Precise estimation of development of active tuberculosis (TB) infection from a latent Mycobacterium tuberculosis H37Rv (MTB) infection within host body signifies an indefinable and serious objective. Most of the infected individuals with excellent immune system are also at risk due to diverse local environmental and systemic factors. Therefore, it becomes mandatory to have a balance between pro and anti-immune regulators to work efficiently in optimized manner. This study thus signifies some essential factors involved in maintaining this balance and rescue host cellular environment from MTB infection. Inside host, MTB develops effective survival strategies, such as hampering of lysosome-phagosome fusion, hampering of phagosome acidification by a secretary protein phosphatase PtpA that binds to H subunit of V-ATPase to block V-ATPase transportation towards phagosome membrane, recruitment of TACO protein on phagosomal membrane to escape their transportation to lysosome. Bacterial infected cell undergoing apoptosis release ATP, UTP, LPC, S1P and several chemokines that intervene signaling of “find me” signal. Eventually, a balance between all these immunological factors must be set for optimized functioning of immune system against MTB. In this review we have elaborated various balancing mechanisms among pro and anti-inflammatory immune components that raise this disease from latent to active form. Through a brief and complete evaluation of different types of immune components that take part in host defense, we aimed to highlight the review with recent documented literatures to have a better and precise understanding of this disease. In conclusion, this review abridges various defensive strategies that moderate the distinct capability to fight MTB infection.
mycobacterium tuberculosis, immune system, granuloma, inflammation, cytokines, phagosome, apoptosis
The host immune system is a shielding system of the body which protects our body from invading harmful organisms and eventually protects us from diseases. Our immune system consists of several organs and tissues such as adenoids, bone marrow, lymph nodes, lymphatic vessels, Peyer’s patches, spleen, thymus, tonsils and blood (immune cells) [1]. Host immune system basically differentiated into innate and adaptive mechanism which then more defines to specific and non-specific system, respectively. Although adaptive system is the specific one but give delayed response. Any shortcoming in host immune system lead to severe diseases such as autoimmune, inflammatory, cancer and so many microbial diseases [1,2]. Immune system interacts and remembers thousands of different opponents and in response produce so many secretions and cells to match up with and eradicate out each one of them. Once the immune cells get the signals, they undergo planned changes and start to secrete effector molecules. These effector molecules decide the growth, behavior and function of the host cell [3]. Despite of being so effective strategies of immune system to fight with several microbial agents, still Mycobacterium tuberculosis H37Rv (MTB) infection dominates over it. MTB survives inside macrophages by secreting certain enzymes which block the “killing pathways of phagocytosis” and cause tuberculosis. Tuberculosis (TB) is an airborne infectious respiratory disease caused by MTB that seriously put a high risk on human health. MTB primarily affects the lungs but also can affect other parts of our body that could define as pulmonary TB and extra-pulmonary TB, respectively. In case of extra-pulmonary tuberculosis, highly vascular areas such as eye, spine, lymph nodes, kidneys, bones are commonly affected [4]. TB is the serious cause of death and the leading cause from the single contagious agent. According to world health organization (WHO) report 2019, approximately 10 million (range, 9.0-11.1 million) people fell ill with TB in 2018 and globally, 1.2 million (range, 1.2-1.3 million) TB deaths occur among HIV uninfected people and approximately 251,000 deaths reported in HIV infected people. Worldwide, the rough calculation is that approximately 10.0 million (range 9.0 - 11.1 million) develops TB disease in 2018: 57% male, 32% female and 11% youngster of all TB cases in 2018. The severity of national epidemics of TB varies among all countries with largest endemic occur in India, Pakistan, Indonesia, China, Philippines [5]. The incidence rate has been dropped down by only 2% per year which is still very less as compare to population of country. Approximately one third of world population has latent tuberculosis infection (LTBI) which is a state of tenacious immune response. In this case, immune system of host becomes unable to recognize, phagocytose the bacterium and did not show any symptoms of the disease. This state promotes bacterial intracellular survival but however, this state can develop active TB disease in the future when immunity would be weak (WHO 2018). Nowadays, a major challenge is to treat TB when there are cases of multidrug resistant tuberculosis (MDR-TB), extremely drug resistant tuberculosis (XDR-TB) and total drug resistant tuberculosis (TDR-TB) [6]. Advancement to treat MDR-TB defined by resistance to rifampicin and isoniazid which is responsible for 3.6% of the new and 20.2 % of the previously treated TB cases is also very slow [7]. Although the vaccination for TB i.e. Bacille Calmette–Guerin (BCG) does offer some defense against MTB the efficiency of BCG is suboptimal and not enough for disease control [8].
TB is spread by close contact with MTB (aerosolized form) infected individuals and bogus air quality exposure. MTB is extremely victorious bacterium and its victory depends on ability to live and develop inside the macrophages, hiding itself from antibodies, complement and other immune cells. When it enter in host body it initiates a series immune response by interacting with macrophage and develops very effective survival strategies, such as (i) the hampering of lysosome-phagosome fusion; (ii) The hampering of phagosome acidification; (iii) the recruitment and retention of tryptophan aspartate containing coat (TACO) protein on phagosome to avert their transportation to lysosome; (iv) the expression of member of host induced PE/PGRS family of proteins ( PE is a family of proteins rich in proline and glutamine and PGRS is a subgroup of PE family rich in glycine and arginine); (v) inhibiting oxidative stress; (vi) inhibiting apoptosis and autophagy collectively using above strategies to escape host immune system [9]. The cell membrane of MTB contains methyl branched chain fatty acid that shield them from host enzyme and make them also to escape from immune response [10]. The review defines and presents a more elaborative file of known and unknown facts and factors about immune system and their working. TB increased accumulation of various immune cells that are listed below in the human lungs which is said to be primary site of infection, concurring with a robust transcriptional response to infection, including a role in orchestrating the recruitment of immune subsets and start defense mechanism.
Host innate immune response is a first line of defense against contagious agents. It comprised of anatomical barriers such as the skin as well as the complement system and several types of innate immune cells. Innate immunity initiates with pattern recognition of microbial structures called pathogen associated molecular patterns (PAMPs) and it leads to initiation of an adaptive immune response in the host [11,12]. PAMPs are recognized by germ line – encoded receptors expressed on immune cells called PRRs (pattern recognition receptors) [11]. PRRs molecules expressed on macrophages and dendritic cells (Antigen presenting cells) recognize non-indigenous ligands during prior immune response [13]. The best familiar PRRs are probably the Toll-like receptors (TLRs). Despite of TLRs there are several other receptors also found, that include: CR3 (complement receptor 3), mannose receptor, scavenger receptors, and DC-SIGN (DC-specific intercellular-adhesion-molecule-3-grabbing non-integrin), and engagement of these leads to expression of immune components that arbitrate an inflammatory response or drive granuloma formation [14,15].
Inflammation is a type of immune response which is partly facilitated by a group of secreted polypeptides known as cytokines. When MTB infects host, it leads to a local inflammatory response which may be condemnatory to the pathological process of TB generation. Once inflammation is induced, it initiates the secretion of pro-inflammatory cytokines, such as: interleukin 1-beta (IL-1β), tumor necrosis factor alpha (TNF-α), gamma interferon (IFN- ϒ) and interleukin-12 (IL-12) etc. [16]. A major pro-inflammatory cytokine IL-1 β plays a major role by participating in host defense mechanism against infection by increasing antimicrobial properties of phagocytes and initiating Th1 and Th17 adaptive immune responses [17]. IL-1β is activated by processing upon gathering of the inflammasome, an exclusive inflammatory caspases-activating complex of the proteins [18]. After stabilizing itself, MTB is also responsible for secretion of some anti-inflammatory cytokines such as IL-10 and TGF-β which adds in the survival of bacilli inside host cellular environment. Anti-inflammatory cytokines such as IL-10 and TGF-β are also produced by macrophages during MTB infection which down regulate the pro-inflammatory cytokines and T-cell multiplication and activation. These pro-inflammatory and anti-inflammatory cytokines stabilize the reciprocation between removal and proliferation of bacterium [19]. Nucleotide – binding oligomerization domain (NOD) recently described as cytosolic regulators of pro-inflammatory response. NOD-2 found in antigen presenting cell, regulate the production of inflammatory arbitrator in response to bacterial surface components [20]. It is reported that MTB suppress the IL-1β production by secreting anti-inflammatory mediators as described above. Rv0198c gene (zmp1) of MTB plays a critical role to hamper the caspase-1- dependent activation and secretion of IL-1β, thus helps in its survival inside macrophage [21]. This local inflammatory response results in the recruitment of macrophages, monocytes and other innate immune cells to the site of infection and form a specialized structure known as granuloma which is also signatory determinator of the malady [22].
Granuloma is an organized structure (size-1-2 mm) formed by highly differentiated immune cells, to provide shelter for bacterium [23]. The central part of the granuloma is enclosed by infected and frothy macrophages, other mononuclear phagocytes and surrounded by lymphocytes [24]. It provides an unsuccessful host immune response which restricts but unable to control MTB infection [25]. Granuloma considered as a classical pathological hallmark of TB and it also provide an immunologic microenvironment in which the bacterium can grow or preserve as latent state therefore attain success to establish a mutual relationship [25]. Granuloma formation is a sequential process which complete in mainly four steps: (i) antigen presenting cells such as macrophages and dendritic cells ( DC) trigger the activation of T-cells (ii) macrophages, dendritic cells and activated lymphocytes start to release cytokines and chemokines (small proteins secreted by cells that influence the immune system) [26] (iii) secreted cytokines and chemokines stabilize the progressive accumulation of immunocompetent cells (mature B and T cells) and emergence of organized structure known as granuloma; (iv) fibrosis is the last step of the granuloma formation [27]. Excessive bacterial growth or continuous replication steps of bacterium leads to the generation of compromised state (necrosis and apoptosis of MTB contained macrophages) which then favors rupturing of the granuloma [28]. These series of events driven by granuloma including necrosis and apoptosis of macrophages lead to transition from latent TB to active TB infection (Figure 1).
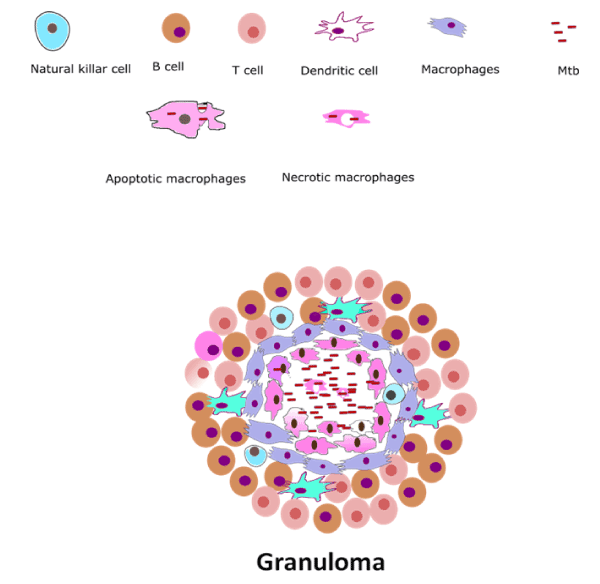
Figure 1. Granulomatous lesion: Granuloma seems to be a self-protective structure that mediates foreign invaders such as bacteria or fungi to keep them far from spreading. On invasion, macrophages, dendritic cells and activated lymphocytes start to release cytokines and these secreted cytokines and chemokines stabilize the progressive accumulation of immunocompetent cells on the site of infection forming granuloma.
Apart from granuloma and inflammatory responses, inhibition of phagosome maturation is a diversion pathway employed by MTB to rescue itself from host immune army. Maturation of phagosome is arrested or manipulated by hampering of phagosome – lysosome fusion which is a most important survival mechanism of MTB inside host macrophages [29]. Hampering of phagosome-lysosome fusion is mainly mediated by IL-10, an anti-inflammatory cytokine resulting in the deactivation of macrophages [30]. Apart from anti-inflammatory effect, IL-10 also blocks the migration of T helper type 1 (Th1) cells and dendritic cells to the site of infection that secrete cytokines such as IFN-gamma, IL-2, and TNF-alpha/beta [30] (Figure 2). These cytokines promote macrophage activation, nitric oxide production and cytotoxic T lymphocyte proliferation leading to the phagocytosis and demolition of microbial pathogens [31]. IL-10 exert their own function through these possible signaling mechanisms, such as Mitogen Activated Protein Kinase p38 (MAPKp38), Extracellular Regulated Kinase 1/2 (ERK1/2), and Signal Transducer and Activator of Transcription 3 (STAT3). IL-10, TNF-α, IL-4 and IFN-γ, production is regulated by Mitogen-activated protein kinase (MAPK) pathway activation [31]. STAT3 inhibits the function of Nf-kB mediator of pro-inflammatory gene induction (Figure 3). In addition to the above functions of IL-10, it also has major effects on the antigen-presenting cells, down regulation of IL-12 production and expression of co-stimulatory molecules in macrophages, thereby reducing the generation of a Th1 response [32-34]. Addition to above mentioned factors, inhibition of phagosome acidification also plays important role to hamper phagosome maturation. V-ATPase (H+-ATPase) a multi subunit protein pump complex that vigorously transports proton using ATP (Adenosine Triphosphate) inside phagosome lumen and acidify the luminal environment. This acidic environment of phagosome inhibits the bacterial growth, enhance the activities of anti-microbial hydrolases and is also crucial for proper vesicular trafficking, directing the fusion of phagosome with lysosome [35]. A secretary protein phosphatase, PtpA binds to H subunits of V-ATPase to block V-ATPase trafficking towards phagosome membrane that eventually blocks the phagosome acidification [35]. A type 1 cytokine IL-27 (Interleukin-27) is a member of the IL-6 sub-family secreted by antigen presenting cells in response to several stimuli and microbial-derived products known to promote Th1 cells differentiation. IL-27 decreases lysosomal acidification in human macrophages by altering the expression of V-ATPase [36]. Apart from inhibition of lysosome fusion and acidification, the TACO/coronin-1 is another factor involved in hampering phagosome maturation. TACO/coronin-1 is a protein which is actin binding plasma membrane associated protein that retains on the phagosomal membrane. TACO/coronin-1 inhibits phagosome–lysosome fusion, as well as in the survival of bacilli within macrophages, which allow escaping bacterial action by preventing lysosomal delivery of mycobacteria [37] (Figure 4). Vitamin D3 and retinoic acid (RA) down regulate the expression of TACO gene by modulation of the VDR/RXR (vitamin ‘D’ receptor/ retinoid-X receptor) response sequence present in the promoter region of TACO gene [38,39]. TACO/coronin-1 is essential to turn on calcium signaling after the entry of mycobacteria into the macrophages, because incorporation of these bacteria is related with a transient increase in intracellular calcium ion concentration [40]. TACO/coronin-1 activates calcineurin, activated calcineurin links up to several downstream signaling cascades ranging from the transfer of transcription factors to the nucleus and cause dephosphorylation of proteins involved in endocytosis [41]. These proteins also modulate the activity of immune system cells, including T and B lymphocytic cells, neutrophils and macrophages [42]. Acquired cellular immune response during MTB infection is widely accepted to consist of T-cells (CD4) and mononuclear phagocytes [43]. Granuloma formation is mediated by T-cells by secreting cytokines such as: INF-ϒ and IL-18R which activate monocytes and macrophages to become microcidal [42].
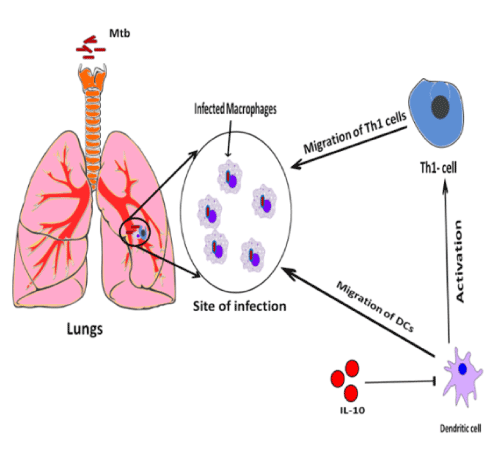
Figure 2. Role of IL-10 in Th 1 and DC migration: Immune system in response to MTB infection, secrete IL-10 as anti-inflammatory response which inhbits macrophage killing and dendritic cells (DC) uptake, processing, presentation, Th1 migration and trafficking of cells from the draining lymph node back to the lungs. IL-10 achieved this effect by inhibiting the dendritic cells (DCs) to further inactivate Th1 cells.
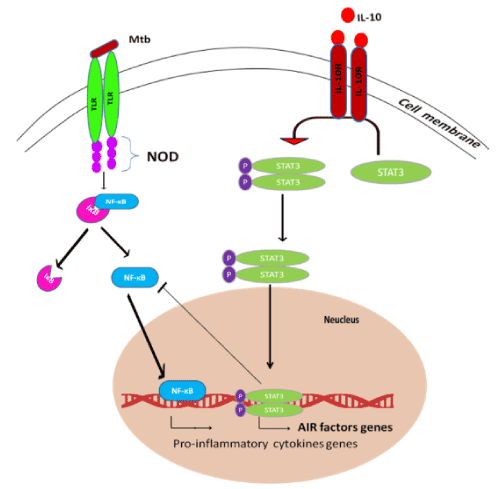
Figure 3. Anti-inflammatory pathway of IL-10: IL-10 prevents up-regulation of many genes in phagocytic and dendritic cells that are normally induced by stimulation via Toll-like receptors (TLRs) or other pattern recognition receptors. Upon binding to its two-chain receptor, IL-10 acts primarily by preventing gene transcription through a mechanism that requires STAT3. STAT3 role is to enhance the expression of specific genes (AIR factors), which in turn repress the expression of pro-inflammatory genes.
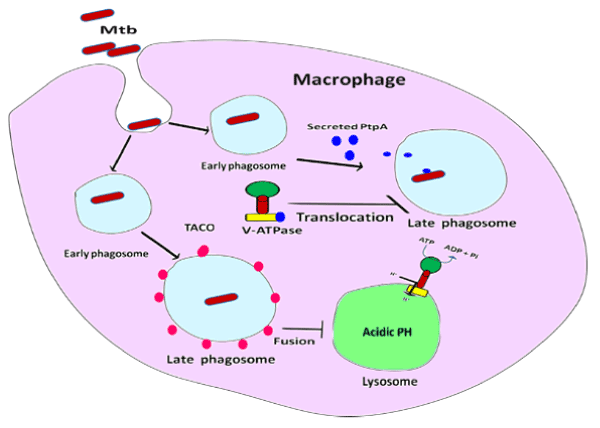
Figure 4. Hampering of phagosome maturation: MTB inside macrophage, inhibits phagosome acidification by secreting PtpA protein in cell cytosol which binds with V-ATPase and inhibits V-ATPase transportation towards phagosomal membrane. TACO/coronin-1 is an actin binding protein which retains on the phagosomal membrane and inhibits phagosome–lysosome fusion.
Cells experiencing stress and need clearance evoke very significant “find me” signals (a signal generated by infected cell to attract phagocytes). “Find me” signals are sensed by macrophages present in vicinity of an infected cell and cause these macrophages to come toward these signals and engulf the cells producing these signals without leaving any remnants behind [44]. Cells undergoing apoptosis release numerous factors such as: Nucleotides (ATP and UTP), Lipids (lysophosphatidylcholine (LPC)) and sphingosine-1-phosphate (S1P), several chemokines and cytokines that mediate signaling cascade of “find me” signal [44,45]. Among all nucleotides, ATP and UTP are well characterized for “find me” signal that released through pannexin-1 channels by macrophages. Liberated ATP and UTP bind to purinergic receptors (P2Y12 and P2Y6) and mediate microglial chemotaxis of the cell toward phagocytes. ATP also elicits inflammatory response by activating NLRP3 inflammasome through its action on P2RX7 (purinergic receptors) that triggers the release of pro-inflammatory cytokines IL-1β and IL-18 which enhance the antimicrobial properties of phagocytes and initiating Th1 and Th17 adaptive immune responses [46]. In addition, apoptotic cell release lipid mediators, apparently before the break down of plasma membrane such as LPC and S1P. On the other side, these lipids will also inhibit the liberation of pro-inflammatory cytokines such as TNF- α and IL-12 by attracting macrophages and enhance the secretion of immunosuppressive factors such as IL-10, PGE2 from the macrophages [47]. Fas/CD95 is a component of TNFR super family which also plays crucial function in regulating cell life span but also in cell –cell communication within immune system [48]. Fas/CD95 stimulate the production of pro-inflammatory cytokines and chemokines such as IL-6, IL-8, Granulocyte-Macrophage Colony-Stimulating Factors (helps make more white blood cells, especially granulocytes, macrophages, and cells that become platelets), CXCL1(recruiting and activating neutrophils for microbial killing at the tissue site), and RANTES (Regulated on Activation, Normal T cell Expressed and Secreted) also called chemokine (C-C motif) ligand 5 (CCL-5) which strengthen immune response by hiring neutrophils, macrophages and basophils to the site of inflammation [47]. TNF- induced proinflammatory cytokines and chemokines triggers the synthesis of Acute Phase Proteins (proteins that change their serum concentration by >25% in response to inflammatory cytokines (IL-1, IL-6, TNF-α)) and also increase the production of macrophages and monocytes from the bone marrow [46]. In case of MTB infection, Mycobacterium impedes the phagosome maturation so that infected cell cannot elicit the “find me” signal due to inhibition of phagosome maturation and compromised host immune system.
Eat-me signal is crucial for the clearance of apoptotic cells to avoid development of autoimmune disease [49]. Recognition of “eat me” signal is mediated by phagocytes that expressed on the surface of apoptotic cells (do not healthy cells) which can later engulf them and actively prevent the inflammation [50]. Phosphotidylseriene (PtdSer) is the well characterized bonafide component of “Eat me” signal which usually resides on the cytosolic leaflet of plasma membrane but in the apoptotic stage, PtdSer flips across the plasma membrane bilayer toward outer side to display on the surface of the apoptotic cell [51]. The externalized PtdSer recognized by its receptor present on phagocytes such as Brain-specific Angiogenesis Inhibitors (BAI), T cell/transmembrane, Immunoglobulin, and Mucin (TIM) and CD300 to mediate efferocytosis (engulfment by phagocytes of dying and dead cells and their debris) [52]. PtdSer binds with Bai-1 receptors and inhibits the levels of IL-1α, IL-6 and TNF, PtdSer binds with Tim-1 and Tim-3 and suppress Nf-kB activation and production of TNF, IL-6, CCL5 and when PtdSer binds with CD300, it increase production of IL-4 and thus potentially endorse M2 polarization in macrophages [51]. M2 polarized macrophages are the alternatively activated macrophages which are immunoregulatory and polarized by Th2 cytokines such as IL-4 and IL-13 which enhance the production of anti-inflammatory cytokines such as IL-10 and TGF-β [53]. MTB induce DCs and macrophages to the release of TNF-α and triggers apoptosis through the classical extrinsic pathway [54]. MTB-infected macrophages are rapidly taken up by uninfected macrophages through efferocytosis. Although apoptosis of MTB-infected macrophages is associated with reduced bacterial growth, the bacteria are relatively resistant to other forms of death. Efferocytosis of an apoptotic macrophage having MTB within the cell further compartmentalizes the bacterium and transport it along with the apoptotic cell debris to the lysosomal content. Killing of MTB only after the efferocytosis, specify that apoptosis itself is not fundamentally bactericidal but requires succeeding phagocytic uptake and lysosomal fusion of the apoptotic body harboring the bacterium [55]. Normal cells mistreat signals use to prevent immune cells from destroying normal cells. These ‘self’ anti-phagocytic signals called “Don’t eat me” signals, express on the surface of the normal cells or cancer cells such as CD-47 and CD-24 [56]. CD-47 binds with signal regulatory protein–a ( SIRP-a) on phagocytic immune cells which results in activation of SIRP-a by which phosphorylation of immunoreceptor tyrosine-based inhibition (ITIM) motifs leading to the recruitment of Src homology phosphatase-1 (SHP-1) and SHP-2 phosphatase preventing myosin-IIA accumulation at the phagocytic synapse preventing phagocytosis and eventually inhibits the engulfment of normal cells [56]. CD-24 is also a novel and prevailing innate immune checkpoint, “Don’t eat me” signal through interaction with the inhibitory receptor sialic acid-binding Ig-like lectin 10 (Siglec-10) that alter the anti-tumor innate immune response [57].
In the brief overview about this report, this can be marked that immunological parameters set an important barrier to protect host cellular environment from unwanted extracellular infections. In addition, from the beginning of 20th century, many immunological components have been tried for immune stimulants for infection. MTB establishes itself as a critical pathogen due to its genetic drift nature that make it drug resistant and forwarding disease to MDR, XDR and TDR level. Pro and anti-inflammatory effect must be ensured to deliver a complete cure to host cellular environment. In most of the cases, they did induce generalized inflammation with significant side effects. Thus, it is a tremendous need to balance out these chemokines and other immunological parameters to inhibit the bacilli at an early stage.
The authors acknowledge financial support from the Department of Science and Technology-SERB, Council of Scientific and Industrial Research-Institute of Genomics and Integrative Biology under the research project GAP0145 (SERB-DST Grant no: EEQ/2016/000514).
There is no conflict of interest
- Goldsby RA, Kindt TJ, Osborne BA, Kuby J (2003) Immunology 5th edition.
- O'Byrne KJ, Dalgleish AG (2001) Chronic immune activation and inflammation as the cause of malignancy. Br J Cancer 85: 473-483. [Crossref]
- US Department of Health and Human Services (2007) Understanding the Immune Systems: How It Work (No. 07-5423). NIH Publication.
- Kandola P, Meena LS (2014) Extra pulmonary tuberculosis: Overview, manifestations, diagnostic and treatment techniques. Adv Mater Rev 1: 13-19.
- Global Tuberculosis Report (2019) World Health Organization.
- Singh R, Rajni, Meena A, Meena LS (2011) Multidrug resistant and Extensively drug resistant TB: A Nuisance to Medical Science. J Bacteriol Parasitol 2: 1-5.
- Rai RC (2014) Convergence of host immune mechanisms in Mycobacterium tuberculosis pathogenesis. Trends Infect Dis 73.
- Berrington WR, Hawn TR (2007) Mycobacterium tuberculosis, macrophages, and the innate immune response: does common variation matter? Immunol Rev 219: 167-186. [Crossref]
- Meena LS, Rajni (2010) Survival mechanisms of pathogenic Mycobacterium tuberculosis H37Rv. FEBS J 277: 2416-2427. [Crossref]
- Zhai W, Wu F, Zhang Y, Fu Y, Liu Z (2019) The immune escape mechanisms of Mycobacterium tuberculosis. Int J Mol Sci 20: 340. [Crossref]
- Kleinnijenhuis J, Oosting M, Joosten LA, Netea MG, Van Crevel R (2011) Innate immune recognition of Mycobacterium tuberculosis. Clin Dev Immunol 405310. [Crossref]
- Bhatt K, Salgame P (2007) Host innate immune response to Mycobacterium tuberculosis. J Clin Immunol 27: 347-362. [Crossref]
- Meena R, Meena LS (2011) Unique characteristic features of Mycobacterium tuberculosis in relation to immune system. Am J Immunol 7: 1-8.
- Gordon S (2002) Pattern recognition receptors: doubling up for the innate immune response. Cell 111: 927-930. [Crossref]
- Das M (2017) The regulatory effects of circulating normal immunoglobulins on autophagy and Th17 response, (Doctoral dissertation, Université Pierre et Marie Curie-Paris VI).
- Saiga H, Shimada Y, Takeda K (2011) Innate immune effectors in mycobacterial infection. Clin Dev Immunol 347594. [Crossref]
- Toossi Z (2000) The inflammatory response in Mycobacterium tuberculosis infection. Inflammation: 139-151.
- Van de V, Frank L, Netea MG, Dinarello CA, Joosten LA (2011) Inflammasome activation and IL-1β and IL-18 processing during infection. Trends Immunol 32: 110-116. [Crossref]
- Master SS, Rampini SK, Davis AS, Keller C, Ehlers S, et al. (2008) Mycobacterium tuberculosis prevents inflammasome activation. Cell Host Microbe 3: 224-232. [Crossref]
- Flynn JL, Chan J, Lin PL (2011) Macrophages and control of granulomatous inflammation in tuberculosis. Mucosal Immunol 4: 271-278. [Crossref]
- Briken V, Ahlbrand SE, Shah S (2013) Mycobacterium tuberculosis and the host cell inflammasome: a complex relationship. Front Cell Infect Microbiol 3: 62. [Crossref]
- Ndlovu H, Marakalala MJ (2016) Granulomas and inflammation: host-directed therapies for tuberculosis. Front Immunol 7: 434. [Crossref]
- Silva Miranda M, Breiman A, Allain S, Deknuydt F, Altare F (2012) The tuberculous granuloma: an unsuccessful host defence mechanism providing a safety shelter for the bacteria? Clin Dev Immunol: 139127. [Crossref]
- Russell DG, Cardona PJ, Kim MJ, Allain S, Altare F (2009) Foamy macrophages and the progression of the human tuberculosis granuloma. Nat Immunol 10: 943-948. [Crossref]
- Ramakrishnan L (2012) Revisiting the role of the granuloma in tuberculosis. Nat Rev Immunol 12: 352-366. [Crossref]
- Kumari P, Meena LS (2014) Factors affecting susceptibility to Mycobacterium tuberculosis: a close view of immunological defence mechanism. Appl Biochem Biotechnol 174: 2663-2673. [Crossref]
- Flynn JL, Chan J, Lin PL (2011) Macrophages and control of granulomatous inflammation in tuberculosis. Mucosal Immunol 4: 271-278. [Crossref]
- Facco M, Miorin M, Agostini C, Semenzato G (2007) Granuloma formation. In Diffuse Parenchymal Lung Disease. Prog Respir Res 36: 87-100.
- Cambier CJ, Falkow S, Ramakrishnan L (2014) Host evasion and exploitation schemes of Mycobacterium tuberculosis. Cell 159: 1497-1509. [Crossref]
- O'Leary S, O'Sullivan MP, Keane J (2011) IL-10 blocks phagosome maturation in Mycobacterium tuberculosis–infected human macrophages. Am J Respir Cell Mol Biol 45: 172-180. [Crossref]
- de Souza AP, Vale VL, Silva Mda C, Araújo IB, Trindade SC, et al. (2014) MAPK involvement in cytokine production in response to Corynebacterium pseudotuberculosis infection. BMC Microbiol 14: 230. [Crossref]
- Hussain T, Shah SZ, Zhao D, Sreevatsan S, Zhou X (2016) The role of IL-10 in Mycobacterium avium subsp. paratuberculosis infection. Cell Commun Signal 14: 29. [Crossref]
- Damsker JM, Hansen AM, Caspi RR (2010) Th1 and Th17 cells. Ann N Y Acad Sci 1183: 211-221. [Crossref]
- Sanjabi S, Zenewicz LA, Kamanaka M, Flavell RA (2009) Anti-inflammatory and pro-inflammatory roles of TGF-β, IL-10, and IL-22 in immunity and autoimmunity. Curr Opin Pharmacol 9: 447-453. [Crossref]
- Wong D, Bach H, Sun J, Hmama Z, Av-Gay Y (2011) Mycobacterium tuberculosis protein tyrosine phosphatase (PtpA) excludes host vacuolar-H+–ATPase to inhibit phagosome acidification. Proc Natl Acad Sci 108: 19371-19376. [Crossref]
- Jungm JY, Robinson CM (2013) Interleukin-27 inhibits phagosomal acidification by blocking vacuolar ATPases. Cytokine 62: 202-205. [Crossref]
- Ferrari G, Langen H, Naito M, Pieters J (1999) A coat protein on phagosomes involved in the intracellular survival of mycobacteria. Cell 97: 435-447. [Crossref]
- Anand PK, Kaul D (2003) Vitamin D3-dependent pathway regulates TACO gene transcription. Biochem Biophys Res Commun 310: 876-877. [Crossref]
- Soni P, Shivangi M, Meena LS (2018) Vitamin D-An Immune Modulator and Growth Inhibitor of Mycobacterium tuberculosis H37Rv. J Mol Biol Biotech 3: 1-2.
- Pieters J (2008) Mycobacterium tuberculosis and the macrophage: maintaining a balance. Cell Host Microbe 3: 399-407. [Crossref]
- Tokarz-Deptuła B, Malinowska M, Adamiak M, Deptuła W (2016) Coronins and their role in immunological phenomena. Cent Eur J Immunol 41: 435-441. [Crossref]
- Orme IM, Andersen P, Boom WH (1993) T cell response to Mycobacterium tuberculosis. J Infect Dis 167: 1481-1497. [Crossref]
- Giacomini E, Iona E, Ferroni L, Miettinen M, Fattorini L, et al. (2001) Infection of human macrophages and dendritic cells with Mycobacterium tuberculosis induces a differential cytokine gene expression that modulates T cell response. J Immunol 166: 7033-7041. [Crossref]
- Medina CB, Ravichandran KS (2016) Do not let death dous part: ‘find-me’signals in communication between dying cells and the phagocytes. Cell Death Differ 23: 979-989. [Crossref]
- Elliott MR, Ravichandran KS (2016) The dynamics of apoptotic cell clearance. Dev cell 38: 147-160. [Crossref]
- Yang Y, Wang H, Kouadir M, Song H, Shi F (2019) Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis 10: 1-11. [Crossref]
- Zitvogel L, Kepp O, Kroemer G (2010) Decoding cell death signals in inflammation and immunity. Cell 140: 798-804. [Crossref]
- Cullen SP, Henry CM, Kearney CJ, Logue SE, Feoktistova M, et al. (2013) Fas/CD95-induced chemokines can serve as “find-me” signals for apoptotic cells. Mol Cell 49: 1034-1048. [Crossref]
- Bell E (2007) Apoptosis: Recognizing'eat-me'signals. Nat Rev Immunol 7: 917.
- Boyle KB, Randow F (2013) The role of ‘eat-me’signals and autophagy cargo receptors in innate immunity. Curr opin microbiol 16: 339-348. [Crossref]
- Li W (2012) Eat‐me signals: Keys to molecular phagocyte biology and “Appetite” control. J Cell Physiol 227: 1291-1297. [Crossref]
- Elliott MR, Koster KM, Murphy PS (2017) Efferocytosis signaling in the regulation of macrophage inflammatory responses. J Immunol 198: 1387-1394. [Crossref]
- Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, et al. (2018) Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol 233: 6425-6440. [Crossref]
- Khan N, Vidyarthi A, Javed S, Agrewala JN (2016) Innate immunity holding the flanks until reinforced by adaptive immunity against Mycobacterium tuberculosis infection. Front Microbiol 7: 328. [Crossref]
- Martin CJ, Booty MG, Rosebrock TR, Nunes-Alves C, Desjardins DM, et al. (2012) Efferocytosis is an innate antibacterial mechanism. Cell Host Microbe 12: 289-300. [Crossref]
- McCracken MN, Cha AC, Weissman IL (2015) Molecular pathways: activating T cells after cancer cell phagocytosis from blockade of CD47 “Don't Eat Me” signals. Clin Cancer Res 21: 3597-3601. [Crossref]
- Bradley CA (2019) CD24—a novel ‘don’t eat me’ signal. Nat Rev Cancer 19: 541-541. [Crossref]