation, senile plaques, NFT and injured neurons may well provoke inflammation in the AD brain [53]. Indeed, both activated microglia and astrocytes cluster at sites of Aβ deposition and express a wide range of inflammatory mediators including cytokines and chemokines and cyclooxygenase [54]. Furthermore, Aβ might directly activate the NADPH oxidase of microglia resulting in generation of superoxide radicals and increased production of hydrogen peroxide [55].
Moreover, the production of ROS/reactive nitration species (RNS) by inflammatory cells is a major mechanism for attacking opsonized targets and activated microglia/astrocytes have the potential to produce large amounts of ROS/RNS by various mechanisms [56]. Activated microglia and astrocytes can produce large amounts of nitric oxide (NO), which in turn can react with superoxide to form peroxynitrite, leaving nitrotyrosine as an identifiable marker. The footprint of excess NO production in AD is confirmed by the increased amounts of nitrotyrosine-modified proteins. Plagues in AD brain have shown the expression of Inducible nitric oxide synthase (iNOS) [57]. Another mechanism producing free radical in microglia involves myeloperoxidase (MPO) and there is evidence that MPO immunoreactivity is present in selective highly activated microglia around amyloid plaques in the AD brain and that Aβ aggregates increase MPO mRNA expression in microglia-like cells in vitro [58]. MPO catalyzes a reaction between hydrogen peroxide and chloride to form hypochlorous acid which can further react with other molecules to generate other ROS including hydroxyl ions. MPO can also catalyse the formation of nitrotyrosine-modified proteins as well as cause advanced glycation end product modifications, both of which are evident in AD [59].
Oxidative stress is crucial in the pathogenesis of various diseases and it leads to production of free radicals. It results in damage to macromolecules in cells [60]. Many studies have reported that the nuclear factor erythroid 2 related factor 2 (Nrf2) is a key regulator of endogenous inducible defense systems in the body and increase the level of many antioxidants, including glutathione-s-transferase [61]. Under oxidative damage conditions, Nrf2 transports to the nucleus, then binds to the antioxidant response element (ARE), and enhances sequence to initiate transcription of cytoprotective genes (Figure 2). In general, Nrf2-ARE activation is a novel neuroprotective pathway that can be considered as a promising therapeutic strategy for the treatment of neurodegenerative disorders, such as AD [62].
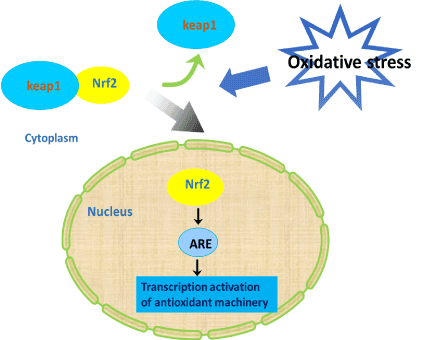
Figure 2. Nrf2 signaling pathway. Under oxidative damage conditions, Nrf2 transports to the nucleus, then binds to the antioxidant response element (ARE), and enhances sequence to initiate transcription of cytoprotective genes
NF-E2 related factor 2 (Nrf2) has been seen to get modulated in various neurodegenerative diseases. Its overexpression is considered as a potential therapeutic target for neurodegenerative disorders such as Amyotrophic lateral sclerosis, Alzheimer’s and Parkinson’s disease [63]. The expression of enzymes involved in phase II detoxification is governed by antioxidant response element (ARE), which is a cis-acting regulatory element. The transcription factor Nrf2 binds to ARE which results in multitude transcription of antioxidant genes. Keap1, a culin 3-based E3 ligase that later degrades Nrf2 and sequesters it in cytoplasm [64]. The endogenous antioxidant capacity of the brain is increased by either disrupting Keap1-Nrf2 interaction or genetic overexpression of Nrf2 that can render protection against oxidative stress in neurodegenerative diseases [65]. Nrf2 is a transcription factor that belongs to Cap ‘n’ collar/basic-leucine zipper family. It regulates the genes containing ARE. It has six erythroid-derived CNC homology protein (ECH) domains. The cytoplasmic association of Keap1 and Nrf2 is governed by a Neh2 domain. Keap1 functions as adaptor for the culin 3-based E3 ligase [66]. Is it necessary to mention that Nrf2 is ubiquitinated and rapidly degraded by ubiquitin-proteasome system under normal condition, as it has half-life ~20 minutes. But under oxidative stress conditions induced by reactive species, toxins or ARE inducers, the association between Nrf2 and Keap1 is disrupted and Nrf2 translocates to the nucleus of the cell, where it binds to small Maf proteins that increase the transcription of genes driven by ARE [67]. In addition, Keap1 also has a role in shifting Nrf2 out of the nucleus by shuffling itself from the cytoplasm to the nucleus. Though the actual mechanism of Keap1-Nrf2 interaction disruption is indescribable, it is reported that direct modification of cysteine thiols groups in Keap1 by ARE inducers leads to the release of Nrf2, thereby increasing its activity [68]. Furthermore, involvement of various kinases through phosphorylation of Nrf2 at serine and threonine residues may also be responsible for translocation of Nrf2 to nucleus. Members of the fibroblast growth factor family have been reported to regulate the transcription of Nrf2, thereby increasing both its mRNA and protein levels. This factor could possibly contribute to the activation of Nrf2 and induction of ARE-driven genes [69].
A multitude of genes that are involved in redox status, detoxification and anti-inflammation are transcribed by activation of Nrf2-ARE pathway. These genes are involved in protection of cell from oxidative burden and cellular injuries in different organs including brain. Nrf2 regulates various antioxidant enzyme systems including peroxiredoxin (Prdx), superoxide dismutase (SOD), catalase (CAT), thioredoxin (Trx), sulfaredoxin (Srx), glutathione peroxidase (Gpx), glutathione reductase (GR), NAD(P)H quinone oxidoreducase (Nqo1) and heme oxygenase 1 (HO-1) and Ferritin.
Though all antioxidant enzymes are essential for brain cells but some antioxidant genes have been reported to have more active roles than others in brain depending on the cell type and disease condition [64]. There is ample evidence in support of the study that the AD brain is under tremendous oxidative stress. Expression of HO-1 gene is significantly increased in post-mortem temporal cortex and hippocampus of AD patients than compared to age-matched normal controls [70]. Furthermore, increase in expression and activity of Nqo1 was found in astrocytes and neurons of AD brain along with predominant cytoplasmic localization of Nrf2 in AD hippocampal neurons [71]. Additionally, increased protein oxidation is common in AD brain when compared to age matched normal controls. A study on aged APP/PS1 AD mouse models showed reduced expression of Nrf2, Nqo1, GCL catalytic subunit (GCLC) and GCL modifier subunit (GCLM) mRNA in addition to decreased Nrf2 protein levels. Another study on triple transgenic AD mouse showed reduced GSH/GSSG ratio [72]. Many studies have focused on neuroprotection role of Nrf2 against ROS generation [73,74].
Antioxidants are the substances which protects cell from damage caused by reactive unstable species known as free radicals. The rate of oxidation is inhibited significantly by antioxidants when present in low concentrations [75]. The antioxidants remove the intermediates of free radical mediated reactions, thereby impeding the chain reactions. They themselves get oxidized in order to stop other oxidation reactions. They help in preventing the growth of many chronic diseases; therefore, antioxidants are emerging as preventive and therapeutic agents [76]. Cellular antioxidants regulate an oxidative phenomenon which is involved in signal transduction, effect gene expression and pathways of cell proliferation and death. Our body possess an intricate natural antioxidant system which prevents us from damage by pro oxidants [77]. Defective endogenous antioxidant system leads to accumulation of free radicals and finally leads to oxidative stress which in turn is involved in pathogenesis of various diseases such as various cancers, diabetes, and neurodegenerative diseases etc [78]. Apart from endogenous systems, there are various dietary sources of antioxidants such as polyphenols which possess antioxidant potential [79].
Our bodies possess natural endogenous defines mechanisms which include both enzymatic antioxidant systems and cellular molecules which protect against free radical induced cellular damage [80]. Enzymatic machinery comprises of superoxide dismutase (SOD), catalase, and glutathione peroxidase which are considered as primary enzymes. They are involved in direct elimination of active oxygen species like superoxide radical and H2O2. In addition, there are various secondary enzymes such as glutathione reductase, glucose-6-phosphate dehydrogenase, and cytosolic GST that function to decrease peroxide levels or to maintain a steady supply of metabolic intermediates like glutathione (GSH) and NADPH for optimum functioning of the primary antioxidant enzymes (Meister, 1992) [81]. Apart from these systems, there are various cellular molecules which act as active antioxidants in our body like GSH, ascorbate (vitamin C), a-tocopherol (vitamin E), β-carotene, NADPH, uric acid, bilirubin, sodium selenite, mannitol, sodium benzoate, the iron-binding protein transferrin, dihydrolipoic acid, melatonin, plasma protein thiol, and reduced CoQ10 which are involved in protecting the body from ROS and their byproducts produced during normal cellular metabolism [82].
Endogenous antioxidants
The biological systems possess natural defense mechanisms to fight against free radical induced cell damage [83].
SOD is an enzyme responsible for the reduction of the superoxide anion which is formed in the body via oxidative phosphorylation, inflammation etc [84]. It is involved in conversion of superoxide anion into a product such as hydrogen peroxide that is metabolized easily to water by glutathione peroxidase (GPx) and catalase (CAT). It acts as a first line of defense in the detoxification of the superoxide anion and seems to be involved in processes of tumor removal or cellular differentiation.
Reactive oxygen species are generated during oxidative metabolism in mitochondria. Coenzyme tend to protect mitochondria from these reactive species. Coenzyme Q is naturally occurring antioxidant. It is currently under investigation in amyotrophic lateral sclerosis and Parkinson disease [85]. It is a biologically active quinone. Furthermore, adenosine triphosphate which is a key source of intracellular energy in the human body is synthesized from CoQ [86]. It helps to neutralize free radicals and it tends to stabilize the cell membrane for most optimal functioning. It is the only known lipid which is produced directly within the body that maintains a redox function.
α-Lipoic Acid
α-Lipoic acid is a mitochondrial coenzyme and possess anti-oxidant action. It results in induction of many antioxidant enzymes [87]. Nrf2 is a transcription factor which is induced by a-Lipoic acid. The lipoic acid is very potent antioxidant as it is able to cross blood brain barrier therefore making it an ideal substance in the treatment of AD. As the formation of beta amyloid leads to pathogenesis of AD by formation of free radicals thereby making it a very important coenzyme [88]. Dihydrolipoic acid (DHLA) the reduced form of lipoic acid and lipoic acid are called “universal antioxidants” as they are involved in neutralization of free radicals.
The glutathione antioxidant system consists of reduced glutathione and the enzyme glutathione reductase. Gluthathione serves as various functions in our cells. It acts as redox buffer of the cell [89]. It is a tripeptide composed of glutamic acid, cysteine, and glycine. It also serves as a reducer, conjugates to drugs to make them more soluble in water. In highly oxidizing environment the role of GSH as a reducing agent is significant. The sulfhydryl group of GSH can be used to reduce peroxides. The resulting form of oxidized GSH consists of two molecules of disulfide linked together (GSSG). GSSG is reduced to two molecules of GSH by the help of Glutathione reductase which uses NADPH as the cofactor. NADPH is produced via pentose phosphate pathway. Glutathione peroxidase is a selenium-dependent enzyme that catalyzes the reduction of hydrogen peroxide (H2O2) or lipoperoxide (L-OOH) using the reduced glutathione (GSH) [90].
Glutathione peroxidases (GPX)
GPX consists of multiple isoenzymes which catalyse the reduction of H2O2 and lipid peroxides utilizing GSH as an electron donor [91]. They are localized both in cytosol and mitochondria. There are five various isoforms of selenium-dependent glutathione peroxidases (GPX1-4 and 6) and three non-selenium congeners (GPX 5, 7 and 8) that have cysteine instead of selenocysteine, in mammals [92].
Catalase
Catalase (CAT) is one of the most abundant enzymes in nature and is widely distributed in the human body [93]. It is a tetrameric enzyme consisting of four identical tetrahedrally arranged subunits of 60 kDa, which contains a single ferriprotoporphyrin group per subunit and has a molecular mass of about 240 kDa. Catalases catalyze the conversion of hydrogen peroxide to water and oxygen, using either an iron or manganese cofactor [94].Here, its cofactor is oxidized by one molecule of hydrogen peroxide and then regenerated by transferring the bound oxygen to a second molecule of substrate [95].
Catalase-Fe (III) + H2O2→ compound I Compound I+H2O2→ catalase-Fe (III) + 2H2O+O2.
Nrf 2
Apart from endogenous enzyme systems, there occur a transcriptional mechanism to reduce cellular oxidative stress. Nrf2 is sequestered in the cytoplasm by interaction with the kelch like enoyl-CoA hydratase-associated protein 1 (ECH) associated protein (Keap1) which leads to its subsequent degradation by the proteasome and ubiquitination. Upon exposure to oxidative stress or electrophiles, Nrf2 translocate to the nucleus where it binds to antioxidant responsive elements (ARE). Nrf2 is released either by phosphorylation of keap 1 or oxidation of sulfhydryl groups on specific cysteines in keap1 [96]. After that Nrf2 is stabilized and translocate from the cytoplasm to the nucleus via bipartite nuclear localization signal where it then transactivates expression of detoxification enzymes, reducing molecules, antioxidant enzymes, and Nrf2 itself [97]. Oxidative damage is prevented by these gene products. When the antioxidant response is no longer needed Nrf is removed from nucleus with the help of nuclear export sequence near its nuclear localization signal.
Nqo1
NAD (P) H:quinone acceptor oxidoreductase 1 (NQO1) ) is a widely-distributed FAD-dependent flavoprotein that promotes obligatory 2-electron reductions of quinones, quinone imines, nitroaromatics, and azo dyes, at rates that are comparable with NADH or NADPH [98]. These reductions depress quinone levels and thereby minimize opportunities for generation of reactive oxygen intermediates by redox cycling, and for depletion of intracellular thiol pools. It is a highly-inducible enzyme that is regulated by the Keap1/Nrf2/ARE pathway. It protects cells from oxidative stress, redox cycling, and neoplastic lesion [99]. Additionally, Nqo1 is also involved in regeneration and α-tocopherol (vitamin E) metabolism.
Apart from endogenous sources there are many dietary natural antioxidants such as vitamin C, vitamin E, and ß-carotene which renders protection against free radicals. Natural dietary antioxidants include Vitamin A, C, and E, carotenoids, flavonoids, and polyphenols [100].
Vitamin C
It is one of the important water soluble antioxidant as it has the capacity to neutralize ROS [101]. It is involved in a number of metabolic reactions in plants and animals. It is a well-known antioxidant that protects the body against oxidative stress. One of the studies showed lower plasma levels of vitamin C in patients with AD despite adequate dietary intake. On the other hand, several studies provided evidence to support a therapeutic role of vitamin C in AD [102]. Combining other antioxidants with vitamin C may prove beneficial for AD prevention by providing a more comprehensive activation of pathways to reduce oxidative stress [103].
Vitamin E
Vitamin E is the major lipid-soluble, chain-breaking, non-enzymatic potent antioxidant in the body [104]. As vitamin E is essential for normal neurological functions, it is potentially useful in preventing and treating neurodegenerative diseases. Vitamin E represents a group of 8 antioxidants (containing 4 tocotrienols and 4 tocopherol. a-tocopherol is the most investigated form of vitamin E in relation to AD and MCI (Mild Cognitive Impairment). Reduced plasma levels of α-tocopherol have been detected in subjects with AD and MCI. It protects membrane fatty acids from lipid peroxidation [105]. The vast majority of literature from animal studies and randomized trials has related a-tocopherol to brain function. Higher levels of α-tocopherol were strongly associated with lower amyloid load as well as with less severe neurofibrillary tangle pathology.
β -carotene
ß-carotene and other carotenoids also provide antioxidant protection to lipid rich tissues. Fruits and vegetables are major sources of carotenoids [106].
Phytonutrients
There are number of other dietary antioxidants which occur in nature other than vitamins, they are collectively known as phytochemicals or phytonutrients. Flavonoids are one such example. They are group of polyphenolic compounds widely distributed in plants and beverages like beer, tea and wine. Flavonoids show anti-inflammatory, anti-allergic, and anti-hepatotoxic activities. Several biological properties are attributed to their antioxidant properties and free radical scavenging capabilities.
Benfotiamine
Benfotiamine is a fat-soluble form of thiamine. It’s considered a potent antioxidant that has been used over the past decade to treat pain symptoms. Benfotiamine is also useful in maintaining brain health. It is used as a broad-spectrum neuroprotector against neuropathies, neuralgia pathologies, and impaired coronary circulation. It is known to reduce amyloid plague as well as tau levels in cortical regions of the transgenic mice brains. Moreover, it increases the phosphorylation level of glycogen synthase kinase-3alpha and -3beta, and reduced their enzymatic activities in the amyloid precursor protein/presenilin-1 transgenic brain [107]. Furthermore, in animal models it appeared to improve the cognitive function and reduce amyloid deposition via thiamine-independent mechanisms, which are likely to include the suppression of glycogen synthase kinase-3 activities. These results suggest that, unlike many other thiamine-related drugs, benfotiamine may be beneficial for clinical Alzheimer's disease treatment. This form of B1 could offer a safe and effective way to reverse memory-destroying plaque in the brain, helping to stop the progression of Alzheimer’s. Clinical trials are also ongoing for this analogy of vitamin B1.
Quercetin is one of the most prominent dietary antioxidant claimed to exert many positive effects on health, including protection against various diseases such as osteoporosis, lung cancer, and cardiovascular disease [108]. It is a member of the flavonoids family. Several in vitro and in vivo studies have provided supportive evidence for neuroprotective effects of quercetin, either against neurotoxic chemicals or in various models of neuronal injury and neurodegenerative diseases. It is currently in Phase II clinical trial [109].
SK-PC-B70M
SK-PC-B70M is an oleanolic-glycoside saponin-enriched fraction derived from the root of Pulsatilla koreana. Recently, it was reported that hederacolchiside-E is an active ingredient of SK-PC-B70M that confers a neuroprotective effect against the cytotoxicity induced by amyloid β (1-42) in SK-N-SH neuroblastoma cells [110].
Curcumin
Curcumin has multiple desirable characteristics for a neuroprotective drug, including anti-inflammatory, antioxidant, and anti-protein aggregate activities [111]. Because of its oral safety, long history of use, and inexpensive cost, curcumin has great potential for the prevention of multiple neurological conditions. It has been found that curcmin reduced oxidative damage, inflammation, and cognitive deficits in rats receiving CNS infusions of toxic Aβ. Curcumin has repeatedly been investigated in clinical trials in AD patients; however, there were no significant positive results reported so far. The mechanism of action is not specified.
Epigallocatechin gallate (EGCG) is the most abundant catechin in green tea, a beverage widely consumed worldwide. It is a component of Japanese green tea extract [112]. It acts as a potent antioxidant. Furthermore, it has also showed anti-inflammatory and antiatherogenic properties in experimental studies conducted in vitro and in vivo [113]. EGCG also attenuated the increase in malondialdehyde levels caused by cerebral ischemia and reduced the formation of post ischemic brain edema and infarct volume. The neuroprotective effect of EGCG against ischemia-induced brain damage was found, in part, due to the modulation of NOS isoforms and preservation of mitochondrial complex activity and integrity. It is likely to have a multitarget mechanism of action similar to most herbal preparations with a strong antioxidant component.
Gingko biloba
Gingko biloba extract is a phytoestrogen that is registered with Ipsen and Schwabe in 1992 as a dietary supplement that improves the cognitive function of patients with senile and peripheral vascular dementia [114]. Ginkgo biloba extract has been therapeutically used for several decades to increase peripheral and cerebral blood flow as well as for the treatment of dementia [115]. The extract contains multiple compounds such as flavonoids and terpenoids that are thought to contribute to its neuroprotective and vasotropic effects.
INM-176
It is another herbal medicine that is based on ferruginosa acid complexes and is used in some food additives [116]. It has an analgesic effect and is indicated to improve the condition of AD patients by improving blood circulation as well as reducing psychiatric symptoms. In primary microglial cells, INM-176 significantly inhibited LPS-induced nitric oxide release and expression of tumor necrosis factor-α and interleukin-1β.
Resveratrol
A widely used herbal medicine, Resveratrol (3, 5, 4’-trihydroxy-trans-stilbene) is a kind of polyphenol produced in several plants, especially grapes skin and seeds, and a phytoalexin against pathogens such as bacteria or fungi [117]. A typical multitargeted medicine and antioxidant, targets monoamine oxidase (MAO-A), beta-secretase, xanthine oxidase, NF-kappaB, and several others enzymes. It has been found to exhibit neuroprotective function in animal models, mostly rat models [118,119]. It has been shown that resveratrol activates Nrf2/ARE signaling pathway [120].
Currently ‘‘only’’ approved treatments by US Food and Drug Administration (FDA), includes five drugs that are used to treat the cognitive manifestations of AD. Acetylcholiesterae inhibitors AChEIs - rivastigmine (Exelon), galantamine (Razadyne, Reminyl), tacrine (Cognex), and Donepzil (Aricept) and NMDA receptor antagonist - memantine (Namenda) that target symptoms at its best [121,122] Reminyl, Exelon and Aricept are effective in the early stages of treatment. Each drug has different mechanism of action and a different way to decrease the breakdown of acetylcholine which is an important neurotransmitter in the brain [123]. In AD there is a decreased level of this neurotransmitter. Memantine (Namenda) is the only drug which is shown to be effective at the later development of the disease [124].
To date, established treatments are only symptomatic in nature, trying to counterbalance the neurotransmitter disturbance of the disease. All the treatments suffer from various side effects. Tacrine (Cognex) is rarely prescribed due to its serious side effects (liver damage). They have all been shown to modestly slow the progression of cognitive symptoms and reduce problematic behaviours in some people, but at least half of the people who take these drugs do not respond to them [125] At the same time antipsychotic and antidepressant treatments are used for the behavioural symptoms of the disease [126].
Novel strategies have been developed to modify the disease process. In this regard major development is targeted to the Aβ and tau based therapeutics, which is a major key to unlock this disease in the near future [127]. New approaches to develop drugs for the treatment of AD that prevent free radical production and hence neurodegeneration including AGE-inhibitors, antioxidants and anti-inflammatory substances are being emphasized. The development of disease-modifying drugs for AD is recognized as a worldwide necessity [128]. These must presumably be drugs that will modify, either by stabilizing or slowing, the molecular pathological steps leading to neurodegeneration and finally dementia.
Natural products and herbal remedies have been a source of many beneficial drugs. About 80% of the world’s population is dependent on plant based medicines [129]. Herbal mixtures might have advantages as they have multiple target approach as compared with the single target [130]. It has been challenging to treat AD. Herbal therapy can be a novel treatment option for AD. Phytotherapy may be a potential corner stone based on which treatment strategies can be streamlined [131]. There is evidence which suggest that herbs or herbal formulations may provide complementary cognitive benefits to the approved drugs, however due to various methodological limitations, their use alone, it remain inconclusive. On the basis of various positive results from clinical trials, herbal therapy formulations may offer certain complementary cognitive benefits to the approved drugs [132,133]. As many drugs are available today for treatment of AD, various plant and their extracts are extensively employed in vivo models and AD patients. Herbal extracts produce a diverse range of natural products including alkaloids, indoles, phytosterols, isoflavonoids which exhibit complex pharmacological properties [134]. Plants provide wealth of bioactive compounds, which exert a substantial strategy for the treatment of neurological disorders such as Alzheimer's disease [135].
The anti-alzhemeric property of phytochemicals has been attributed to them by either of the following mechanisms:
Antioxidative and radical scavenging activities
Acetylcholiesterase inhibition
Anti-inflammatory activity
Modulation of antioxidant machinery
Plants with traditional knowledge of antioxidant and anti-alzhemeric activity have been studied [136]. Some of the examples are: Withania somnifera showed antioxidant activity in Alzheimer’s disease by increasing the levels of the major free-radical scavenging enzymes like superoxide dismutase, catalase and glutathione peroxidase in the frontal cortex and striatum [137]. Curcuma longa (Turmeric) has shown potent antioxidant and anti-inflammatory activity. It has shown to activate Nrf2/ARE signaling [138]. Antioxidant activity is shown by Centella asiatica. It reduces brain lipid peroxidation (LPO) and protein carbonyl levels [139]. It also reversed Aβ pathology [140].
Bacopa monneri shows anti-alzhemeric activity by possessing antioxidant activity by increasing levels of superoxide dismutase, catalase and glutathione peroxidase in the prefrontal cortex, striatum, and hippocampus [141]. Ocimum sanctum inhibits lipid peroxide generation thereby showing antioxidant activity 159.The seeds of Cassia obtisufolia showed neuroprotective role in mice via attenuation of Ca2+ ion dysregulation. Moreover they inhibit AChE [142]. The neuroprotective role of extracts of Cassia obtisufolia might be due to group of phenolic compounds i.e., flavonoids [143]. Dried ginger showed Ca2+ antagonistic activity and butylcholinesterase inhibition activity which are effective in AD treatment [144]. Aqueous and ethyl acetate extract of Convolvulus pluricaulis has shown memory enhancement effect. Various secondary metabolites have been isolated from it such as terpenoids, anthocyanins, steroids which are responsible for its effects [145]. Zeatin shows protective role against Aβ induced neurotoxicity in neuronal cell line PC12 and ameliorate scopolomine induced amnesia in mice models [146]. Gingko biloba showed various health benefits to the patients of AD. It possess various antioxidants. It has various effects like antiamyloid aggregation. There are few substantial studies which support amelioration of AD. The clinical trials have been promising. Clinical evaluation of EGb761 is widely used for dementia. In many countries and is used as dietary supplement on large scale in US for memory enhancement. It has been found to improve AD symptoms both in vivo and in vitro studies [147]. Desmodium gangeticum commonly known as Salparni has been used in ayurveda extensively for the amelioration of neurological symptoms. Various in vivo studies have been carried down. Furthermore, it also showed potent antioxidant property [148]. Rosmarinic acid which is isolated from Salvia officinalis attenuates many processes proved by reactive oxygen species, lipid peroxidation, DNA fragmentation, Caspase 3 activation and Tau protein hyperphosphorylation. Other pharmacological activities include antioxidant, anti-inflammatory, AChE inhibition yet the mode of action is elusive. It effectively inhibits hall mark events of AD-like formation of fibrils from Aβ, destabilization preformed Aβ fibrils in vitro and tau hyperphosphorylation [149]. Melissa officinalis extract has been proven to ameliorate mild to moderate AD. It might present a natural treatment for AD by amelioration of cognition [150]. Huperzine A extracted from the serrate clubmoss herb is a potent, reversible and selective inhibitor of acetylcholinesterase. From various trails, it seems to have some beneficial effects on improvement of cognitive function [151,152]. Panaxsaponin main ingredient of Panaxi ginseng enhance cognitive performance. It decrease level of Aβ and repair damaged neurons. Medhya Rasayanas or drugs from Ayurveda considerably ameliorate memory and intellect. In vivo studies on rats evaluated that the oral dose of Trasina, a herbal formulation, once daily 21 days effectively ameliorate colchinine induced effects. Herbal formulation may have advantages with multiple target regulation as compared with the single target antagonist. There have been few clinical trials examining the efficacy and safety of herbal formulations in AD patients.
As oxidative stress plays a crucial part in pathogenesis of AD, it remains a challenge to design some sort of treatment intervention because it lacks a typical treatment target. To combat oxidative stress involve augmenting antioxidant defences eg: with nutritional supplements or vitamins. Various epidemiological, clinical and basic researches provide a strong support for antioxidant treatment in AD. It is plausible that one single antioxidant may not be sufficient to resist the oxidative damage since the oxidative stress is modulated by a complex system of endogenous and exogenous antioxidants. In this regard, the combinatory approach of antioxidants would be necessary to be studied in the treatment of neurodegenerative diseases. Antioxidant therapy can provide treatment option. The integrated approach is needed to combat the pathogenesis of AD. Several substances are considered as therapeutic candidates for oxidative stress; however, further preclinical and clinical studies are required before clinical application of these substances.
The authors acknowledge the support from University Grants Commission.
- Szymanska R, Pospisil P, Kruk J (2016) Plant-derived antioxidants in disease prevention. Oxidative Medicine and Cellular Longevity 2017: 5092754.
- Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O (2012) Oxidative stress and antioxidant defense. World Allergy Organ J 5: 9-19. [Crossref]
- Rhee SG, Yang KS, Kang SW, Woo HA, Chang TS (2005) Controlled elimination of intracellular H (2) O (2): regulation of peroxiredoxin, catalase, and glutathione peroxidase via post-translational modification. Antioxid Redox Signal 7: 619-26.
- Hussain T, Tan B, Yin Y, Blachier F, Tossou MC, et al. (2016) Oxidative stress and inflammation: what polyphenols can do for us? Oxid Med Cell Longev 2016: 7432797. [Crossref]
- Sies H, Berndt C, Jones DP (2017) Oxidative stress. Annu Rev Biochem 86: 715-748. [Crossref]
- Ma Q (2013) Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 53: 401-426. [Crossref]
- Rahal A, Kumar A, Singh V (2014) Oxidative stress, prooxidants, and antioxidants: the interplay. BioMed Research International 2014: 761264.
- Kashou AH, Agarwal A (2011) Oxidants and antioxidants in the pathogenesis of HIV/AIDS. The Open Reproductive Science Journal 3: 154-161.
- Iranshahy M, Iranshahi M, Abtahi SR, Karimi G (2018) The role of nuclear factor erythroid 2-related factor 2 in hepatoprotective activity of natural products: A review. Food Chem Toxicol 120: 261-276.
- Beckhauser TF, Francis-Oliveira J, De Pasquale R (2016) Reactive oxygen species: Physiological and physiopathological effects on synaptic plasticity. J Exp Neurosci 10: 23-48. [Crossref]
- Morgan MJ, Kim YS, Liu ZG (2008) TNFalpha and reactive oxygen species in necrotic cell death. Cell Res 18: 343-349. [Crossref]
- Wang X, Michaelis EK (2010) Selective neuronal vulnerability to oxidative stress in the brain. Frontiers in Aging Neuroscience 2: 12.
- Pereira CF, Oliveira CR (2000) Oxidative glutamate toxicity involves mitochondrial dysfunction and perturbation of intracellular Ca2+ homeostasis. Neurosci Res 37: 227-236.
- Oliveira AI, Pinho C, Sarmento B, Dias ACP (2016) Neuroprotective activity of hypericum perforatum and its major components. J Front Plant Sci 7: 1004.
- Kumar A, Singh A, Ekavali (2015) A review on Alzheimer's disease pathophysiology and its management: an update. Pharmacol Rep 67: 195-203. [Crossref]
- Ding X, Zhang M, Gu R, Xu G, Wu H (2017) Activated microglia induce the production of reactive oxygen species and promote apoptosis of co-cultured retinal microvascular pericytes. Graefes Arch Clin Exp Ophthalmol 255: 777-788.
- Kumar V, Khan AA, Tripathi A, Dixit PK, Bajaj UK (2015) Role of oxidative stress in various diseases: relevance of dietary antioxidants. The Journal of Phytopharmacology 4: 126-132.
- Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, et al. (2005) Global prevalence of dementia: a delphi consensus study. Lancet 366: 2112-2117. [Crossref]
- Kalaria RN, Maestre GE (2008) Alzheimer’s disease and vascular dementia in developing countries: prevalence, management, and risk factors. Lancet Neurology 7: 812-826.
- Van Cauwenberghe C, Van Broeckhoven C, Sleegers K (2016) The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genetics in Medicine 18: 421-430.
- Bekris LM, Yu CE, Bird TD, Tsuang DW (2010) Genetics of Alzheimer disease. J Geriatr Psychiatry Neurol 23: 213-227. [Crossref]
- Bagyinszky E, Youn YC, An SSA, Kim S (2014) The genetics of Alzheimer’s disease. Clinical Interventions in Aging 9: 535-551.
- Chen X, Guo C, Kong J (2012) Oxidative stress in neurodegenerative diseases. Neural Regeneration Research 7: 376-385.
- Mohandas E, Rajmohan V, Raghunath B (2009) Neurobiology of Alzheimer's disease. Indian J Psychiatry 51: 55-61. [Crossref]
- Harman D (1992) Free radical theory of aging. Mutat Res 275: 257-266. [Crossref]
- Wang X, Wang W, Li L, Perry G, Lee H, et al. (2014) Oxidative stress and mitochondrial dysfunction in alzheimer’s disease. Biochimica et Biophysica Acta 1842: 1240.
- Gella A, Durany N (2009) Oxidative stress in Alzheimer disease. Cell Adh Migr 3: 88-93. [Crossref]
- Su B, Wang X, Nunomura A, Moreira PI, Lee H, Perry G, et al. (2008) Oxidative stress signaling in alzheimer’s disease. Current Alzheimer Research 5: 525-532.
- Sasaki N, Fukatsu R, Tsuzuki K, Hayashi Y, Yoshida T, et al. (1998) Advanced glycation end products in alzheimer’s disease and other neurodegenerative diseases. The American Journal of Pathology. 153: 1149-1155.
- Ott C, Jacobs K, Haucke E, Navarrete Santos A, Grune T, et al. (2014) Role of advanced glycation end products in cellular signaling. Redox Biology 2: 411-429.
- Lobo V, Patil A, Phatak A, Chandra N (2010) Free radicals, antioxidants and functional foods: Impact on human health. Pharmacognosy Reviews 4: 118-126.
- Nita M, Grzybowski A (2016) The role of the reactive oxygen species and oxidative stress in the pathomechanism of the age-related ocular diseases and other pathologies of the anterior and posterior eye segments in adults. Oxidative Medicine and Cellular Longevity 2016: 3164734.
- Huang WJ, Zhang X, Chen WW (2016) Role of oxidative stress in Alzheimer's disease. Biomed Rep 4: 519-522. [Crossref]
- Simic G, Babic Leko M, Wray S, Harrington C, Delalle I, et al. (2016) Tau protein hyperphosphorylation and aggregation in alzheimer’s disease and other tauopathies, and possible neuroprotective strategies. Biomolecules 6: 6.
- Kins S, Kurosinski P, Nitsch RM, Gotz J (2003) Activation of the ERK and JNK signaling pathways caused by neuron-specific inhibition of pp2a in transgenic mice. The American Journal of Pathology 163: 833-843.
- Greifenhagen U, Frolov A, Bluher M, Hoffmann R (2016) Plasma proteins modified by advanced glycation end products (ages) reveal site-specific susceptibilities to glycemic control in patients with type 2 diabetes. The Journal of Biological Chemistry 291: 9610-9616.
- Pfeiffer CJ (1968) A mathematical evaluation of the thymic weight parameter. Toxicology and Pharmacology 13: 220-227. [Crossref]
- Nisha A, Muthukumar SP, Venkateswaran G (2009) Safety evaluation of arachidonic acid rich Mortierella alpina biomass in albino rats-a subchronic study. Regulatory Toxicology and Pharmacology 53: 186-194. [Crossref]
- Zhu X, Lee H, Perry G, Smith MA (2007) Alzheimer disease, the two-hit hypothesis: An update. Molecular Basis of Disease. Biochimica et Biophysica Acta (BBA). 1772: 494-502.
- Kalyanaraman B, Cheng G, Hardy M, Ouari O, Lopez M, et al. (2018) A review of the basics of mitochondrial bioenergetics, metabolism, and related signaling pathways in cancer cells: Therapeutic targeting of tumor mitochondria with lipophilic cationic compounds. Redox Biology 14: 316-327.
- Lee J, Boo JH, Ryu H (2009) The failure of mitochondria leads to neurodegeneration: Do mitochondria need a jump start? Adv Drug Deliv Rev 61: 1316-1323. [Crossref]
- Aliev G, Palacios HH, Gasimov E, Obrenovich ME, Morales L, et al. (2010) Oxidative stress induced mitochondrial failure and vascular hypoperfusion as a key initiator for the development of Alzheimer disease. Pharmaceuticals 3: 158-187.
- Trimmer PA, Keeney PM, Borland MK, Simon FA, Almeida J, et al. (2004) Mitochondrial abnormalities in cybrid cell models of sporadic Alzheimer's disease worsen with passage in culture. Neurobiol Dis 15: 29-39. [Crossref]
- Aliev G, Obrenovich ME, Smith MA, Perry G (2003) Hypoperfusion, Mitochondria Failure, Oxidative Stress, and Alzheimer Disease. Journal of Biomedicine and Biotechnology 3: 162-163.
- Su B, Wang X, Nunomura A, Moreira PI, Lee H, Perry G, et al. (2008) Oxidative stress signaling in alzheimer’s disease. Current Alzheimer Research 5: 525-532.
- Castellani RJ, Rolston RK, Smith MA (2010) Alzheimer disease. Dis Mon 56: 484-546. [Crossref]
- Skjorringe T, Moller LB, Moos T (2012) Impairment of interrelated iron- and copper homeostatic mechanisms in brain contributes to the pathogenesis of neurodegenerative disorders. Front Pharmacol 3: 169.
- Maynard CJ, Bush AI, Masters CL, Cappai R, Li QX (2005) Metals and amyloid-ß in Alzheimer’s disease. International Journal of Experimental Pathology 86: 147-159.
- Cheignon C, Tomas M, Bonnefont-Rousselot D, Faller P, Hureau C, et al. (2018) Oxidative stress and the amyloid beta peptide in Alzheimer's disease. Redox Biol 14: 450-464. [Crossref]
- Gaeta A, Hider RC (2005) The crucial role of metal ions in neurodegeneration: the basis for a promising therapeutic strategy. British Journal of Pharmacology 146: 1041-1059.
- Carrillo-Mora P, Luna R, Colin-Barenque L (2014) Amyloid beta: multiple mechanisms of toxicity and only some protective effects? Oxid Med Cell Longev 2014: 795375. [Crossref]
- Nunomura A, Tamaoki T, Tanaka K, Motohashi N, Nakamura M, Hayashi T, et al. (2010) Intraneuronal amyloid ß accumulation and oxidative damage to nucleic acids in Alzheimer disease. Neurobiology of Disease. 37: 731-737.
- Shi Q, Gibson GE (2007) Oxidative stress and transcriptional regulation in Alzheimer’s disease. Alzheimer Disease and Associated Disorders 21: 276-291.
- Laurent C, Buée L, Blum D (2018) Tau and neuroinflammation: What impact for Alzheimer's disease and Tauopathies? Biomed J 41: 21-33. [Crossref]
- Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, et al. (2015) Neuroinflammation in Alzheimer's disease. Lancet Neurol 14: 388-405. [Crossref]
- Della Bianca V, Dusi S, Bianchini E, Pra ID, Rossi F (1999) ß-Amyloid Activates the O. Forming NADPH Oxidase in Microglia, Monocytes, and Neutrophils. A Possible Inflammatory Mechanism Of Neuronal Damage in Alzheimer’s Disease. The Journal of Biological Chemistry 274: 15493-15499.
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, et al. (2000) Inflammation and Alzheimer's disease. Neurobiol Aging 21: 383-421. [Crossref]
- Calingasan NY, Park LCH, Calo LL, Trifiletti RR, Gandy SE, et al. (1998) Induction of nitric oxide synthase and microglial responses precede selective cell death induced by chronic impairment of oxidative metabolism. The American Journal of Pathology 153: 599-610.
- Gellhaar S, Sunnemark D, Eriksson H, Olson L, Galter D (2017) Myeloperoxidase-immunoreactive cells are significantly increased in brain areas affected by neurodegeneration in Parkinson’s and Alzheimer’s disease. Cell and Tissue Research 369: 445-454.
- Pravalika K, Sarmah D, Kaur H, Wanve M, Saraf J, et al. (2018) Myeloperoxidase and neurological disorder: A Crosstalk. ACS Chem Neurosci 9: 421-430. [Crossref]
- Li W, Yang S (2016) Targeting oxidative stress for the treatment of ischemic stroke: Upstream and downstream therapeutic strategies. Brain Circ 2: 153-163.
- Nguyen T, Nioi P, Pickett CB (2009) The Nrf2-Antioxidant response element signaling pathway and its activation by oxidative stress. The Journal of Biological Chemistry. 284: 13291-13295.
- Niture SK, Khatri R, Jaiswal AK (2014) Regulation of Nrf2-an update. Free Radic Biol Med 66: 36-44. [Crossref]
- Gazaryan IG, Thomas B (2016) The status of Nrf2-based therapeutics: current perspectives and future prospects. Neural Regen Res 11: 1708-1711.
- Joshi G, Johnson JA (2012) The Nrf2-ARE pathway: a valuable therapeutic target for the treatment of neurodegenerative diseases. Recent Pat CNS Drug Discov.7: 218-29.
- Cullinan SB, Gordan JD, Jin J, Harper JW, Diehl JA (2004) The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol Cell Biol 24: 8477-8486. [Crossref]
- Ma Q (2013) Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 53: 401-426. [Crossref]
- Nguyen T, Sherratt PJ, Nioi P, Yang CS, Pickett CB (2005) Nrf2 controls constitutive and inducible expression of ARE-driven genes through a dynamic pathway involving nucleocytoplasmic shuttling by Keap1. J Biol Chem 280: 32485-32492.
- Canning P, Sorrell FJ, Bullock AN (2015) Structural basis of Keap1 interactions with Nrf2. Free Radic Biol Med 88: 101-107. [Crossref]
- Vargas MR, Pehar M, Cassina P, Martinez-Palma L, Thompson JA, et al. (2005) Fibroblast growth factor-1 induces heme oxygenase-1 via nuclear factor erythroid 2-related factor 2 (Nrf2) in spinal cord astrocytes: consequences for motor neuron survival. J Biol Chem 280: 25571-25579.
- Schipper HM, Bennett DA, Liberman A, Bienias JL, Schneider JA, et al. (2006) Glial heme oxygenase-1 expression in Alzheimer disease and mild cognitive impairment. Neurobiol Aging 27: 252-261.
- Ramsey CP, Glass CA, Montgomery MB, Lindl KA, Ritson GP, et al. (2007) Expression of Nrf2 in neurodegenerative diseases. J Neuropathol Exp Neurol 66: 75-85. [Crossref]
- Kanninen K, Malm TM, Jyrkkänen HK, Goldsteins G, Keksa-Goldsteine V, et al. (2008) Nuclear factor erythroid 2-related factor 2 protects against beta amyloid. Mol Cell Neurosci 39: 302-313. [Crossref]
- Wang Z, Guo S, Wang J, Shen Y, Zhang J, et al. (2017) Nrf2/HO-1 mediates the neuroprotective effect of mangiferin on early brain injury after subarachnoid hemorrhage by attenuating mitochondria-related apoptosis and neuroinflammation. Scientific Reports 7 : 11883.
- Velagapudi R, El-Bakoush A, Olajide OA (2018) Activation of Nrf2 pathway contributes to neuroprotection by the dietary flavonoid tiliroside. Mol Neurobiol 55: 8103-8123. [Crossref]
- Halliwell B (1996) Antioxidants in human health and disease. Annu Rev Nutr 16: 33-50. [Crossref]
- Hill MF (2008) Emerging role for antioxidant therapy in protection against diabetic cardiac complications: experimental and clinical evidence for utilization of classic and new antioxidants. Curr Cardiol Rev 4: 259-268.
- Irshad M, Chaudhuri PS (2002) Oxidant-antioxidant system: role and significance in human body. Indian J Exp Biol 40: 1233-1239. [Crossref]
- Bhattacharya A, Chatterjee A, Ghosal S, Bhattacharya SK (1999) Antioxidant activity of active tannoid principles of emblica officinalis (amla). Indian J Exp Biol 37: 676-680. [Crossref]
- Scalbert A, Manach C, Morand C, Rémésy C, Jiménez L (2005) Dietary polyphenols and the prevention of diseases. Crit Rev Food Sci Nutr 45: 287-306. [Crossref]
- Ghareeb MA (2014) Antioxidant and cytotoxic activities of tectona grandis linn leaves. International Journal of Phytopharmacology5: 143-157.
- Meister A (1992) On the antioxidant effects of ascorbic acid and glutathione. Biochem Pharmacol 44: 1905-1915. [Crossref]
- Vendemiale G, Grattagliano I, Altomare E (1999) An update on the role of free radicals and antioxidant defense in human disease. Int J Clin Lab Res 29: 49-55. [Crossref]
- Poljsak B, Suput D, Milisav I (2013) Achieving the balance between ros and antioxidants: when to use the synthetic antioxidants. Oxidative Medicine and Cellular Longevity 2013: 956792.
- Marklund SL (1984) Properties of extracellular superoxide dismutase from human lung. Biochem J 220: 269-272. [Crossref]
- Wadsworth TL, Bishop JA, Pappu AS, Woltjer RL, Quinn JF (2008) Evaluation of coenzyme Q as an antioxidant strategy for Alzheimer's disease. J Alzheimers Dis 14: 225-234. [Crossref]
- Beal MF (2004) Therapeutic effects of coenzyme Q10 in neurodegenerative diseases. Methods Enzymol 382: 473-487. [Crossref]
- Shay KP, Moreau RF, Smith EJ, Smith AR, Hagen TM (2009) Alpha-lipoic acid as a dietary supplement: molecular mechanisms and therapeutic potential. J Biochim Biophys Acta 60: 1790-1149.
- Packer L, Witt EH, Tritschler HJ (1995) Alpha-Lipoic acid as a biological antioxidant. Free Radic Biol Med 19: 227-250. [Crossref]
- Kerksick C, Willoughby D (2005) The Antioxidant Role of Glutathione and N-Acetyl-Cysteine Supplements and Exercise-Induced Oxidative Stress. J Int Soc Sports Nutr 2: 38-44.
- Presnell CE, Bhatti G, Numan LS, Lerche M, Alkhateeb SK, et al. (2013) Computational insights into the role of glutathione in oxidative stress. Curr Neurovasc Res 10: 185-194. [Crossref]
- Jurkovic S, Osredkar J, Marc J (2008) Molecular impact of glutathione peroxidases in antioxidant processes. J Biochemia Medica 18: 162-74.
- Dayer R, Fischer BB, Eggen RIL, Lemaire SD (2008) The Peroxiredoxin and glutathione peroxidase families in chlamydomonas reinhardtii. Genetics 179: 41-57.
- Díaz A, Muñoz-Clares RA, Valdés VJ, Hansberg W (2005) Functional and structural analysis of catalase oxidized by singlet oxygen. Biochimie 87: 205-214. [Crossref]
- Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, et al. (2007) Free radicals and antioxidants in normal physiological functions and human disease. The International Journal of Biochemistry & Cell Biology 39: 44-84.
- Tiedge M, Lortz S, Drinkgern J, Lenzen S (1997) Relation between antioxidant enzyme gene expression and antioxidative defense status of 1nsulin producing cells. Diabetes 46: 1733-1742.
- Muiswinkel V, Freek L, Kuiperij HB (2005) The Nrf2-ARE signaling pathway: promising drug target to combat oxidative stress in neurodegenerative disorders cns & neurological disorders. Drug Targets 4: 267-81.
- Li W, Khor TO, Xu C, Shen G, Jeong WS, et al. (2008) Activation of Nrf2-antioxidant signaling attenuates NF kappaB-inflammatory response and elicits apoptosis. Biochem Pharmacol 76: 1485-1489.
- Dinkova-Kostova AT, Talalay P (2010) NAD (P) H:quinone acceptor oxidoreductase 1 (NQO1), a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector. Archives of Biochemistry and Biophysics. 501: 116-123.
- Atia A, Alrawaiq N, Abdullah A (2014) A Review of NAD (P) H: Quinone Oxidoreductase 1 (NQO1); A multifunctional antioxidant enzyme. Journal of Applied Pharmaceutical Science 4: 118-122.
- Landete JM (2013) Dietary intake of natural antioxidants: vitamins and polyphenols. Crit Rev Food Sci Nutr 53: 706-721. [Crossref]
- Riviere S, Aragon IB, Nourhashemi F, Vellas B (1998) Low plasma vitamin C in Alzheimer patients despite an adequate diet. International journal of geriatric psychiatry 13: 749-754.
- Huang J, May JM (2006) Ascorbic acid protects SH-SY5Y neuroblastoma cells from apoptosis and death induced by beta-amyloid. Brain Res 1097: 52-58. [Crossref]
- Padayatty SJ, Katz A, Wang Y, Eck P, Kwon O, et al. (2003) Vitamin C as an antioxidant: evaluation of its role in disease prevention. J Am Coll Nutr 22: 18-35. [Crossref]
- Burton GW, Traber MG (1990) Vitamin E: antioxidant activity, biokinetics, and bioavailability. Annu Rev Nutr 10: 357-382. [Crossref]
- Engelhart MJ, Ruitenberg A, Meijer J, Kiliaan A, van Swieten JC, et al. (2005) Plasma levels of antioxidants are not associated with Alzheimer's disease or cognitive decline. Dement Geriatr Cogn Disord 19: 134-139. [Crossref]
- Fiedor J, Burda K (2014) Potential role of carotenoids as antioxidants in human health and disease. Nutrients 6: 466-488. [Crossref]
- Pan X (2010) "Powerful beneficial effects of benfotiamine on cognitive impairment and beta-amyloid deposition in amyloid presursor protein/presenilin-1 transgenic mice." Brain 133: 1342-1351.
- Boots AW, Haenen GR, Bast A (2008) Health effects of quercetin: from antioxidant to nutraceutical. Eur J Pharmacol 585: 325-337. [Crossref]
- Costa LG, Garrick JM, Roquè PJ, Pellacani C (2016) Mechanisms of neuroprotection by quercetin: counteracting oxidative stress and more. Oxid Med Cell Longev 2016: 2986796. [Crossref]
- Seo JS, Kim TK, Leem YH, Lee KW, Park SK, et al. (2009) SK-PC-B70M confers anti-oxidant activity and reduces Abeta levels in the brain of Tg2576 mice. Brain Res 1261: 100-108. [Crossref]
- Cole GM, Teter B, Frautschy SA (2007) Neuroprotective effects of curcumin. Adv Exp Med Biol 595: 197-212. [Crossref]
- Ou HC, Song TY, Yeh YC (2010) “EGCG protects against oxidized LDL-induced endothelial dysfunction by inhibiting LOX-1-mediated signaling,” Journal of Applied Physiology 108: 1745-1756.
- Forester SC, Lambert JD (2011) The role of antioxidant versus pro-oxidant effects of green tea polyphenols in cancer prevention. Mol Nutr Food Res 55: 844-854. [Crossref]
- Ahlemeyer B, Krieglstein J (2003) Neuroprotective effects of ginkgo biloba extract. Cell Mol Life Sci 60: 1779-1792. [Crossref]
- Luo Y (2001) Ginkgo biloba neuroprotection: Therapeutic implications in Alzheimer's disease. J. Alzheimers Dis 3: 401-407.
- Park SJ, Jung HJ, Son MS, Jung JM, Kim DH, et al. (2012) Neuroprotective effects of INM-176 against lipopolysaccharide-induced neuronal injury. Pharmacol Biochem Behav 101: 427-33.
- De la Lastra CA, Villegas I (2007) Resveratrol as an antioxidant and pro-oxidant agent: mechanisms and clinical implications. Biochem Soc Trans 35: 1156-1160.
- Ma T, Tan MS, Yu JT, Tan L (2014) Resveratrol as a therapeutic agent for Alzheimer's disease. Biomed Res Int 2014: 350516. [Crossref]
- Jia Y, Wang N, Liu X (2017) Resveratrol and Amyloid-Beta: Mechanistic Insights. Nutrients 9. [Crossref]
- Kode A, Rajendrasozhan S, Cato S, Yang SR, Megson IL, et al. (2008) Resveratrol induces glutathione synthesis by activation of Nrf2 and protects against cigarette smoke-mediated oxidative stress in human lung epithelial cells. Am J. Physiol Lung Cell. Mol. Pathol 294: L478-488.
- Desai P, Shete H, Adnaik R, Disouza J, Patravale V (2005) Therapeutic targets and delivery challenges for Alzheimer’s disease. World J. Pharmacol 4: 236-264.
- Yiannopoulou KG, Papageorgiou SG (2013) Current and future treatments for Alzheimer’s disease. Therapeutic Advances in Neurological Disorders 6: 19-33.
- Onor ML, Trevisiol M, Aguglia E (2007) Rivastigmine in the treatment of Alzheimer's disease: an update. Clin Interv Aging 2: 17-32. [Crossref]
- Tampi RR, van Dyck CH (2007) Memantine: efficacy and safety in mild-to-severe Alzheimer’s disease. Neuropsychiatric Disease and Treatment 3: 245-258.
- Casey DA, Antimisiaris D, O'Brien J (2010) Drugs for Alzheimer's disease: are they effective? P T 35: 208-211. [Crossref]
- Ballard C, Corbett A (2010) Management of neuropsychiatric symptoms in people with dementia. CNS Drugs 24: 729-739. [Crossref]
- Aprahamian I, Stella F, Forlenza OV (2013) New treatment strategies for Alzheimer's disease: is there a hope? Indian J Med Res 138: 449-460. [Crossref]
- Gurib-Fakim A (2006) Medicinal plants: traditions of yesterday and drugs of tomorrow. Mol Aspects Med 27: 1-93. [Crossref]
- Tian J, Shi J, Zhang X, Wang Y (2010) Herbal therapy: a new pathway for the treatment of Alzheimer's disease. Alzheimers Res Ther 2: 30. [Crossref]
- Singhal AK, Naithani V, Bangar OP (2012) Medicinal plants with a potential to treat Alzheimer and associated symptoms. Int J Nutr Pharmacol Neurol Dis 2: 84-91.
- Fu LM, Li JT (2009) A systematic review of single chinese herbs for Alzheimer’s disease treatment. Evid Based Complement Alternat Med 2011: 640284
- Kennedy DO, Wightman EL (2011) Herbal extracts and phytochemicals: plant secondary metabolites and the enhancement of human brain function. Advances in Nutrition 2: 32-50.
- Obulesu M, Rao DM (2011) Effect of plant extracts on Alzheimer's disease: An insight into therapeutic avenues. J Neurosci Rural Pract 2: 56-61. [Crossref]
- Yuan H, Ma Q, Ye L, Piao G, et al. (2016) The traditional medicine and modern medicine from natural products. Molecules 21. [Crossref]
- Bhattacharya A, Ghosal S, Bhattacharya SK (2001) Anti-oxidant effect of withania somnifera glycowithanolides in chronic footshock stress-induced perturbations of oxidative free radical scavenging enzymes and lipid peroxidation in rat frontal cortex and striatum. J Ethnopharmacol 74: 1-6. [Crossref]
- Scapagnini G, Vasto S, Abraham NG, Caruso C, Zella D, et al. (2011) Modulation of Nrf2/ARE pathway by food polyphenols: a nutritional neuroprotective strategy for cognitive and neurodegenerative disorders. Mol Neurobiol 44: 192-201. [Crossref]
- Meena H, Pandey HK, Pandey P, Arya MC, Ahmed Z (2012) Evaluation of antioxidant activity of two important memory enhancing medicinal plants baccopa monnieri and centella asiatica. Indian Journal of Pharmacology. 44: 114-117
- Amala S, Yong P, Joseph F (2012) Centella asiatica extract improves behavioral deficits in a mouse model of Alzheimer's disease: Investigation of a possible mechanism of action. Int J Alzheimer Dis 38: 974-976.
- Simpson T, Pase M, Stough C (2015) “Bacopa monnieri as an antioxidant therapy to reduce oxidative stress in the aging brain,” Evidence-Based Complementary and Alternative Medicine 2015: 615384
- Joshi RK, Setzer WN, Da Silva JK (2017) Phytoconstituents, traditional medicinal uses and bioactivities of Tulsi (Ocimum sanctum Linn.): A review. American Journal of Essential Oils and Natural Products. 5: 18-21
- Benjamin DD, William GL, Riedel ARG, Kim DH, Ryu JHD, et al. (2008) The seed extract of cassia obtusifolia offers neuroprotection to mouse hippocampal cultures. J Pharmacol Sci 107: 380-392
- Ghayur MN, Gilani AH, Ahmed T, Khalid A, Nawaz SA, et al. (2008) Muscarinic, Ca(++) antagonist and specific butyrylcholinesterase inhibitory activity of dried ginger extract might explain its use in dementia. J Pharm Pharmacol 60:1375-83
- Bihaqi SW, Singh AP, Tiwari M (2011) In vivo investigation of the neuroprotective property of convolvulus pluricaulis in scopolamine induced cognitive impairments in wistar rats. Indian J Pharmacol 43: 520-525.
- Choi SJ, Jeong CH, Choi SG, Chun JY, Kim YJ, et al. (2009) Zeatin prevents amyloid beta-induced neurotoxicity and scopolamine-induced cognitive deficits. J Med Food 12: 271-277. [Crossref]
- Joshi H, Parle M (2006) Antiamnesic effects of desmodium gangeticum in mice. Yakugaku Zasshi 126: 795-804. [Crossref]
- Iuvone T, Filippis D, Esposito G, D'Amico A, Izzo AA (2006) The spice sage and its active ingredient rosmarinic acid protect pc12 cells from amyloid-ß peptide-induced neurotoxicity. Journal of Pharmacology and Experimental Therapeutics 317: 1143-1149.
- Ozarowski M, Przemyslaw M, Anna Piasecka A (2016) Influence of the melissa officinalis leaf extract on long-term memory in scopolamine animal model with assessment of mechanism of action. Evidence-Based Complementary and Alternative Medicine 2016: 9729818.
- Zhang Z, Wang X, Chen Q, Shu L, Wang J, Shan G (2002). Clinical efficacy and safety of huperzine Alpha in treatment of mild to moderate Alzheimer disease, a placebo-controlled, double-blind, randomized trial. Zhonghua Yi Xue Za Zhi 82: 941-944. [Crossref]
- Lee ST, Chu K, Sim JY, Heo JH, Kim M (2008) Panax ginseng enhances cognitive performance in Alzheimer disease. Alzheimer Dis Assoc Disord 22: 222-226.
- Bhattacharya SK, Kumar A (1997) Effect of Trasina, an ayurvedic herbal formulation, on experimental models of Alzheimer's disease and central cholinergic markers in rats. J Altern Complement Med 3: 327-336. [Crossref]
- Persson J, Bringlov E, Nilsson LG, Nyberg L (2004) The memory-enhancing effects of ginseng and ginkgo biloba in healthy volunteers. Psychopharmacology 172: 430-434.