Background: Left ventricular diastolic dysfunction that may develop during the early stages of type 2 diabetes mellitus (T2DM) is an important predictor of heart failure and long-term mortality. However, there is limited prospective evidence to demonstrate the molecular mechanisms of diastolic dysfunction in diabetic patients. Nonhuman primates have physiological, metabolic, biochemical and genetic similarity to humans that facilitate the study of diabetes and cardiovascular disease. This study investigated the pathogenesis of left ventricular diastolic dysfunction in cynomolgus monkeys (Macaca fascicularis) with spontaneous T2DM.
Methods: Four cynomolgus monkeys with T2DM and left ventricular diastolic dysfunction and two nondiabetic healthy controls were analysed in this study. mRNA, protein and phosphoprotein expression profiles were generated from the isolated left ventricle of each monkey and analysed using a systematic approach integrating transcriptomics, proteomics and phosphoproteomics.
Results: Differential expression of 1,404 mRNAs, 528 proteins, and 709 phosphoproteins was observed in the left ventricles of T2DM monkeys compared with controls. Functional analysis of these dysregulated molecules showed that inflammation and immune response and the calcium signalling pathway were prominently altered cellular processes in T2DM monkeys. According to IPA pathway analysis, reduced PP1, SERCA2a, and phosphorylated SERCA2a expression were the main effectors leading to impaired Ca2+ homeostasis in the cardiac sarcoplasmic reticulum and cytoplasm in T2DM monkeys with left ventricular dysfunction. Targeting the new identified phosphorylated SERCA2a might be a new potential therapy for cardiovascular dysfunction in diabetic patients.
Type 2 diabetes mellitus, Left ventricular diastolic dysfunction, Diabetic cardiomyopathy, Calcium signalling, SERCA2a
LVDD: Left ventricular diastolic dysfunction; DDD: diabetes-associated left ventricular diastolic dysfunction; NDC: Nondiabetic control; IPA: Ingenuity Pathway Analysis
Type 2 diabetes mellitus (T2DM) affects 90-95% of diabetic individuals and is a major health problem with increasing incidence worldwide [1]. Cardiovascular disease is the most severe T2DM comorbidity, with cardiovascular complications accounting for approximately half of diabetes-associated deaths [2]. The most notable causes of cardiovascular disease in patients with T2DM are coronary artery disease and stroke [2]; however, T2DM-related processes have also been hypothesized to cause cardiovascular disease by directly affecting the structure and function of the myocardium [3-5], resulting in a condition known as “diabetic cardiomyopathy”. Diabetic cardiomyopathy is, therefore, one of the key determinants in understanding and controlling the adverse effects of T2DM onset and progression.
Structural changes in diabetic cardiomyopathy is characterised by diastolic dysfunction early in the disease’s progression that leads to a loss of contractile (systolic) function over time [6]. In particular, left ventricular diastolic dysfunction (LVDD), an important predictor of heart failure and long-term mortality, is already present in pre-diabetic patients [7-9]. Because most diastolic dysfunction occurs in asymptomatic, normotensive patients with well-controlled diabetes and preserved left ventricular ejection fraction (i.e., ejection fraction >50%) [10], diastolic dysfunction incidence has been underestimated until recently.
Diabetic cardiomyopathy pathophysiology is complex and multifactorial: cardiac histopathological analysis has revealed perivascular and interstitial fibrosis, myocardial hypertrophy, increased apoptosis and upregulation of oxidative stress [11]. Specific therapeutic strategies for diabetic cardiomyopathy are currently undefined because the pathological mechanisms are poorly understood. Several underlying mechanisms have been proposed, including autonomic dysfunction, metabolic derangements, Ca2+ homeostasis abnormalities, and structural protein alterations [12]. In addition, little is known about the mechanisms underlying the transition from a well-compensated cardiomyopathy to heart failure.
Cynomolgus monkeys develop spontaneous diabetes and exhibit clinical features, including insulin resistance/hyperinsulinemia, hyperglycaemia, dyslipidaemia, and pancreatic pathology, that are similar to those observed in humans [13]. Approximately 30% of cynomolgus monkeys >15 years-of-age have basal and/or postprandial hyperinsulinemia, which precedes the development of overt T2DM [14]; therefore, we consider them ideal models for studying human T2DM. It is also anticipated that they also share similar mechanisms underlying cardiac dysfunction in human.
Here we aimed to investigate the molecular mechanisms underlying cardiac dysfunction in four cynomolgus monkeys with T2DM and LVDD and two healthy, nondiabetic controls. We conducted a systematic transcriptomic, proteomic and phosphoproteomic analysis in left ventricles isolated from all animals and compared the differentially regulated pathways and cellular processes. Finally, we identified that SERCA2a (cardiac sarcoplasmic reticulum Ca2+-ATPase) was phosphorylated at a novel site (Ser626) and reduced protein phosphatase expression in the left ventricle of T2DM monkeys with LVDD. We propose that these two events might have an important role in maintaining the dynamic equilibrium of calcium in the sarcoplasmic reticulum and cytoplasm.
Animals selection and tissue sample collection
In humans, the American Diabetes Association criteria for diabetes are FBG (fasting blood glucose) ≥126 mg/dL and HbA1c (haemoglobin A1c) ≥6.5% [15]. In normal nonhuman primates, however, the FBG is typically ~20 mg/dL lower than that in healthy humans [16], and the mean HbA1c in nonhuman primates is ≤5.0% [17,18]. We thus defined T2DM in the cynomolgus monkeys as HbA1c >5.0% and/or FBG ≥106. Among the echocardiography indices, peak early (E) and late (A) diastolic filling velocities and the E/A ratio measured by pulsed wave Doppler are commonly used to assess diastolic function in the heart of monkeys. The Doppler pattern of impaired left ventricle relaxation was defined as an E/A ratio <1, and a restrictive filling pattern was characterized as an E/A ratio >2, which was associated with aging (>10 years old) [19-21].
According to the upper mentioned criteria, the cynomolgus monkeys with diabetes and LVDD were selected from CrownBio animal facility (Crown Bioscience Inc. in Taicang, Jiangshu Province, China) according to their clinical chemistry data and echocardiographic examinations. The four cynomolgus monkeys with spontaneously developed diabetes and LVDD were designated Y04, A01, O09 and F02 and two nondiabetic healthy monkeys were designated E05 and B03. The left ventricle tissues were collected from these six monkeys, snap-frozen in liquid nitrogen and stored at -80°C until use.
Histopathological examination of the heart tissues
Left ventricles were fixed in 10% neutral buffered formalin, trimmed, paraffin-embedded into blocks and cut into sections of 5-μm thickness for histochemical staining. Tissue sections were stained with haematoxylin and eosin (H&E; Haematoxylin, GHS132-1L, Sigma; Eosin Y solution, alcoholic, HT110132-1L, Sigma-Aldrich) and Masson’s trichrome stain (MT; Trichrome stain-connective tissue stain, ab150686, Abcam) following the manufacturer’s instructions. Specimens were sent for histopathological examination by a ACVP—boarded veterinary pathologist. Images of the stained sections were captured by an OlympusBX53digital microscope with cellSens digital imaging software (Olympus, Tokyo, Japan).
RNA isolation, cDNA library preparation, and Illumina sequencing
Total RNA was isolated from the snap frozen left ventricle tissues using an RNeasy Mini Kit (Qiagen, Germany), according to the manufacturer’s instructions. Paired-end cDNA libraries were synthesized using a TruSeq® RNA Sample Preparation Kit (Illumina, USA) following the manufacturer’s protocol. Purified libraries were quantified with a Qubit® 2.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA) and validated on an Agilent 2100 bioanalyzer (Agilent Technologies, USA) to confirm the insert size and to calculate the concentration. Clusters were generated by cBot with the library diluted to 10 pM and then sequenced on an Illumina HiSeq 2500 platform (Illumina, USA). Library construction and RNA-seq were performed at Shanghai Biotechnology Corporation (Shanghai, China).
RNA-seq data and differentially expressed genes (DEGs) analysis
Sequenced raw reads were trimmed using Trimadap (https://github.com/lh3/trimadap) to remove adapters and filtered using the following criteria by Seqtk (https://github.com/lh3/seqtk) to remove low-quality reads: (1) base quality <20 at the 3’ end of the reads; (2) reads <25 bp; and (3) rRNA sequences. HISAT 2.0.4 [22] was used to map the cleaned reads to the Macaca fascicularis (Macaca_fascicularis_5.0) reference genome with a maximum of two mismatches. After genome mapping, FPKM (fragments per kilobase per million mapped reads) values for each RNA-seq sample were calculated using StringTie 1.3.0 [23,24]. For differential gene expression analysis, DESeq [25] was used to normalize the counts of a gene in each sample and to determine the ratio of the fold change (FC) and p-values for each transcript. Differentially expressed genes (DEGs) were filtered using the following criteria: FPKM ≥3 for at least one of the two individuals under comparison, FC ≥2 or ≤0.5.
Protein extraction and digestion
For protein extraction, left ventricle tissues were homogenized twice for 1 min each in SDT buffer (4% SDS, 100 mM DTT, 150 mM Tris-HCl, pH 8.0) using ceramic beads (MP 6540-424) and an MP homogenizer. Debris was removed by centrifugation at 14,000 × g for 40 min and then the homogenates were filtered using 0.22-μm filters. After quantification with a BCA Protein Assay Kit (Bio-Rad, USA), the supernatants were stored at -80°C for proteomic sample preparation. For each sample, 200 μg protein was reduced with UA buffer (8 M urea, 150 mM Tris-HCl, pH 8.0) by repeated ultrafiltration (Microcon units, 10 kD) and then alkylated with iodoacetamide for 30 min at room temperature in the dark. The samples were then digested with trypsin (Promega) overnight at 37°C.
Tandem mass tag (TMT) labelling and strong cation exchange fractionation
Trypsin-digested peptides (100 µl) were labelled using TMT reagent, according to the manufacturer’s protocol (Thermo Fisher Scientific, Waltham, MA, USA), and pooled in equal ratios. Then, a Pierce High pH Reversed-phase Fractionation Kit (Thermo Fisher Scientific, Waltham, MA, USA) was used to fractionate the pooled peptides into 10 fractions by increasing acetonitrile step-gradient elution, according to the manufacturer’s instructions. Each fraction was dried under vacuum and resolved in 0.1% formic acid for LC-MS/MS analysis.
Enrichment of phosphorylated peptides by TiO2 beads
The combined labelled peptides were dissolved in DHB buffer (0.5% DHB, 37% ACN, and 2.6% TFA) and incubated with TiO2 5-μm beads (GL Biosciences, Tokyo, Japan) for 40 min at room temperature with gentle rotation. The beads were sequentially washed three times with Washing buffer 1 (30% ACN and 3% TFA), three times with Washing buffer 2 (80% ACN and 0.3% TFA), and finally eluted with Elution buffer (40% ACN and 15% NH3•H2O). The eluted fraction was dried under vacuum and resolved in 0.1% formic acid for LC-MS/MS analysis.
LC-MS/MS
The NanoLC-MS/MS experiments were performed on a Q Exactive mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) coupled to an Easy-nLC 1000 (Thermo Fisher Scientific, Waltham, MA, USA). The peptide mixture was loaded onto a reversed-phase trap column (Thermo Fisher Scientific Acclaim PepMap 100, 100 μm×2 cm, nanoViper C18) connected to a C18-reversed-phase analytical column (Thermo Fisher Scientific EASY column, 10-cm long, 75-μm inner diameter, 3-μm resin) in buffer A (0.1% formic acid) and separated with a linear gradient of buffer B (84% acetonitrile and 0.1% formic acid) at a flow rate of 300 nL/min controlled by IntelliFlow technology. For peptides, the 1.5-h gradient was determined as follows: 0-55% buffer B for 80 min, 55-100% buffer B for 5 min, and then 100% buffer B for a 5-min hold. For phosphopeptides, the 4-h gradient was determined as follows: 0-55% buffer B for 220 min, 55-100% buffer B for 8 min, and then 100% buffer B for a 12-min hold. Mass spectrometry data were acquired using a data-dependent top-10 method, dynamically choosing the most abundant precursor ions from the survey scan (300–1800 m/z) for high-energy collision dissociation fragmentation. The automatic gain control target was set to 1.0×106, and the maximum injection time was set to 50 ms. The dynamic exclusion duration was 60.0 s. Survey scans were acquired at a resolution of 70,000 at m/z 200. The resolution for high-energy collision dissociation spectra was set to 35,000 at m/z 200, and the isolation width was 2 m/z. The normalized collision energy was 30. The underfill ratio, which specifies the minimum percentage of the target value likely to be reached at maximum fill time, was defined as 0.1%. The instrument was run with peptide recognition mode enabled. Sample preparation and LC-MS/MS analysis were performed at Shanghai Applied Protein Technology Co., Ltd. (Shanghai, China).
LC-MS/MS data, differentially expressed protein (DEP) and differentially expressed phosphopeptide analysis
MS/MS spectra were searched using the MASCOT engine (Matrix Science, London, UK; version 2.2) embedded into Proteome Discoverer 1.4, and the database searches were carried out against a target and decoy separated Macaca fascicularis database downloaded from UniProtKB (uniprot_Macaca_fascicularis_30673_171228.fasta). The enzyme used was trypsin, which allowed up to two missed cleavages per peptide. Cys carbamidomethylation and TMT 10-plex Lys and N-terminus labelling were set as fixed modifications, while Ser/Thr/Tyr oxidation and phosphorylation were specified as variable modifications. Precursor mass tolerance was set to 20 ppm, and fragment mass tolerance was set to 0.1 Da. Only proteins with at least two unique peptides and with a peptide false discovery rate ≤0.01 were selected for further quantitative analysis. The intensity of each peptide was normalized to the protein median intensity, and the protein ratios were calculated as the median of only unique peptides of the protein. DEPs and differentially expressed phosphopeptides were defined by FC ≥1.2 or ≤0.83. Confident localization of phosphorylation sites was determined by phosphoRS integrated in the Proteome Discoverer 1.4 workflow with a phosphoRS score >50 and phosphoRS site probabilities >75% [26-28].
Bioinformatic analysis
Principal component analysis (PCA) and hierarchical heatmap cluster of the analysed genes, proteins and phosphoproteins were generated with plotPCA and pheatmap in the R package on FPKM values for mRNA and normalized intensities derived from mass spectrometry for proteins and phosphopeptides, respectively. For the functional annotation, the gene or protein sequences were converted to a human gene symbol by identifying homologue sequences with the NCBI BLAST [29] and UniProtKB tools [30]. Gene ontology (GO) analysis on DEGs, DEPs, and differentially expressed phosphoproteins (DEPPs) was performed using the Protein Analysis Through Evolutionary Relationship classification system (version 14.1; http://www.pantherdb.org). The most significantly enriched ontologies were presented in a bar chart format. DEG, DEP and DEPP lists were uploaded into IPA software (Qiagen, Redwood City, CA) and characterized by core analysis and biomarker filter analysis to gain insight into the molecular mechanisms of the genes or proteins through annotation of biological functions, identification of interaction networks and canonical pathways, and the discovery of potential biomarkers.
Parallel reaction monitoring (PRM) and phosphopeptide PRM validation
To verify the protein expression levels obtained by LC-MS/MS analysis, the expression levels of selected proteins were further quantified by PRM mass spectrometry analysis at Shanghai Applied Protein Technology Co., Ltd. (Shanghai, China). Briefly, peptides were prepared according to the TMT protocol. Phosphopeptides were enriched using a High-SelectTM Fe-NTA Phosphopeptide Enrichment Kit (Thermo Fisher Scientific, Waltham, MA, USA) and resolved in 0.1% formic acid. Next, 10 fmol of a heavy isotope-labelled peptide, DSPSAPVNVTVR (V is heavy isotope-labeled peptide site), was spiked into each sample as an internal standard. Tryptic peptides or phosphopeptides were loaded on C18 stage tips for desalting prior to reversed-phase chromatography on an Easy-nLC 1200 system (Thermo Fisher Scientific, Waltham, MA, USA) and then separated using a Thermo Fisher Scientific EASY column at a speed of 300 nL/min. The 2-h gradient was determined as follows: 2-8% buffer B for 5 min, 8-23% buffer B for 85 min, 23-40% buffer B for 15 min, and then 40-100% buffer B, and 100% buffer B was held for 15 min. PRM mass spectrometric analysis was performed on a Q Exactive HF mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). Methods optimized for collision energy, charge state, and retention times for the most significantly regulated peptides were generated experimentally using unique peptides of high intensity and confidence for each target protein. The mass spectrometer was operated in positive ion mode with the following parameters: the mass spectrometry 1 scan was acquired at a 60,000 resolution (at 200 m/z), an automatic gain control target value of 3.0×106, and a 200-ms maximum ion injection time. Full mass spectrometry scans were followed by 20 PRM scans at 30,000 resolution (at m/z 200) with automatic gain control 3.0×106 and a maximum injection time of 120 ms. The targeted peptides were isolated with a 1.6 Thomson window. Ion activation/dissociation was performed at a normalized collision energy of 27 in an high-energy collision dissociation collision cell. The raw data were analysed using Skyline 3.5.0 (MacCoss Laboratory, University of Washington) [31], where signal intensities for individual peptide sequences for each of the significantly altered proteins were quantified relative to each sample and normalized to the reference standard.
Real-time quantitative reverse transcription PCR (qRT-PCR)
cDNA was synthesized using 1.0 μg extracted total RNA and a SuperScript™ IV First-Strand Synthesis System (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. qRT-PCR was performed using Applied Biosystems TaqMan® Gene Expression Assay primers (Thermo Fisher Scientific, Waltham, MA, USA). Assay IDs included Mf00892884_m1 (RYR2), Mf02828564_m1 (MYL2), Mf04389914_g1 (NPPB), Mf01564006_m1 (ATP2A2), and Mf04354341_g1 (ACTB). ACTB (β-Actin) was quantified as an internal control, and 2-Δ ΔCt was used to analyse the differential expression. qRT-PCR was carried out using TaqMan™ Fast Advanced Master Mix (Thermo Fisher Scientific, Waltham, MA, USA) on a QuantStudio™ 7 Flex Real-Time PCR system (Thermo Fisher Scientific, Waltham, MA, USA). All reactions were carried out in technical duplicates for each biological replicate.
Statistical analysis
Statistical analysis was performed with SigmaPlot 12.5 (Systat Software, lnc., San Jose, California, USA). The pairwise correlation matrix and the correlation of the abundance of mRNAs and proteins were used a Spearman correlation analysis.
Cohort characteristics and T2DM status
According to the defined criteria (see Methods section), we collected six left ventricles from four cynomolgus monkeys with spontaneous T2DM and two nondiabetic controls provided by Crown Bioscience. The four cardiac tissues from two males (A01 and O09) and two females (Y04 and F02) had diabetes-associated LVDD (DDD), and two cardiac tissues from two males (E05 and B03) were nondiabetic controls (NDCs). We confirmed the T2DM status in each monkey by clinical biochemical analysis (Table 1). We detected significant increases in FBG and HbA1c in three T2DM monkeys who had hyperglycaemia. The remaining monkey with T2DM had a marked increase in insulin concentration despite normal FBG and slightly higher HbA1c, indicating a prediabetic state with compensatory hyperinsulinemia. The lipid profiles, including total triglyceride, total cholesterol, and low-density lipoprotein levels were higher, and high-density lipoprotein levels were slightly lower in all four T2DM monkeys compared to the two NDCs (Figure 1), resulting in a higher LDL/HDL ratio. Therefore, it is indicated that hyperlipidaemia was observed in DDD monkeys and was closely associated with abnormalities in left ventricular function. The accompanying dyslipidaemia in our NHP model of dysmetabolism seems to contribute, at least in part, to the pathogenesis of cardiac dysfunction.
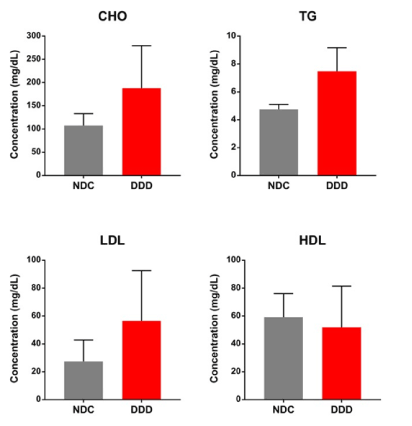
Figure 1. Lipid profiles were analysed in DDD monkeys, comparing to the NDCs. Cholesterol (CHO), triglyceride (TG), low-density lipoprotein (LDL), and high-density lipoprotein (HDL) serum concentrations in two control (NDC) and four T2DM monkeys (DDD). The data represent the means ±SD
Table 1. Individual clinical biochemistry characteristics in monkeys with diabetes-associated LVDD (DDD) and nondiabetic controls (NDC) (BW: body weight; FBG: fasting blood glucose; HbA1c: haemoglobin A1c; LVE/LVA: peak early (E) and late (A) diastolic filling velocities of the left ventricle)
Group |
Sample |
Sex |
Age (years) |
BW (kg) |
Insulin (μIU/mL) |
FBG (mg/dL) |
HbA1c (%) |
LVE (m/sec) |
LVA (m/sec) |
E/A ratio |
DDD |
Y04 |
F |
23 |
4.4 |
10.8 |
232.0 |
7.2 |
0.39 |
0.64 |
0.61 |
F02 |
F |
20 |
5.4 |
72.2 |
262.8 |
9.0 |
0.88 |
0.41 |
2.12 |
A01 |
M |
22 |
8.3 |
193.0 |
385.0 |
8.5 |
0.68 |
0.70 |
0.97 |
O09 |
M |
15 |
14.1 |
578.8 |
65.9 |
5.2 |
1.38 |
0.59 |
2.34 |
NDC |
B03 |
M |
10 |
6.2 |
74.8 |
62.0 |
5.0 |
0.83 |
0.54 |
1.53 |
E05 |
M |
14 |
10.5 |
40.4 |
102.6 |
4.5 |
0.96 |
0.66 |
1.45 |
Histopathologic analysis of cardiac structure and function
We next aimed to identify cardiac phenotype in the DDD and NDC monkeys by histopathological examination. Compared with a representative nondiabetic monkey (B03), H&E stained left ventricle sections from a representative diabetic monkey (A01) with severe hyperglycaemia (FBG of 385.0 mg/dL and HbA1c of 8.5%), showed multifocal myocardial degeneration and necrosis characterized by loss of striations and homogenous cytoplasmic eosinophilia and/or loss of cardiomyocytes nuclei. Many of the degenerated or necrotic cardiomyocytes were actively phagocytized by infiltrated macrophages (Figures 2A and 2B). Moreover, H&E and Masson’s trichrome staining highlighted extensive areas of cardiac fibrosis; these areas were randomly distributed in the left ventricle sections as well as in the inflammatory foci (Figures 2C and 2D). These histopathological evaluations indicated that the heart had a combination of subacute and chronic inflammation as well as fibrosis.
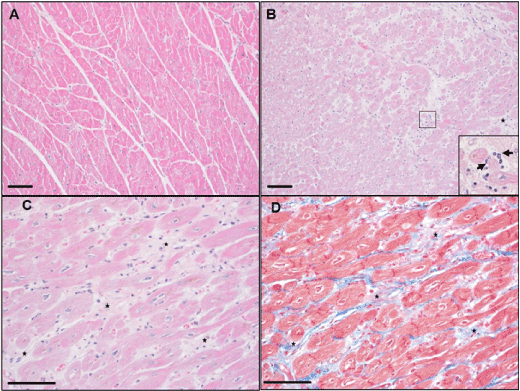
Figure 2. Histopathologic images captured of the left ventricle of representative nondiabetic and diabetic cynomolgus monkeys. A, Normal microscopic morphology of the left ventricle of monkey B03 by H&E staining; B, Myocardial foci with degeneration and necrosis of cardiomyocytes and infiltrated macrophages (arrow) phagocytizing apoptotic and/or necrotizing cells (insert) by H&E staining. The lesions were characterized by multifocal to coalescing loss of striations and homogenous eosinophilia of the cardiomyocytes (degeneration) and some myofibers with karyorrhexis or karyolysis (necrosis). There were multifocal infiltrations of primarily macrophages and a few other mononuclear cells; B-C, Multifocal to coalescing fibrosis of the myocardium (star) with infiltrating mononuclear cells and macrophages by H&E staining; D, Multifocal to coalescing deposit of the collagen (blue) replacing the necrotizing cells and/or filling in the acellular areas due to the loss of necrotic cells in the left ventricle (star) by Masson’s trichrome staining. Scale bar = 100 μm
Transcriptome, proteome and phosphoproteome analysis of left ventricle tissues
To provide insight into the molecular mechanisms of LVDD in cynomolgus monkeys with T2DM, we investigated the changes occurring in the gene and protein/phosphoprotein expression profiles by RNA-seq and TMT-based LC-MS/MS, respectively, of left ventricle tissues in DDD and NDC. For transcriptomic data analysis, we filtered genes with FPKM values ≥3 for at least one of the DDD and NDC individuals being compared. As a result, we identified 10,511 genes. To obtain an overview of genome-wide gene expression alterations, we performed PCA to assess intergroup and intragroup variations using all identified genes. PCA revealed that the first two principal components clearly separated the DDD and NDC groups (Figure 3A left). A pairwise correlation matrix also clearly indicated the differences between the two groups (Figure S1A). Then, we filtered a total of 1,404 genes as DEGs by FC ≥2 or ≤0.5. Among these genes, 910 (65%) and 494 (35%) were upregulated and downregulated in the DDD group compared with those in the NDC group, respectively (Table S1). We then constructed a heatmap based on these DEGs to generate a dendrogram for overlapping gene expression patterns across the six subjects (Figure 3B left). Here we found that the patterns of altered gene expression were highly consistent within individuals in each group but were markedly distinct between groups.
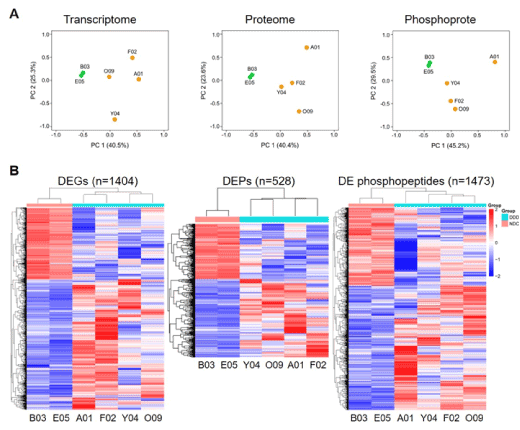
Figure 3. Transcriptome, proteome, and phosphoproteome data analysis. A, PCA of all identified genes, proteins, and phosphopeptides. Orange dots represent diabetic monkeys and the green dots represent the nondiabetic control monkeys. The labels on each dot represent the monkey ID; B, Heatmap analysis of the differentially expressed genes (DEGs), differentially expressed proteins (DEPs) and differentially expressed (DE) phosphopeptides. Each row represents one gene, protein, or phosphopeptide. Each column represents one sample. Samples were clustered based on their expression similarities. Red bars represent higher and blue bars represent lower expressions
For the proteomic data analysis, we identified a total of 2,994 proteins with unique peptides ≥2 and with a peptide false discovery rate ≤0.01, and as with the transcriptomic data analysis, the six proteomes were clearly separated based on different groups, as visualized by the PCA plot (Figure 3A middle). Further filtering by FC ≥1.2 or ≤0.83 resulted in 528 DEPs. Among these proteins, 273 (59%) and 189 (41%) proteins were upregulated and downregulated in the DDD group, respectively (Table S2). Heatmap analysis of these DEPs showed that the proteins from these two groups were well distinguished (Figure 3B middle).
We also analysed the phosphoproteome since phosphorylation is one of the commonest cells signalling mechanisms, involved in the regulation of many molecular pathways [26,32]. We enriched for 3,067 phosphopeptides originating from 1,179 phosphoproteins with very high confidence, based on their phosphorylated site probability of modification >75% and phosphoRS score >50. The PCA plot also clearly separated the DDD and NDC groups (Figure 3A right). Further analysis revealed that 1,413 (46.1%), 1,292 (42.1%), and 362 (11.8%) of the 3,067 phosphopeptides were singly, doubly, and multiply phosphorylated, respectively (Figure S1B). Among the identified phosphorylated sites, 4,665 sites (90.6%) were serine (Ser) residues, 458 sites (8.9%) were threonine (Thr) residues, and 26 sites (0.5%) were tyrosine (Tyr) residues (Figure S1C). Finally, we found that 1,473 significantly differentially expressed phosphopeptides from 709 DEPPs were filtered with FC ≥1.2 or ≤0.83, including 904 (61%) upregulated and 569 (39%) downregulated phosphopeptides when comparing DDD with NDC monkeys (Table S3). Heatmap analysis of these altered phosphopeptides showed a similar clustering pattern to the transcriptomic and proteomic data, further verifying the reproducibility of these results (Figure 3B right).
Comparison with the gene and protein expression patterns
Since mRNA abundances are generally believed to be adequate for simply predicting protein presence/absence, we firstly compared the gene and protein expression patterns. To ensure comparable and compensate for the lack of functional annotation for cynomolgus monkey genes and proteins, the human genes and proteins were assigned by one-to-one orthologue matching between the human and cynomolgus monkey sequences using the NCBI BLAST [29] and UniProtKB tools [30]. After this step, 9,316 identified genes, including 1,183 DEGs; 2,718 identified proteins, including 479 DEPs; and 1,114 identified phosphoproteins, including 669 DEPPs, aligned with the human gene symbols (Tables S1-S3). We then performed a correlation analysis between the transcriptomic and proteomic data; globally, the expression levels of both detected proteins and their corresponding mRNAs had a moderate positive correlation (r=0.4, p<0.001) (Figure 4A). A higher correlation was observed between the DEGs and their corresponding proteins (r=0.50, p<0.001), as well as the DEPs and their corresponding mRNAs (r=0.49, p<0.001) (Figures 4B and 4C). In summary, the gene expression pattern was highly correlated with the protein expression pattern.
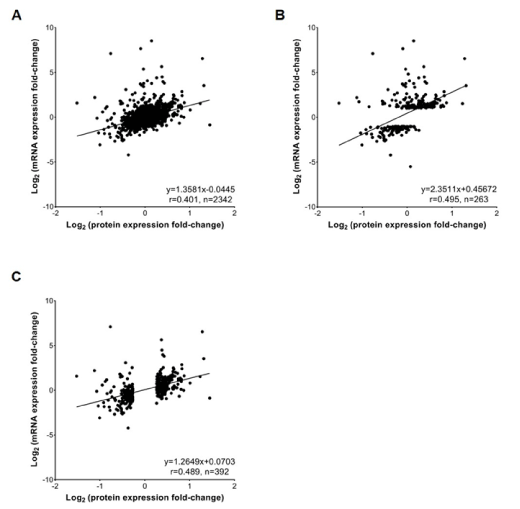
Figure 4. Correlation analysis of mRNA and protein abundance. A, Scatterplots and correlation coefficients between all detected proteins and mRNAs; B, Scatterplots and correlation coefficients between the differentially expressed genes (DEGs) and their corresponding proteins; C, Scatterplots and correlation coefficients between the differentially expressed proteins (DEPs) and their corresponding mRNAs. The linear regression equation, correlation coefficient (r) and the number of quantified gene products (n) for each comparison are indicated
GO enrichment analysis of the correlated DEGs, DEPs and DEPPs
To gain insight into the functional characteristics of all the altered genes, proteins and phosphoproteins in DDD monkeys, we performed GO term analysis. GO terms were enriched in cellular component, biological process and molecular function (Figure 5). The biological process analysis indicated that the majority of the DEGs, DEPs and DEPPs were engaged in “cellular process” and “metabolic process”. For the molecular function category, GO terms including “binding” and “catalytic activity” captured the predominant unctions of the DEGs, DEPs and DEPPs. In the cellular component, GO terms including “cell” and “organelle” were enriched in the DEGs, DEPs and DEPPs. Overall, the DEGs, DEPs and DEPPs displayed similar GO annotation patterns.
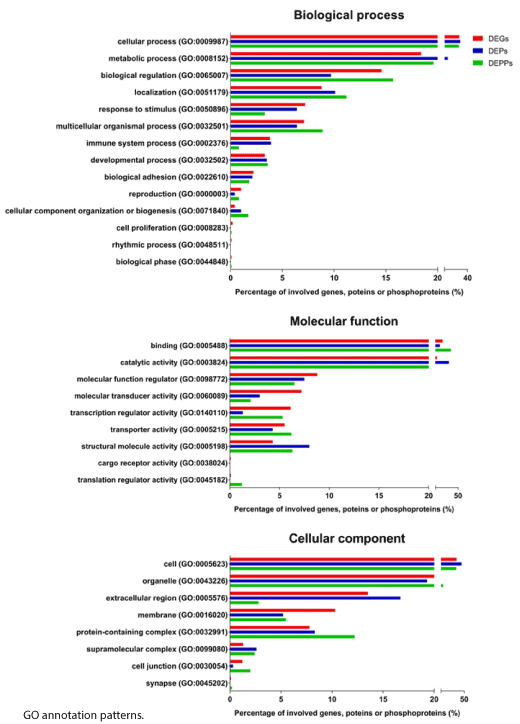
Figure 5. GO term enrichment analysis for biological process, molecular function, and cellular component assigned to the differentially expressed genes (DEGs) (red), differentially expressed proteins (DEPs) (blue), and differentially expressed phosphoproteins (DEPPs) (green)
Pathway and network analysis of the DEGs, DEPs, and DEPPs
To further obtain functional insights into diastolic dysfunction pathogenesis in T2DM monkeys, we conducted an IPA core analysis for a systematic bioinformatic analysis. By providing an unbiased and statistically feasible identification of biological processes, pathways and networks, the application of IPA helps to discover the disease pathogenesis and new therapeutic targets [33]. The DEGs, DEPs and DEPPs were firstly categorized to related canonical pathways. The top 10 overlapping significantly enriched categories of canonical pathways (p-value ≤0.05) involved in differentially expressed mRNAs and proteins are listed in Figure 6A and Table S4. The most enriched pathways were mainly related to inflammation and the immune response, cardiovascular signalling, and lipid metabolic pathways. The top 10 overlapping significantly enriched categories of canonical pathways (p-value ≤0.05) involved in differentially expressed proteins and phosphoproteins are listed in Figure 6B and Table S4. The predominantly enriched pathway categories were the intracellular and second messenger signalling, immune response, cardiovascular signalling, cellular growth and development, and metabolites and biosynthesis. Notably, calcium signalling pathway was significantly activated both in protein and phosphoprotein levels, suggesting that this pathway in left ventricle tissues may play an important role in the pathogenesis of LVDD.
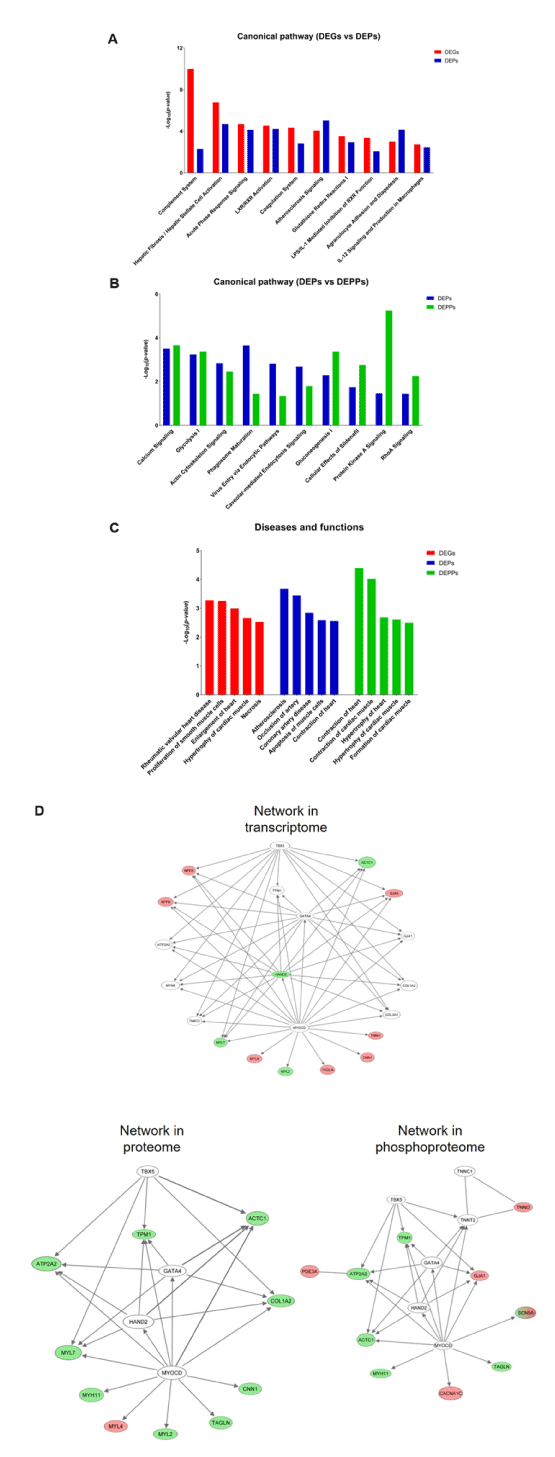
Figure 6. IPA core analysis of the differentially expressed genes (DEGs), differentially expressed proteins (DEPs), and differentially expressed phosphoproteins (DEPPs) identified between DDD and NDC monkeys. A, Comparison of the top 10 significantly enriched canonical pathways between the DEGs and DEPs; B, Comparison of the top 10 significantly enriched canonical pathways between the DEPs and DEPPs; C, Comparison of the top 5 classified diseases and biofunctions among the DEGs, DEPs, and DEPPs. Each bar chart shows the -log10(p-value) calculated by IPA; D, The network associated with cardiovascular system development and function and cardiovascular disease. Molecules labelled red and green show up- and downregulated mRNAs (transcriptome), proteins (proteome), and phosphoproteins (phosphoproteome) in DDD monkeys. Molecules labelled in white were not identified in the current data
In addition to canonical pathways, we categorized the DEGs, DEPs and DEPPs based on diseases and biofunctions. Similar as the results from canonical pathway analysis, the top 5 significantly enriched categories of disease and biofunctions involved in DEGs, DEPs, and DEPPs are listed in Figure 6C and Table S5. Here, the remarkable related diseases included “hypertrophy of heart”, “necrosis”, and “contraction of heart”, which were closely correlated to the results of histopathology examinations (Figure 2).
Finally, we used an IPA network to analyse gene networks, which were built to connect key molecules. The top network correlated with the DEGs, DEPs, and DEPPs was the same, and mainly related diseases and functions are associated with cardiovascular system development and function and cardiovascular disease (Figure 6D). It is worth noting that most of the key molecules in this network were vastly involved the calcium signalling pathway, which function in contraction of heart (see Discussion section). Therefore, IPA indicated that the calcium signalling pathway may be the potential molecular mechanisms of LVDD in T2DM monkeys.
Differential gene, protein and phosphopeptide expression validation by qRT-PCR and PRM
Our IPA indicated that the calcium signalling pathway was significantly dysregulated in DDD monkeys (Figure 6B and 6D). We confirmed pathway-related mRNA and/or protein
changes including the decreased RyR2 (ryanodine receptor 2), SERCA2a (sarcoplasmic reticulum Ca2+ -ATPase), MYL2 (myosin regulatory light chain 2), CASQ2 (calsequestrin-2), and TPM1 (tropomyosin alpha-1 chain) (Figure 7A and 7B), biomarker -ndphosphopeptides expression changes including the decreased of phosphorylated SERCA2a, TPM1, and CACNB2 (voltage-dependent L-type calcium channel subunit β-2) (Figure 7B and 7C) in DDD monkeys. The mRNA level of a hypertrophic and heart failure BNP (B-type natriuretic peptide) [34]was also quantified (Figure 7A), because the IPA disease and biofunctions analysis involved in DEGs identified “hypertrophy of heart” in DDD monkeys (Figure 6B). The results showed that the expression changes of all the key molecules associated with the calcium signalling pathway and BNP were consistent with the transcriptome, proteome and/or phosphoproteome data. We presume that these observations in our cohort could be responsible for diabetic cardiomyopathy in T2DM monkeys. These results further support the reliability of the omics datasets.
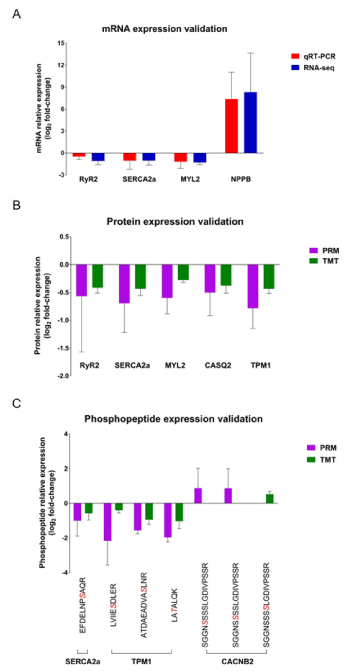
Figure 7. Validation of calcium signalling pathway-related molecules. A, qRT-PCR analysis (red) of the mRNA expression changes in left ventricle tissues from four DDD monkeys and two NDCs, compared with RNA-seq data (blue); B-C, PRM protein and phosphopeptide quantities (purple) of the candidate proteins and phosphoproteins in the same tissues, compared with TMT data (green). The sequence denoted in red italics indicates the phosphosites. The data represent the mean ± SD
Potential biomarker investigations of DEGs and DEPs
Besides the biomarker BNP, we wanted to explore other potential biomarkers to assess whether the biomarkers strongly associated with disease processes in DDD monkeys. To do this, the DEGs and DEPs were imported to IPA biomarker module that has the capacity to generate a list of candidate biomarkers across different diseases. We filtered candidate biomarkers for T2DM and LVDD using the terms “metabolic disease” and “cardiovascular disease” using the IPA biomarker filters tool. This approach resulted in 22 biomarkers for these two diseases based on changes in mRNA expression levels (Table 2). All of these biomarkers were upregulated in the left ventricle tissues in DDD monkeys, with the exception of FABP3 (FABP3, fatty acid-binding protein 3), which is an early biomarker for acute myocardial infarction [35]. The up-regulated biomarkers included: clinical, diagnostic and prognostic biomarkers for heart failure (e.g. NPPA [ANP, atrial natriuretic peptide], NPPB [BNP], and EDN1 [ET-1, endothelin-1]) [36,37]; molecules involved in cardiac remodelling (e.g. ICAM1 [ICAM1, intercellular adhesion molecule 1]) [38], and biomarkers for LVDD and fibrosis in hypertension (e.g. TIMP1 [TIMP1, intercellular adhesion molecule 1]) [39]. These data were consistent with histopathological observations of multifocal myocardial degeneration and cardiac fibrosis. Further 11 biomarkers were identified based on changes in protein expression levels, including biomarkers for cardiovascular risk (e.g. ApoA1 [apolipoprotein A1], ApoB [apolipoprotein B], and ApoE [apolipoprotein E]) [40,41]and enzymes involved in energy homeostasis in the heart (e.g. CKM [creatine kinase M]) [42]. These findings were highly correlated with the lipid profiles detected in the blood chemistry (Figure 1). We also identified other up and downregulated cardiovascular disease biomarkers involved in inflammation, extracellular matrix turnover and remodelling, and neurohormonal activation (Table 2). Together, all the identified biomarkers in DDD monkeys indicated the potential pathogenesis of LVDD.
Table 2. Potential biomarkers for metabolic disease and cardiovascular disease identified based on the DEGs or DEPs by IPA biomarker analysis. DEGs: differentially expressed genes; DEPs: differentially expressed proteins; ND: none detected; DDD: diabetes-associated left ventricular diastolic dysfunction; NDC: nondiabetic control. aMf: Macaca fascicularis, bHomo: Human, cLog2 fold change of DDD compared with NDC
Mf a Gene ID |
Mf Protein ID |
Homo b Gene Symbol |
Biomarkers |
mRNAs c |
Proteins c |
102142791 |
G7NWW7 |
FABP3 |
Fatty acid-binding protein 3 |
-1.21 |
-0.21 |
102115953 |
G8F4Z7 |
NPPB |
Natriuretic peptide B |
11.11 |
ND |
102116318 |
A0A2K5VBH4 |
NPPA |
Natriuretic peptide A |
7.08 |
ND |
102115765 |
A0A2K5U5J5 |
EDN1 |
Endothelin 1 |
2.52 |
ND |
102138837 |
G7PZ70 |
ICAM1 |
Intercellular adhesion molecule 1 |
1.56 |
0.38 |
102144412 |
A0A2K5WNU0 |
TIMP1 |
Metallopeptidase inhibitor 1 |
2.21 |
ND |
102123471 |
A0A2K5X7X6 |
ADIPOQ |
Adiponectin, C1Q and collagen domain containing |
4.40 |
ND |
102144139 |
A0A2K5TWD7 |
IGFBP3 |
Insulin-like growth factor binding protein 3 |
3.61 |
ND |
102119749 |
P79182 |
IL1B |
Interleukin 1 beta |
3.22 |
ND |
102135739 |
P61274 |
CCL2 |
C-C motif chemokine ligand 2 |
2.68 |
ND |
101866035 |
A0A2K5UU26 |
ITGAM |
Integrin subunit alpha M |
2.11 |
ND |
102128018 |
A0A2K5URS7 |
IGF1 |
Insulin-like growth factor 1 |
1.84 |
ND |
102129342 |
B3Y6B7 |
CD14 |
CD14 molecule |
1.80 |
ND |
102124562 |
A0A2K5WKS0 |
NCF1 |
Neutrophil cytosolic factor 1 |
1.75 |
ND |
102124268 |
A0A2K5WXU9 |
CXCL12 |
C-X-C motif chemokine ligand 12 |
1.61 |
ND |
101865088 |
G7NZ62 |
PROS1 |
Protein S |
1.54 |
ND |
102141631 |
A0A2K5TMC4 |
CSF1 |
Colony-stimulating factor 1 |
1.25 |
ND |
102132631 |
A0A2K5X4G5 |
VWF |
Von Willebrand factor |
1.18 |
ND |
102145462 |
A0A2K5VX93 |
PLAT |
Plasminogen activator, tissue type |
1.09 |
ND |
101925978 |
G8F351 |
APOA1 |
Apolipoprotein A1 |
1.91 |
-0.34 |
102121781 |
G7PLQ1 |
APOB |
Apolipoprotein B |
ND |
0.28 |
102138471 |
A2V9Z3 |
APOE |
Apolipoprotein E |
0.32 |
0.68 |
102146019 |
G7PXW6 |
CKM |
Creatine kinase, M-type |
-0.70 |
-0.37 |
102133993 |
G7NUT6 |
PLA2G2A |
Phospholipase A2 group IIA |
3.54 |
1.32 |
101926221 |
G8F2E1 |
AGT |
Angiotensinogen |
-0.01 |
0.87 |
102115978 |
G7PH52 |
CST3 |
Cystatin C |
0.19 |
0.30 |
102141875 |
G7PV74 |
SELP |
Selectin P |
3.08 |
-0.43 |
102124806 |
G7PGE5 |
LBP |
Lipopolysaccharide-binding protein |
ND |
0.78 |
In this study, we employed for the first time, a systematic approach based on transcriptomics, proteomics and phosphoproteomics analysis of left ventricle tissues from T2DM cynomolgus monkeys to understand the molecular changes underlying diastolic dysfunction. We firstly found impaired calcium signalling pathway in our DDD monkeys. These data may be utilized to elucidate the potential mechanism(s) for the increased cardiac disease observed in diabetic patients.
Diastolic dysfunction can also be diagnosed by ANP and BNP measurements in daily clinical practice [43]. ANP is mainly synthesized in the atria and stored in granules., while BNP is synthesized primarily by the ventricles and is minimally stored in granules. Consistent with the echocardiography data, we found that the mRNA levels of NPPA (135-fold change) and NPPB (2210-fold change) were dramatically increased in monkeys with LVDD compared to the NDCs (Table 2 and S1), suggesting a high risk of heart failure in DDD monkeys. While we could confirm this increase in NPPB by qRT-PCR (Figure 7A), the ANP and BNP protein were not detected in our proteome analysis, perhaps because of their short half-lives [44,45].
Another structural hallmark of the diabetic myocardium is the development of interstitial and/or perivascular fibrosis. Numerous histopathologic studies have demonstrated cardiac fibrosis in diabetic patients. The same morphology was also identified in the DDD monkeys (Figure 2). The key determinants of cardiac fibrosis are an activated transforming growth factor-β signalling cascade and accumulation of increased extracellular matrix proteins composed of collagen and fibronectin [46]. In our study, we found that the mRNA expression level of transforming growth factor-β receptor 3 and the protein levels of various collagens and fibronectin were upregulated in DDD monkeys (Table S1 and S2). In the meanwhile, the increased mRNA expression levels of EDN1, angiotensinogen, connective tissue growth factor, and platelet-derived growth factor were observed in DDD monkeys as well (Table S1), which have been demonstrated to activate collagen production, leading to increased myocardial stiffness and impaired left ventricle relaxation with subsequent compromise in the efficiency of left ventricle contraction [47]. The molecular evidence highly correlated with the cardiac fibrosis observed in these monkeys at the microscopic level.
Our study revealed several signalling pathways associated with diabetic cardiomyopathy. In the IPA, the calcium signalling pathway involving 14 proteins and 18 phosphoproteins in left ventricle tissues seemed to be significantly perturbed (Table S6). We validated the expression of key proteins in the calcium signalling pathway by PRM analysis, including RyR2 , SERCA2a, MYL2, CASQ2, and TPM1, and the phosphorylation levels of SERCA2a, TPM1, and CACNB2; the data were highly consistent with our TMT analysis (Figure 7B and 7C). Consistent data were obtained at the mRNA level, as verified by qRT-PCR (Figure 7A). These data have not only proved that the calcium signalling pathway may play an important role in pathogenesis of diabetic cardiomyopathy, but also indicate that our RNA-seq and mass spectrometry-based proteome/phosphoproteome results are technically reliable.
Cardiac contractility is triggered by a Ca2+-induced Ca2+-release mechanism during the excitation-contraction coupling [48]. Activation of voltage-dependent L-type calcium channels by membrane depolarization enables external Ca2+ to enter into the cytoplasm. A local increase in Ca2+ influx activates the RyR2 channel on the sarcoplasmic reticulum membrane, thereby triggering the release of Ca2+ from the sarcoplasmic reticulum. In this study, RyR2 expression was reduced in DDD monkeys (Figure 7A and B), with higher phosphorylation at Ser2792 and Ser2798 (Table S6). These sites are phosphorylated by PKA (protein kinase A) and CaMKII (Ca2+/calmodulin-dependent protein kinase), respectively. These data are highly consistent with observations made in T2DM (db/db) mice (Ser2807 in mice) and in an heart failure canine model (Ser2808 and Ser2814 in dogs) [49,50]. Moreover, RyR2 was markedly hyperphosphorylated at the CaMKII-dependent site Ser2815 (Ser2798 in monkeys) in patients with heart failure, but there was no significant regulation of PKA-dependent phosphorylation [51]. Recent studies clarified that RyR2 dysregulation contributes to the altered Ca2+ regulatory phenotype of heart failure; however, PKA-mediated phosphorylation of RyR2-Ser2808 has little or no role in these alterations [52]. We detected two phosphopeptides by mass spectrometry technology. One peptide was identified at the PKA-dependent site Ser2792, and the other peptide was identified at both the PKA-dependent site Ser2792 and the CaMKII-dependent site Ser2798. Because both phosphopeptides were upregulated in DDD monkeys, distinguishing which phosphopeptide is responsible for Ca2+ impaired release is difficult. Thus, the precise phosphosites and the exact mechanism underlying the synergy between these different modifications require follow-up studies.
Moreover, previous studies demonstrated that PKA and CaMKII directly phosphorylate not only RyR2 but also voltage-dependent L-type calcium channels [53]. Our phosphoproteomics and PRM validation also revealed upregulated one of the voltage-dependent L-type calcium channels - CACNB2 phosphorylation in the DDD group (Figure 7C). Although we did not detect increased PKA and CaMKII expression, we did find decreased levels of the CaMKII isoform CaMK2B and increased CaMK2D phosphorylation at Thr287 (in mice). The different functions of phosphorylated CaMK2B and CaMK2D are unclear. Our interpretation is that hyperphosphorylated RyR2 and CACNB2 are not regulated by increased PKA and CaMKII but are mediated by reduced protein phosphatases such as PP1 and protein phosphatase type-2A, which could dephosphorylate regulatory Ca2+-handling proteins [54]. In support of this proposal, we found decreased protein and phosphorylation levels of protein phosphatase 1 regulatory subunit 3A - a regulatory PP1 subunit (Table S6). Taken together, our results indicate that abnormalities in the kinase/phosphatase balance in cellular microdomains might contribute to impaired Ca2+-handling and contractility in diabetic cardiomyopathy.
Diastolic relaxation occurs as Ca2+ is released from the myofilaments, pumped back into the sarcoplasmic reticulum via SERCA2a and extruded from the cell via the sarcolemmal Na+/Ca2+ exchanger [55]. Most functional studies indicate depressed SERCA2a activity and protein levels in diabetic cardiomyopathy, resulting in Ca2+ overload in the cytosol and impaired relaxation, which correlates with the clinical findings of diastolic dysfunction [56]. We observed SERCA2a downregulation in samples from the DDD monkeys, which is highly consistent with the previous studies. SERCA2a activity is directly regulated by its inhibitor protein phospholamban. Phospholamban phosphorylation by CaMKII and PKA causes it to disassociate from SERCA2a, enhancing SERCA2a activity [57]. It has been also reported that SUMOylation and acetylation are critical post-translational modifications, which modulate SERCA2a activity [58]. They indicated that decreased SUMOylation and increased acetylation of SERCA2a were observed in human and animal heart failure models and correlated with the reduced activity of SERCA2a. Moreover, SERCA2a phosphorylation at Ser38, which is part of the CaMKII consensus site, has been identified [59,60]; however, Ser38-phosphorylated SERCA2a has not been detected in the sarcoplasmic reticulum vesicles, stimulated cardiac myocytes or in in vivo animal models [61,62]. Ours is the first study, however, to report of a new phosphosite, SERCA2a Ser626 in DDD monkeys, of which we found the phosphorylation to be downregulated despite no changes in phospholamban expression, and its potential correlation with cardiac contraction and relaxation. According to the PhosphoSitePlus database (http://www.phosphosite.org/), the same site was identified by mass spectrometry techniques in humans and rodents. Further investigation is necessary to characterize its function in diabetic cardiomyopathy.
The present study provides an integrated data analysis for understanding the underlying molecular mechanisms of LVDD in cynomolgus monkeys with spontaneous T2DM. Nevertheless, several limitations need to be noted when interpreting the data. First, the sample size was small, especially for the control animals, and the age of the selected controls in this study was slightly younger than that of the T2DM monkeys. The potential age-associated impact on our omics data is uncertain; thus, a future study that includes a larger number of diseased and age-matched control monkeys is needed to buffer the individual variation between monkeys. Second, we could not validate some of the novel phosphosites in calcium signalling pathway-related proteins, even though these phosphosites were first identified in our phosphoproteome analysis. Therefore, additional confirmation of the cellular targets for novel phosphorylation is needed to obtain a comprehensive understanding of the responses to calcium signalling pathway dysregulation. Finally, our echocardiographic examinations and microscopic evaluations would suggest that our tissue samples were probably obtained from monkeys in the relatively early stages of cardiovascular dysfunction. The molecular pathogenesis of the disease stage transition from diabetic cardiomyopathy to heart failure remains to be elucidated. Improving our understanding of obesity to diabetes to heart failure progression in monkeys would provide unprecedented information for understanding disease progression in humans.
In summary, our omics analysis provides a large dataset to interrogate the molecular mechanism of spontaneously occurring cardiovascular dysfunctions in the cynomolgus monkey with T2DM. Strong correlations among the clinical features, histopathological observations, and omics data demonstrated that the selected monkeys had T2DM and heart dysfunctions and that the molecular data generated from these monkeys were reliable. From our initial analysis, we found that reduced expression of PP1, which is known to dephosphorylate RyR2 and CACNB2, resulted in RyR2 and CACNB2 hyperphosphorylation. In addition, we observed decreased SERCA2a expression and phosphorylation, which is anticipated to lead to sarcoplasmic reticulum Ca2+ leakage from the RyR2 and consequent sarcoplasmic reticulum Ca2+ load depletion. Finally, abnormal excitation–contraction coupling alterations reduced Ca2+ availability, leading to depressed contractile function in T2DM monkeys (Figure 8). Ours is believed to be the first study to identify phosphorylated SERCA2a during LVDD in vivo. Targeting phosphorylated SERCA2a might constitute an innovative therapeutic approach to treating cardiovascular dysfunction in patients with diabetes.
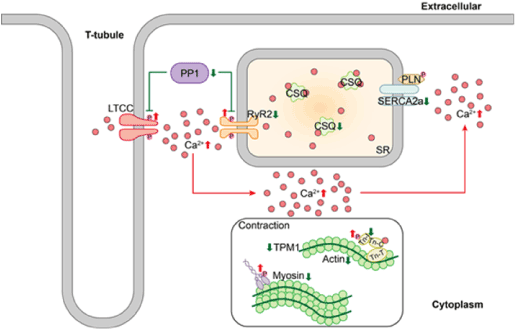
Figure 8. Impaired Ca2+-handling and contractility in diabetic cynomolgus monkeys with diastolic dysfunction. Downregulated PP1 expression induces RyR2 and LTCC hyperphosphorylation. This hyperphosphorylation event is accompanied by decreased SERCA2a expression, leading to SR Ca2+ leakage from RyR2 and consequent SR Ca2+ load depletion. The arrows indicate activation, and the T bars indicate repression. The up/down arrows indicate upregulation or downregulation, respectively
The author contributions are as follows: (1) study conception and design (YL, GC, LGH); (2) sample collection and selection (GC, DD); (3) omics data analysis and interpretation (YL, LJ); (4) laboratory data acquisition and analysis (YL, CQ, LGH); and (5) drafting of the manuscript (YL, LJ, CQ, LGH). All authors critically revised the manuscript, gave final approval and agreed to be accountable for all aspects of the work ensuring integrity and accuracy. All authors read and approved the final manuscript.
We are grateful to Crown Bioscience Company for providing animal tissue samples and Shanghai OE Biotechnology Company for their valuable contributions to omics data analysis.
Funding was received from Amgen.
Ethics approval and consent to participate: All the animal used in this work and the experimental protocol and procedures were approved by the Institutional Animal Care and Use Committee of Crown Bioscience, Inc. and in accordance with the guidelines of Association for Assessment and Accreditation of Laboratory Animal Care.
The authors declare that they have no competing interests.
All authors have read and agree to the publication of the manuscript.
The datasets used and analysed during the current study are available from the corresponding author on reasonable request.
- Seferovic PM, Petrie MC, Filippatos GS, Anker SD, Rosano G, et al. (2018) Type 2 diabetes mellitus and heart failure: a position statement from the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail 20: 853-872.
- Einarson TR, Acs A, Ludwig C, Panton UH (2018) Prevalence of cardiovascular disease in type 2 diabetes: a systematic literature review of scientific evidence from across the world in 2007–2017. Cardiovascular Diabetology 17: 83.
- Seferović PM, Paulus WJ (2015) Clinical diabetic cardiomyopathy: a two-faced disease with restrictive and dilated phenotypes. European Heart Journal 36: 1718-1727.
- Boudina S, Abel ED (2005) Diabetic cardiomyopathy revisited. Circulation 115: 3213-3223.
- Fang ZY, Prins JB, Marwick TH (2004) Diabetic cardiomyopathy: evidence, mechanisms, and therapeutic implications. Endocrine Reviews 25: 543-567.
- Murtaza G, Virk HUH, Khalid M, Lavie CJ, Ventura H, et al. (2019) Diabetic cardiomyopathy-A comprehensive updated review. Progress in Cardiovascular Diseases.
- Paulus WJ, Tschöpe C, Sanderson JE, Rusconi C, Flachskampf FA, et al. (2007) How to diagnose diastolic heart failure: a consensus statement on the diagnosis of heart failure with normal left ventricular ejection fraction by the Heart Failure and Echocardiography Associations of the European Society of Cardiology. European heart journal 28: 2539-2550.
- Kane GC, Karon BL, Mahoney DW, Redfield MM, Roger VL, et al. (2011) Progression of left ventricular diastolic dysfunction and risk of heart failure. JAMA 306: 856-863.
- Mogelvang R, Sogaard P, Pedersen SA, Olsen NT, Marott JL, et al. (2009) Cardiac dysfunction assessed by echocardiographic tissue Doppler imaging is an independent predictor of mortality in the general population. Circulation 119: 2679-2685.
- Zabalgoitia M, Ismaeil MF, Anderson L, Maklady FA (2001) Prevalence of diastolic dysfunction in normotensive, asymptomatic patients with well-controlled type 2 diabetes mellitus. The American journal of cardiology 87: 320-323.
- Huynh K, Bernardo BC, McMullen JR, Ritchie RH (2014) Diabetic cardiomyopathy: mechanisms and new treatment strategies targeting antioxidant signaling pathways. Pharmacology & therapeutics 142: 375-415.
- Falcao-Pires I, Leite-Moreira AF (2012) Diabetic cardiomyopathy: understanding the molecular and cellular basis to progress in diagnosis and treatment. Heart Fail Rev 17: 325-344.
- Wagner JE, Kavanagh K, Ward GM, Auerbach BJ, Harwood HJ, et al. (2006) Old world nonhuman primate models of type 2 diabetes mellitus. ILAR J 47: 259-271.
- Wagner JD, Cline JM, Shadoan MK, Bullock BC, Rankin SE, et al. (2001) Naturally occurring and experimental diabetes in cynomolgus monkeys: a comparison of carbohydrate and lipid metabolism and islet pathology. Toxicologic pathology 29: 142-148.
- American Diabetes A (2018) Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2018. Diabetes Care 41: S13-S27.
- Hansen BC, Tigno XT (2007) The Rhesus Monkey (Macaca mulatta) Manifests All Features of Human Type 2 Diabetes. Animal Models of Diabetes: Frontiers in Research. 2007: 251.
- Marigliano M, Casu A, Bertera S, Trucco M, Bottino R, et al. (2011) A1C Percentage in Nonhuman Primates: A Useful Tool to Monitor Diabetes before and after Porcine Pancreatic Islet Xenotransplantation. J Transplant 2011: 965605.
- McTighe MS, Hansen BC, Ely JJ, Lee DR (2011) Determination of Hemoglobin A1c and Fasting Blood Glucose Reference Intervals in Captive Chimpanzees (Pan troglodytes). Journal of the American Association for Laboratory Animal Science 50: 165-170.
- Galderisi M (2006) Diastolic dysfunction and diabetic cardiomyopathy: evaluation by Doppler echocardiography. Journal of the American College of Cardiology 48: 1548-1551.
- Danzmann LC, Bodanese LC, Köhler I, Torres MR (2008) Left atrioventricular remodeling in the assessment of the left ventricle diastolic function in patients with heart failure: a review of the currently studied echocardiographic variables. Cardiovascular ultrasound 6: 56.
- Nagueh SF, Smiseth OA, Appleton CP, Byrd BF, Dokainish H, et al. (2016) Recommendations for the Evaluation of Left Ventricular Diastolic Function by Echocardiography: An Update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J Am Soc Echocardiogr 29: 277-314.
- Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast-spliced aligner with low memory requirements. Nature methods 12: 357.
- Pertea M, Pertea GM, Antonescu CM, Chang T-C, Mendell JT, et al. (2015) StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature biotechnology 33: 290.
- Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL, et al. (2016) Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nature protocols 11: 1650.
- Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome biology 11: R106.
- Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, et al. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127: 635-648.
- Taus T, Köcher T, Pichler P, Paschke C, Schmidt A, et al. (2011) Universal and Confident Phosphorylation Site Localization Using phosphoRS. Journal of Proteome Research 10: 5354-5362.
- Beausoleil SA, Villen J, Gerber SA, Rush J, Gygi SP, et al. (2006) A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat Biotechnol 24: 1285-1292.
- Coordinators NR (2017) Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res 45: D12-D7.
- Pundir S, Martin MJ, O'Donovan C (2017) UniProt Protein Knowledgebase. Methods Mol Biol 1558: 41-55.
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, et al. (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26: 966-968.
- Pawson T, Scott JD (2005) Protein phosphorylation in signaling--50 years and counting. Trends Biochem Sci 30: 286-290.
- Krämer A, Green J, Pollard Jr J, Tugendreich S (2013) Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 30: 523-530.
- Vuolteenaho O, Ala-Kopsala M, Ruskoaho H (2005) BNP as a biomarker in heart disease. Advances in clinical chemistry 40: 1-36.
- Catalucci D, Latronico MVG, Condorelli G (2015) FABP3 as Biomarker of Heart Pathology 439-454.
- Januzzi JL (2013) Natriuretic peptides as biomarkers in heart failure. Journal of Investigative Medicine 61: 950-955.
- Gaggin HK, Truong QA, Gandhi PU, Motiwala SR, Belcher AM, et al. (2017) Systematic Evaluation of Endothelin 1 Measurement Relative to Traditional and Modern Biomarkers for Clinical Assessment and Prognosis in Patients with Chronic Systolic Heart Failure: Serial Measurement and Multimarker Testing. Am J Clin Pathol 147: 461-472.
- Salvador AM, Nevers T, Velazquez F, Aronovitz M, Wang B, et al. (2016) Intercellular Adhesion Molecule 1 Regulates Left Ventricular Leukocyte Infiltration, Cardiac Remodeling, and Function in Pressure Overload-Induced Heart Failure. J Am Heart Assoc 5: e003126.
- Lindsay MM, Maxwell P, Dunn FG (2002) TIMP-1: a marker of left ventricular diastolic dysfunction and fibrosis in hypertension. Hypertension 40: 136-141.
- Bennet AM, Di Angelantonio E, Ye Z, Wensley F, Dahlin A, et al. (2007) Association of apolipoprotein E genotypes with lipid levels and coronary risk. JAMA 298: 1300-1311.
- Andrikoula M, McDowell IFW (2008) The contribution of ApoB and ApoA1 measurements to cardiovascular risk assessment. Diabetes, Obesity and Metabolism 10: 271-278.
- Turko IV, Murad F (2003) Quantitative protein profiling in heart mitochondria from diabetic rats. Journal of Biological Chemistry 278: 35844-35849.
- Bhalla MA, Chiang A, Epshteyn VA, Kazanegra R, Bhalla V, et al. (2004) Prognostic role of B-type natriuretic peptide levels in patients with type 2 diabetes mellitus. J Am Coll Cardiol 44: 1047-1052.
- Potter LR, Yoder AR, Flora DR, Antos LK, Dickey DM, et al. (2009) Natriuretic peptides: their structures, receptors, physiologic functions and therapeutic applications. In: cGMP: Generators, Effectors and Therapeutic Implications. Springer; 341-366.
- Kimura K, Yamaguchi Y, Horii M, Kawata H, Yamamoto H, et al. (2007) ANP is cleared much faster than BNP in patients with congestive heart failure. European journal of clinical pharmacology 63: 699-702.
- Edgley AJ, Krum H, Kelly DJ (2012) Targeting Fibrosis for the Treatment of Heart Failure: A role for transforming growth factor‐β. Cardiovascular therapeutics 30: e30-e40.
- Leask A (2010) Potential therapeutic targets for cardiac fibrosis: TGFβ, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circulation research 106: 1675-1680.
- Fabiato A (1983) Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am J Physiol 245: C1-14.
- Belevych AE, Terentyev D, Terentyeva R, Nishijima Y, Sridhar A, et al. (2011) The relationship between arrhythmogenesis and impaired contractility in heart failure: role of altered ryanodine receptor function. Cardiovascular research 90: 493-502.
- Pereira L, Ruiz-Hurtado G, Rueda A, Mercadier JJ, Benitah JP, et al. (2014) Calcium signaling in diabetic cardiomyocytes. Cell Calcium 56: 372-380.
- Fischer TH, Herting J, Tirilomis T, Renner A, Neef S, et al. (2013) Ca2+/calmodulin-dependent protein kinase II and protein kinase A differentially regulate sarcoplasmic reticulum Ca2+ leak in human cardiac pathology. Circulation 128: 970-981.
- Dobrev D, Wehrens XH (2014) Role of RyR2 phosphorylation in heart failure and arrhythmias: Controversies around ryanodine receptor phosphorylation in cardiac disease. Circulation research 114: 1311-1319.
- Wehrens XH, Marks AR (2004) Novel therapeutic approaches for heart failure by normalizing calcium cycling. Nature Reviews Drug Discovery 3: 565.
- El-Armouche A, Boknik P, Eschenhagen T, Carrier L, Knaut M, et al. (2006) Molecular determinants of altered Ca2+ handling in human chronic atrial fibrillation. Circulation 114: 670-680.
- Tamargo J, Lopez-Sendon J (2011) Novel therapeutic targets for the treatment of heart failure. Nature reviews Drug discovery 10: 536.
- Netticadan T, Temsah RM, Kent A, Elimban V, Dhalla NS, et al. (2001) Depressed levels of Ca2+-cycling proteins may underlie sarcoplasmic reticulum dysfunction in the diabetic heart. Diabetes 50: 2133-2138.
- Frank KF, Bölck B, Brixius K, Kranias EG, Schwinger RH, et al. (2002) Modulation of SERCA: implications for the failing human heart. Basic research in cardiology 97: I72-I78.
- Gorski PA, Jang SP, Jeong D, Lee A, Lee P, et al. (2019) Role of SIRT1 in Modulating Acetylation of the Sarco-Endoplasmic Reticulum Ca2+-ATPase in Heart Failure. Circulation research 124: e63-e80.
- Xu A, Hawkins C, Narayanan N (1993) Phosphorylation and activation of the Ca (2+)-pumping ATPase of cardiac sarcoplasmic reticulum by Ca2+/calmodulin-dependent protein kinase. Journal of Biological chemistry 268: 8394-8397.
- Toyofuku T, Kurzydlowski KC, Narayanan N, MacLennan DH (1994) Identification of Ser38 as the site in cardiac sarcoplasmic reticulum Ca (2+)-ATPase that is phosphorylated by Ca2+/calmodulin-dependent protein kinase. Journal of Biological Chemistry 269: 26492-26496.
- Rodriguez P, Jackson WA, Colyer J (2004) Critical evaluation of cardiac Ca2+-ATPase phosphorylation on serine 38 using a phosphorylation site-specific antibody. Journal of Biological Chemistry 279: 17111-17119.
- Valverde CA, Mundiña‐Weilenmann C, Said M, Ferrero P, Vittone L, et al. (2005) Frequency‐dependent acceleration of relaxation in mammalian heart: a property not relying on phospholamban and SERCA2a phosphorylation. The Journal of physiology 562: 801-813.