Inherited cardiomyopathies (CMs) are a major cause of heart diseases in all age groups, which severely burdens patients as well as their family members. Data from the past two decades has identified defects in several genes especially those encoding sarcomeric proteins as an important cause of familial hypertrophic, dilated or restrictive CMs. A growing number of families with inherited CMs have also provided a unique resource for studies on the pathogenicity of genetic defects in CMs. Studies on imaging, genetics, and genomics have identified molecular triggers of CMs. However, the functional consequences of genetic mutations and the exact details of the signalling pathway leading to hypertrophy, dilation and/or contractile impairment remain to be elucidated. Despite marked improvement in prophylactic and therapeutic agents, morbidity and mortality rates of inherited CMs have not reduced significantly. However, increased focus on genetic studies have the potential to influence the shift from the current symptomatic to novel aetiologic-specific treatment for various forms of inherited CMs. This review summarizes published findings of inherited CMs with a focus on genetic aetiology, pathogenesis, genetic sequencing and treatment.
dilated cardiomyopathy, familial cardiomyopathies, genetic cardiomyopathies, hypertrophic cardiomyopathy, inherited cardiomyopathies
Cardiomyopathies (CMs) are a difficult and complicated group of myocardial diseases, as well as a leading cause of heart failure (HF) and/or sudden cardiac death (SCD). Over the past two decades, the morbidity and mortality rates of CMs have remained virtually unchanged [1,2]. Contrastingly, the understanding of the major forms of CMs have significantly improved partly because of advancements made on diagnostics and therapeutics of HF particularly in non-invasive cardiac imaging, genetics and genomics [3]. The classical classification of CMs based on morphological and functional criteria categorizes CMs into five types: (i) dilated cardiomyopathy (DCM); (ii) hypertrophic cardiomyopathy (HCM); (iii) restrictive cardiomyopathy (RCM); (iv) arrhythmogenic ventricular cardiomyopathy (AVC); and (v) left ventricular non-compaction (LVNC) [4-7]. This morphological and functional classification is crude but practical and very useful in clinical settings. Despite considerable heterogeneity in DCM, HCM and RCM, and other CM types, morphological and functional classification has been useful in predicting and delineating treatment options for each category [5,7].
With considerable advances in genetics and genomics, the clinical use of molecular genetics has further refined the traditional morphological and functional categories into two clinically significant sub-groups – genetic (inherited or familial form) and acquired (non-inherited or secondary form). However, molecular genetics as a basis of classification for CMs does not supersede clinical classification because of variability of mutations within the same gene can manifest as different disorders [3,7]. All inherited CMs express genetic heterogeneity, and within each disease category, multiple genes and several different mutations may exist. In the past, these genetic mutations were uncommon, but at present, technical advances have allowed routine genetic testing of families leading to the discovery of many more previously unidentified mutations [8]. The degree of genetic variability among the CMs determines the extent to which a final common pathway of pathogenesis is clinically identifiable for each condition. Tobin and colleagues [9,10] proposed the concept of the “final common pathway”. The concept postulates that genetic and mechanistic causes of CMs often follow a disturbance in a particular disease-specific final common pathway. This review focuses on the molecular genetic basis of CMs including pathogenesis, presentation, genetic sequencing, and clinical management.
A precise understanding of the cardiomyocyte cellular physiology is essential to appreciate the pathogenicity of genetic mutations leading to CM. The primary cardiac function is to propel oxygenated blood to the peripheral body tissues to meet their metabolic demands and to eliminate cellular waste products from tissues. The systemic circulation system (arterial and venous systems) provide the conduits for delivery of oxygenated blood to the peripheral tissues. It is vital to understand the mechanisms of the function of the normal heart as a prelude to comprehend the effect of pathologic genetic mutations on cardiac contractility. Consequently, this section provides a discussion on the cardiomyocyte physiology to understand normal cardiac contraction and relaxation.
Microscopic anatomy
The myocardium is a highly organized cardiac muscle tissue composed of several cell types including the smooth muscle cells, cardiomyocytes and fibroblasts. The cardiomyocyte is the fundamental contractile cell of the myocardium. The myocardium, same to skeletal muscle, appears striated because of the organization of muscle tissue into sarcomeres. However, the myocardium differs from the skeletal muscle in a few ways. The myocardium consists of tubular cardiomyocytes, in turn, composed of tubular myofibrils (repeating pattern of sarcomeres). Intercalated disc transmit electrical action potential between sarcomeres [10,11].
Sarcomere structure: The sarcomere is the basic unit of muscle tissue found in both the myocardium and skeletal muscle. Microscopically, the sarcomere appear as striation with alternating dark and light bands. T-tubules bind sarcomeres to the sarcolemma (a plasma membrane) and increase the rate of depolarisation within the sarcomere. Individual sarcomeres is composed of long, contractile proteins that slide over each other during myocardial relaxation and contraction. The two key fibrous proteins in the cardiomyocyte are myosin, which forms a thick flexible filament, and actin, which forms a thin, more rigid filament. The structure of myosin, a long fibrous tail and a globular head, allows it to bid to actin. The myosin head also binds to ATP, the primary source of energy for cardiac contraction. The actin molecules attach to the Z-disc, at the border of the sarcomere [10]. Both the myosin and actin form the myofibrils, which are repeating patterns of the molecular structure of the sarcomere.at the molecular level. The activity of myofibril leads to muscle contraction. Mysin binds to ATP dissociating from the actin within the myofibril, causing a contraction. The contraction of the myocardium is a complex process that occurs in the presence of calcium influx and the stimulus of electrical impulses. The cardiomyocytes also feature regulatory proteins troponin and tropomyosin, which also play a role in myocardial contraction [10,11].
Intercalated discs: Generally, an intercalated disc is the junction that binds cells across a gap. In the myocardium, intercalated discs are specialized attachment sites (gaps) that bind the cardiomyocytes together and prevent them from pulling apart. They transmit contractile force of one cardiomyocyte axially to the next. They also permit the transmission of electrical impulses (action potentials) and calcium ions between the cardiomyocytes during muscle contraction [12]. The intercalated disc connect the cardiomyocytes to the syncytium (a multi-nucleated muscle cell) to support the rapid spread of electrical impulses allowing synchronized contraction of the myocardium. Observed under light microscopy, intercalated discs appear as dense staining cross-bands – thin lines that divide adjacent cardiomyocytes and run perpendicular to the direction of the muscle fibres [10,11].
Mechanism of contraction
The widely accepted mechanism of contraction of both the normal and diseased myocardium is the actin-myosin interaction. Also, involved in cardiomyocyte contraction are the troponin-tropomyosin complex, calcium ions and other regulatory factors. The proposed mechanisms for myocardial contraction are the sliding window, the swinging cross-bridge hypothesis and the leverarm hypotheses. However, the sliding window theory remains the most widely cited and accepted mechanism to explain myocardial contraction.
Actin-myosin interaction: Bowditch et al. [13] provided the seminal description of several important aspects of cardiac contractility that continue to underlie current studies on cardiac contraction. His publication in 1871, “Properties of excitability of cardiac muscle fibres”, raised several fundamental issues. Janssen et al. [14] cited the Bowditch et al. [13] study and indicated that, in principle, the myocardium in contrast to skeletal muscle has an all-or-nothing excitation mode, which when stimulated at a fixed frequency and amplitude, contraction occurs, and the force of contraction of the myocardium increases with the frequency of stimulation [14]. The focus of the study was not only on the strength of contraction but also on the speed of both contraction and relaxation, which are both critical in governing contractility. Eight decades later, Huxley et al. [15] proposed the theory of the sliding filament, which remains the accepted mechanism for the process of muscle contraction.
The sliding filament theory posits that the myocardium generates mechanical force by the sliding movement of the thick filaments (myosin) over the thin filaments (actin) mediated by the cyclical attachment and detachment of the myosin cross-bridges to actin. The cross-bridges are composed of myosin heavy chain molecules that protrude from the thick filament. Muscle myosin contains two myosin heavy chains and each chain has a head with an actin binding the site and ATPase site. The heads hinge to a long rod that contains an elastic element, and binds the myosin light chains. The initial step of the sliding filament theory consists of myosin strongly bounded to actin, with myosin head orientated in a 45 degrees position relative to its tail. ATP binding occurs at the ATPase site causing a rapid dissociation of myosin from actin and the formation of ADP and P. Actin recombines weakly with the myosin-ADP-P complex, with myosin head angled at 90 degrees. The release of ADP and P allows the strong actin binding to regain its position. This last step achieves the power stroke through a rowing like motion of the myosin head as it slides down the actin filament. Each cross-bridge cycle leads to hydrolysis of one ATP molecule [13,15].
The swinging cross-bridge hypothesis is a subsequent modification of the sliding filament theory. The hypothesis postulates that the energy from the hydrolysis of ATP is converted to the mechanical swinging motion of the myosin head while bound to actin. The light-chain binding region acts as a lever arm that amplifies movement near the catalytic site. The swinging movement of the myosin head is a critical factor for the progression along actin [16,17]. However, findings that myosin VI takes larger steps along the actin filaments than early interpretation of its structure appear to allow challenges the swinging cross-bridge hypothesis. It is now known that myosin VI operates by an unusual ~180 degrees arm swing and achieves its large step size using special morphological features in its tail domain [17].
The leverarm model is a subsequent modification of the swinging cross-bridge model. The model hypothesizes that the rotation of the myosin tail, which acts as a lever, amplifies morphological changes in the catalytic domain of the myosin head [15,18,19]. The pivoting movement of the myosin tail (at the hinge region) produces the power stroke and not the movement of the myosin head (at the point of actin attachment). The lever arm rotation has been attributable to the transition between open and closed conformation of the myosin head, which influence nucleotide affinity and hydrolysis determined by acting binding. In a subsequent proposal, altered orientation of both the myosin head and the alpha helical tail may contribute to the overall displacement of actin [11]. However, there is conflicting data for the extent of actin displacement, ranging from 4-5 nm to 15-30 nm [15,20]. These conflicting findings may be attributable to the number of attached states because of the possibility that a single myosin head can attach at two points on an actin molecule, or one or two myosin heads may attach at any point in time. Differences in compliance and load in the different hypothesized mechanisms of myocardial contraction may also contribute to different extent of actin displacement.
Role of troponin-tropomyosin complex: Troponin is a protein complex and a component of thin filaments alongside actin and tropomyosin. Calcium ions (Ca2+) bind to troponin to trigger the production of muscular force. Thus, troponin-tropomyosin complex is a Ca2+ sensitive switch responsible for the regulation of the actin-myosin interaction. The backbone of the thein filament consists of a double helical array of globular actin molecules. Tropomyosin proteins assemble as alpha-helical coiled-coil dimers lying in a head-to-tail orientation within the major groove of the actin filaments. Troponin T predominantly, and troponin I less commonly, anchors the troponin complex (troponins T, I and C). Troponin C interacts with both troponins T and I. During diastole, the binding of troponin I to actin-tropomyosin inhibits actin-myosin interaction. Ca2+/troponin C binding weaken troponin I/actin-tropomyosin and strengthens troponin I/troponin C interaction. These alterations leads to the release of the thin filament from its inhibitory state and promotes actin-myosin interaction and force generation. Reduced Ca2+ concentration results in the dissociation of C2+ from troponin C and restores the relaxed state [21].
Role of calcium ions: Calcium ions (C2+) play a critical role in myocardial contraction. Cardiac muscle fibres undergo synchronized contraction controlled by calcium-induced calcium release (CICR) mechanism conducted through the intercalated discs. The gap junctions of intercalated discs control the coordinated contraction of the cardiomyocytes spread electrical impulses. The contraction occurs through a phenomenon known as excitation contraction coupling (ECC), meaning the process of converting electrical impulses from the neurons into a mechanical response that causes muscle movement. Action potentials are the electrical stimulus that evokes the mechanical response in ECC. In the myocardium, ECC depends on a mechanism referred to as CICR that involves the direct entry of CA2+ into the cytoplasm. Influx of Ca2+ triggers a further release of ions into the cytoplasm. The underlying mechanism for CICR are receptors located in the cardiomyocyte, which bind to CA2+ when the CA2+ ion channel opens during depolarization leading to the release of more CA2+ into the cardiomyocyte. Same to skeletal muscles, the influx of sodium ions (Na+) leads to an initial depolarization. However, in the myocardium, the influence of Ca2+ sustains the depolarization making it persist longer. The CICR mechanisms appear to create a plateau phase, in which the cell’s charge stays depolarized (positive) shortly before it becomes more negative as it repolarizes because of potassium (K+) influx. In contrast, skeletal muscles repolarizes immediately.
The actual myocardial mechanical contraction occurs because of the sliding filament model of contraction. As discussed previously, the model postulates that myosin filaments slide along the actin filaments shortening or lengthening the muscle fibre for contraction and relaxation. The contraction pathway is composed of five phases [10-12].
- The conduction of action potential induced by the pacemaker cells in the sinoatrial (SA) and atrioventricular (AV) nodes to contractile cardiomyocytes occurs through gap junctions.
- The conduction of action potential between sarcomeres activates calcium channels in the T-tubules causing an influx of Ca2+ ions into the cardiomyocyte.
- In the cytoplasm, Ca2+ binds to cardiac troponin C moving the troponin complex away from the actin-binding site, consequently freeing the actin and making it available for myosin binding to initiate contraction.
- The myosin head binds to ATP and pulls the actin filaments towards the centre of the sarcomere to cause muscle contraction.
- The sarcoplasmic reticulum then removes intracellular calcium decreasing the concentration of intracellular calcium and returning the troponin complex to its inhibiting position on the active site of actin. Consequently, these actions end contraction as the actin filaments return to their initial position, relaxing the muscle.
Other regulatory factors: Intrinsic sarcomere components (myosin light chains (MLC) and cardiac myosin binding protein C) act as regulatory factors in myocardium contraction. Essential and regulatory MLC bind to the alpha-helical lever arm of the myosin cross-bridge resulting in force production achieved through modulating cross-bridge kinetics [22,23]. The cardiac myosin binding protein C binds to the S2 segment near the lever arm of the myosin head and is involved in the adrenergic regulation of cardiac contractile function [24,25]. Ultrastructural studies reveal phosphorylation of the cardiac myosin binding protein C by cAMP-dependent protein kinase extends myosin cross-bridge from the backbone of the thick filament as well as alters their orientation [26]. Functional studies further show that the phosphorylation status of the cardiac myosin binding protein C determines the stiffness and attachment rates of cross-bridges and sensitivity of Ca2+ force production [26,27]. In addition to intrinsic sarcomere components, several other extrinsic factors such as neurohormonal, endocrine and hemodynamic factors are involved in myocardial contractility in vivo [9].
Inherited cardiomyopathies
The past two decades has seen the identification of many genes whose mutations are responsible for the development of different types of CM. Towbin et al. [9,28] developed the “final common pathway” hypothesis for inherited cardiovascular disease (Figure 1). The hypothesis states that genes encoding proteins with similar functions or involved in the same pathway are responsible for the development for a particular disease or syndrome phenotype. The authors identified structural and functional similarities in proteins encoded by genes whose disruption leads to a somewhat predictable gross clinical phenotype. The authors found out that, causative genes for inherited arrhythmic disorders encode for ion channels, those for HCM encode for sarcomeric proteins, and those for AVC encode cell-to-cell junction proteins. In addition, protein altered by the mutated gene directly disrupts the normal function of the structures in which the protein is integrated such as mostly the sarcomere in HCM when the mutated gene encodes for a sarcomeric protein. In some instances, the mutated gene disrupts a binding pattern protein causing downstream disturbance of the final common pathway, such as a Z-disk protein disrupting the cell-to-cell junction through maladaptive binding to desmin, which negatively interacts with desmsomal protein causing AVC [9,28]. The main types of inherited CMs are HCM, DCM, AVC and RCM
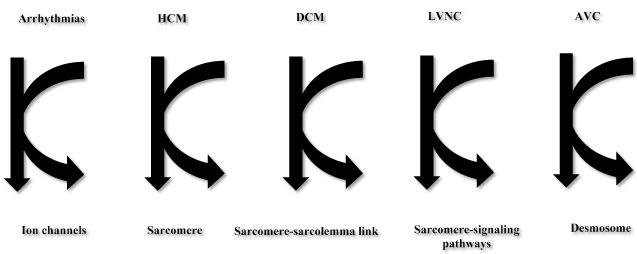
Figure 1. Final common pathway hypothesis for arrhythmias, HCM, DCM, LVNC, AVC Adapted from Towbin, 2014 [9]
Hypertrophic cardiomyopathy
Clinical presentation: Inherited HCM is a primary myocardial disease inherited in an autosomal dominant pattern and characterized by unexplained thickening (hypertrophy) of the left ventricle (LV), and sometimes the right ventricle (RV), often with a predominant involvement of the inter-ventricular septum. The disorder may also reveal histological features of cardiomyocyte hypertrophy, myofibrillar disarray, and interstitial fibrosis [3,4]. HCM is among the most prevalent inherited cardiac disorder affecting approximately 1 in every 500 individuals [29]. HCM has other various names including hypertrophic obstructive CM and idiopathic sub-aortic stenosis derived from the textbook features of asymmetric septal hypertrophy and LV outflow tract obstruction. These features of HCM refer to severe symptomatic patients seen in tertiary hospital referral centres [11]. Epidemiological data however demonstrate a wide heterogeneity of clinical presentation with varying severity and prognosis in community populations suggesting the use of the term HCM as more appropriate to describe this patient cohort as well as preferred by both the WHO, AHA and ESC consensus reports on CM [3-6].
Natural history: HCM has a variable history and a wide spectrum presentation. Some patients may remain asymptomatic throughout life; others may develop progressive disease in the presence or absence of HF; and others may succumb to SCD. Longitudinal echocardiographic studies document LV remodelling progresses with age. Individuals during adolescent and early adulthood who have not undergone genotype investigation demonstrate progressive LV wall thickening [30]. Some patients with long standing disease may develop age-associated decline in LV wall thickness associated with cardiomyocyte loss and fibrosis. Indeed, 10-20% of HCM patients may develop DCM, and 16% atrial fibrillation, in which LA enlargement is a significant risk factor. HCM is a frequent cause of SCD in young adults, particularly competitive athletes but the prevalence varies between <1% in the general community to 306% in tertiary referral hospitals.
The cause of SCD in HCM patients is multifactorial encompassing bradyarrhythmias secondary to sinus node and AV conduction abnormalities; tachyarrhythmias induced by re-entrant depolarization pathways associated with myofibrillar disarray and fibrosis, abnormal Ca2+ homeostasis, myocardial ischemia, LV diastolic dysfunction or LV outflow tract obstruction (LVOTO) [11]. Despite several clinical parameters, SCD in HCM patients lacks a single identifiable risk factor but young age at diagnosis, history of syncope, severity of cardiac symptoms, LV wall thickness, LV outflow tract gradient, LA size, and atrial fibrillation are potential risk factors in HCM individuals who have not undergone genotype evaluation. Genotyped individuals, however, have significant prognostic variability among the different HCM causative genes and between different mutations in the same gene. The underlying mechanism in which HCM gene mutations may influence prognosis remains unknown [11].
Clinical evaluation: Since its recognition as a genetic disorder in the late 1950s, numerous clinicopathological studies on HCM have greatly improved our current understanding of this myocardial disorder. More importantly, in the past three decades, molecular genetic studies have provided an even deeper insight into the pathogenesis of HCM that informed new perspectives for the diagnosis and management of HCM patients. Current understanding of HCM indicate HCM individuals exhibit considerable variability in clinical presentation. Genotype positive individuals may be asymptomatic or present with a range of cardiac symptoms from palpitations and dizziness to syncope and SCD. Genotype-phenotype studies reveal the age of onset of symptoms varies between different HCM disease genes. In patients with β-MHC mutations, symptoms presents in the first two decades of life, while those with cardiac myosin binding protein C mutations remain asymptomatic until the fifth or sixth decade of life [30].
The definitive diagnosis of HCM rests on non-invasive cardiac imaging. The preferred initial non-invasive imaging modality for the diagnosis of HCM is transthoracic echocardiography. The cardinal diagnostic feature is asymmetric hypertrophy of the interventricular septum in the presence or absence of LVOTO and systolic anterior motion of the mitral valve. The obstructive form affects less than 25% of the HCM population. Studies of family members of HCM patients documented that the distribution and severity of LV hypertrophy (LVH) vary considerably. Thus, in 2007, the ESC [7] and suggested that asymmetric hypertrophy was no longer the sole reliable pathognomonic diagnostic feature for HCM. In addition to LVH, diagnosis require the exclusion of secondary aetiologies of LVH such as hypertension or aortic stenosis, which may coexist with HCM in older patients.
Distinguishing HCM from physiological LVH is difficult more so in competitive athletes. The extent of LVH also varies among the causative mutational genes. Individuals with β-MHC gene mutations often develop moderate to severe LVH with a high degree of disease penetrance, while individuals with cardiac troponin T gene mutations often have mild or clinically undetectable LCH [31,32]. Other genetic mutations leading to the development of unusual forms of hypertrophy include cardiac troponin I gene mutations causing hypertrophy localized to the LV apex [33], and cardiac actin and MLC gene mutations causing hypertrophy of the mid-cavity [22,34]. The extent of LVH also varies among members of a single family with the same genetic mutation. Variability in both clinical expression and LVH among HCM patients may be a consequence of modifying roles of additional genetic and environmental factors including pressure, exercise, diet and body mass [11].
The current use of genetic testing for HCM is the identification of families with a detectable genetic cause of the disease and screening of at-risk family members. Genetic testing is also useful in ruling out non-genetic cardiac conditions such as athlete’s heart although only possible after the detection of a pathogenic variant. Inherited HCM lacks clear genotype-phenotype correlations, which limits the utility of genetic findings in guiding clinical management. A notable limitation is enzyme replacement therapy for storage disorders, which can present as isolated LVH. Emerging genotype data shows a potential use in guiding therapeutic decisions in individuals with pre-clinical disease. Animal studies suggest that calcium channel blockers (diltiazem) may delay the clinical progression of disease, and clinical trials are ongoing [9,11].
Genetics of HCM: Over the past three decades, molecular genetic studies have provided valuable insights into the pathogenesis of HCM as well as new perspectives for diagnosis and management. To date, over 20 genes have been discovered, mostly affecting the sarcomere although other mutations may affect genes encoding proteins of the Z-disk or intracellular calcium modulators (Figure 2). Of genetic causes of HCM, eight causative genes encode sarcomeric proteins with a majority (about 80%) mutations identified in the β-Myosin heavy chain 7 (MYH7) and myosin binding protein (MYBPC3) genes [3,35-37]. The typical type of mutations in the sarcomeric genes are single nucleotide substitutions. The mutation protein incorporates into the sarcomere exerting a poison peptide effect except MYBPC3 genes where deletion or insertions lead to a frameshift leading to haploinsufficiency [38]. Besides mutations in the sarcomeric genes, mutations in the Z-disk and other non-sarcomeric encoding genes may cause HCM [35,36,39,40]. Mutations in protein titin and its interactive Z-disk proteins – MLP, ZASP, telethonin, nexilin, myopalladin, myozenin-2, α-actinin 2, CARP and vinculin are non-sarcomeric genetic causes of HCM [40]. The specific mechanisms for Z-disk and calcium modulator genes remains unclear.
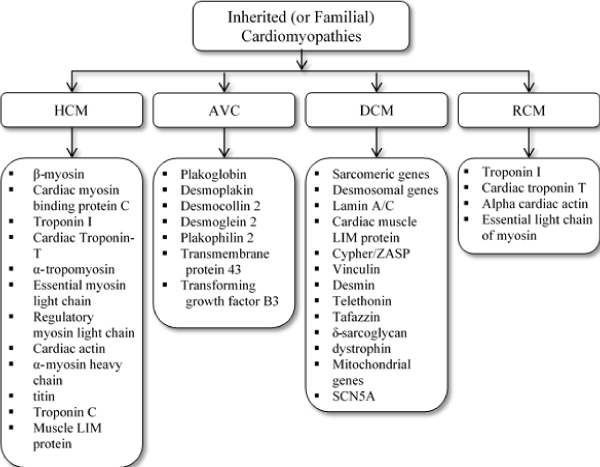
Figure 2. Classification of cardiomyopathies and the associated major genetic mutations
Recent clinical guidelines for inherited forms of HCM recommend testing for five genotypes – MYBPC3, MYH7, Cardiac troponin I, type 3 (TNN13), Cardiac troponin T, type 2 (TNNT2), and α-Tropomyosin 1 (TPM1), although current next generation sequencing (NGS) techniques have facilitated a wide availability of a much broader testing panels from commercial laboratories [41]. At present, the primary use of genetic testing in HCM patients is the identification of families with detectable genetic cause of disease and screening at-risk family members. The main constraint in the use of genetic testing in guiding clinical management of HCM is limited genotype-phenotype correlations. Despite the identification of ~1,000 variants for HCM, most of them are unique variants and only identifiable through comprehensive genetic testing.
Variants in sarcomeric gene mutations occur in up to 60-70% of genotyped HCM patients and 60% of sporadic HCM [36,42]. Genotype-phenotype of HCM patients. However, genotype-phenotype correlations for HCM remains incompletely defined partly because of unique variants of the mutations. Findings that a specific mutation in the sarcomeric proteins leads to a specific phenotype are inconclusive because of heterogeneous variability in the pattern and extent of LVH, and outcomes in HCM patients including first-degree relatives [43]. The heterogeneity has complicated correlations between genotype, and phenotype or outcomes in HCM patients. A recent systematic review and meta-analysis including 2,459 patients from 53 families with gene mutation in the sarcomere, reported sarcomeric mutation with younger age of presentation (< 45 years), family history of HCM, family history of SCD and greater LV wall thickness (≥20 mm) as positive predictors of mutation, whereas hypertension as a negative predictor [44-47].
Dilated cardiomyopathy
Clinical presentation: Inherited DCM is a myocardial disease and a common cause of HF defined by the presence of LV systolic dysfunction with LV dilatation on the absence of coronary artery disease (CAD) or other causes such as hypertension or valvular pathology [7]. The RB may be involved but it is not necessary for diagnosis. DCM may result from a wide variety of causes that lead to cardiomyocyte injury and dysfunction. In about 50% of the cases, a specific precipitation cause cannot be identified (idiopathic DCM). The incidence of idiopathic DCM is 5-8 cases per 100,000 per year [48]. Although traditional perception was that DCM is a sporadic non-genetic disorder, recent studies of large families with DCM demonstrate that inherited gene defects are an important aetiologic agent of inherited or familial DCM.
DCM is the most common CM, affecting approximately 50% of CMs [3]. The reported annual incidence ranges from 2 to 8 in 100,000 individuals, 0.57 per 10,000 per year in children. Its prevalence is about 1 in 2,500 individuals [3,49]. The proportion of patients with a genetic cause varies significantly because of the different methods of detection and diagnostic criteria adopted by individual studies. On the one hand, studies based solely on positive clinical history of DCM in relatives of probands yielded a relatively low prevalence (< 10%) of inherited diseases. On the other hand, studies based on first-degree relatives of probands evaluated with physical examination, 12-lead ECG and transthoracic echocardiography yielded a higher prevalence (up to 35%) [50-52]. However, the reported prevalence potentially underestimates the true prevalence of inherited DCM. A greater majority of DCM studies adopted the diagnostic criteria proposed by the World Health Organization (WHO) – the presence of both ventricular dilation and contractile dysfunction [4].
Natural history: The natural history of inherited DMC shows variation between and within family members. Individuals with DCM may express a relatively benign course, develop progressive HF (sometimes requiring heart transplantation) or succumb to SCD. Severe HF or ventricular arrhythmias are the cause of premature deaths in DCM patients. Sudden death can occur at any age independent of ventricular function. HF is an independent prognostic marker, with a 5-year survival at 50% after the diagnosis of HF [48-52]. In a sub-group of DCM families with coexisting conduction system disease, progressive AV conduction disturbances may occur in the fourth decade of life and subsequent development of DCM. In these patients, permanent cardiac pacemakers may be necessary for high-grade AV conduction block. Thromboembolic events in individual with inherited DCM may be a consequence of marked blood stasis in the ventricles or coincide with the onset of atrial arrhythmias. Genotype-phenotype correlations in familial DCM remain to be determined. Despite proposals of multiple clinical parameter as predictors of mortality, family genotype may be the strongest predictor of patient outcomes.
Clinical evaluation: The most frequently observed inheritance fashion for inherited DCM is autosomal dominant although cases of autosomal recessive, X-linked and maternal modes of inheritance have been documented. Inherited DCM expresses considerable clinical variability with several distinct phenotypes: DCM alone; DCM with conduction system disease such as bradycardia, AV conduction block or atrial arrhythmias; DCM with skeletal myopathy in the absence or presence of conduction system disease; or DCM with skeletal with sensorineural deafness. In some families, DCM may manifest as part of a larger multi-system inherited disorder. The relative prevalence of these phenotypic groups remains undetermined. Besides family history, no single pathognomonic feature can reliably differentiate inherited from secondary DCM (due to other cardiac or systemic diseases).
Inherited DCM can be asymptomatic in some individuals usually detected incidentally as cardiomegaly on routine chest radiography. Clinical presentation in a majority of DCM individuals are symptoms due to the LV failure or cardiac arrhythmias such as weakness, fatigue, dyspnoea, orthopnoea, palpitations or exercise intolerance. Other symptoms such as right HF such as peripheral oedema and abdominal dimension associated with hepatic congestion and ascites may occur during disease progression. Although diagnosis of inherited DCM is at an earlier age compared to non-inherited forms, the presence of relatively malignant phenotype in some families or a more intensive screening of at high-risk family members may explain the early diagnosis [53].
The non-invasive cardiac imaging modality of choice for the diagnosis of inherited and non-inherited DCM is transthoracic echocardiography. Two-dimensional (2D) echocardiographic imaging allows quantitative assessment of ventricular structure and systolic function, and the evaluation of valvular pathology and atrial size. Tissue Doppler echocardiography allows the assessment of ventricular diastolic function and the severity of mitral and tricuspid valve regurgitation. Individuals who echocardiography is technically difficult or inconclusive, gated radionuclide scans may be used to quantify ventricular volumes and contractile performance.
Supplementary tests include cardiac catheterization for the assessment of ventricular function, particularly in DCM patients at risk of CAD. Endomyocardial biopsy generally has non-specific findings such as myocyte hypertrophy, necrosis, nuclear abnormalities and interstitial fibrosis. ECG is an important initial test for DCM patients, which may reveal abnormalities conduction system such as sinus tachycardia, poor R-wave progression in the pre-cordial leads, intraventricular conduction delays or non-specific ST segment and T-wave changes. Inherited DCM in the presence of conduction system disease may present with sinus bradycardia, AV conduction block and atrial flutter or fibrillation. Advance HF may also be a cause of conduction abnormalities and atrial tachyarrhythmias. Ambulatory ECG monitoring in DCM patients often reveals ventricular ectopic beats or non-sustained ventricular tachycardia [11].
The use of genetic testing for DCM in clinical setting has increased in the past three decades. However, due to significant locus and allelic heterogeneity, the variant spectrum and detection rates of DCM remain less defined compared to HCM. Pathogenic variants occur in 17% to 30% of individuals with DCM after sequencing up to 20 genes. In contrast to HCM, most DCM genes contribute only a small percentage of all pathogenic variants [9,11]. Clear genotype-phenotype correlations are very rare except variants in the LMNA and SCN5A genes typically associated with DCM and conduction system disease [9]. The electrophysiological presentation appear prior to the inset of DCM and may represent the only cardiac manifestations. Recent evidence indicate desmosomal genes known to cause AVC, may be involved in the aetiology of DCM. Novel genes for DCM continue to be discovered with recent additions including titin (TTN). TTN contributes up to 25% of inherited and 18% of sporadic DCM cases, making it the most mutated gene in DCM as well as elevating the detection rate of genetic testing panels of HCM.
The current consensus guidelines are yet to incorporate these recent additions and recommend comprehending testing for the LMNA and SCN5A in DCM patients with co-occurring cardiac conduction disease and/or a family history of premature CAD. Despite a wide genetic heterogeneity, Genetic testing is important for the diagnosis of DCM since it allows for informed family evaluation if a pathogenic variant is identified in the probands. Additionally, evidence is accumulating on patients with asymptomatic systolic dysfunction may benefit from
Genetics of DCM: The inherited forms of DCM usually results from mutations in the genes encoding cytoskeletal and sarcomeric proteins [28,35,36,54]. Inherited DCM may also manifest indirectly in the setting of neuromuscular diseases such as Emery-Dreifuss muscular dystrophy, Barth syndrome, myofibrillar myopathy, limb-girdle muscular dystrophy, and Duchenne/Becker muscular dystrophy (DMD/BMD) [55]. Inherited form of DCM accounts for 30 to 50% of all the reported DCM cases transmitted predominantly in an autosomal dominant pattern, and less commonly X-linked, autosomal recessive, and mitochondrial inheritance patterns [48,56-59]. The typical characteristics of DCM is LV dilatation accompanied by systolic dysfunction (reduced myocardial contractility) and is the most common indication for heart transplantation [3,48,60].
The X-linked forms of DCM disproportionally affects males in their twenties and early thirties, typically exhibiting elevated serum creatinine-kinase muscle isoforms (CK-MM). The disease exhibits a rapid progression from HF to death due to arrhythmias and is a leading indication for cardiac transplantation [59,61]. In contrast, female carriers develop a benign and gradually progressive disease usually in the fifth decade of life. Towbin et al. [61] identified mutation in the dystrophin gene as a cause of familial DCM, and demonstrated a significant reduction or the absence of dystrophin protein in the heart [61-63]. Dystrophin links the sarcomere to the sarcolemma and extracellular matrix [59]. Mutation in the dystrophin gene causes DMD or BMD, which are skeletal myopathies presenting in males early in life marked with elevated CK-MM and DCM manifesting between 10 and 25 years of age [59-66].
The prevalence and mechanisms of DCM phenotypes varies based on the mutant gene. The most prevalent form of inherited DCM displays an autosomal dominant pattern of transmission [28,54,58]. Compared with inherited HCM, which mainly affects the sarcomere, inherited DCM exhibits a considerably higher degree of genetic heterogeneity with more than 40 genes implicated. These genes mostly encode cytoskeletal, sarcomeric or Z-disk proteins, although ion channel-encoding and desmosome-encoding genes may also be involved [35,36,67,68] (Figure 2). Cytoskeletal proteins cause defects of force of transmission, whereas sarcomeric proteins cause defects of force generation resulting in the DCM phenotype [69,70]. Mutations in genes encoding desmosomal proteins disrupt the links between the intercalated disks, Z-disk and sarcomere. Gene mutations may also disrupt protein-binding patterns resulting in different phenotypes and severity [71,72].
Arrhythmogenic ventricular cardiomyopathy
Clinical presentation: Arrhythmogenic ventricular cardiomyopathy (AVC) is a primary heart muscle disorder characterized by cardiomyocyte loss caused by necrosis and/or apoptosis with fibrofatty replacement of the myocardium. The disorder constitutes a hereditary CM with an autosomal dominant pattern of inheritance although a recessive cardiocutaneous disorder also occurs [73]. Previously, this disorder was termed arrhythmogenic right ventricular cardiomyopathy or dysplasia because the initial description suggested pathological disease process affected solely the RV and any associated disease of the LV was considered an exclusion criterion for diagnosis in the original Task Force Criteria for diagnosis. Evidence that is more recent suggests a possible involvement of the LV often associated with severe disease and a worse prognosis [74] and renamed AVC in consensus statement [3].
Classically, the AVC is characterized by dilated RV with fibrofatty involvement with no or minimal involvement of the LV leading to chamber dilation and wall thinning. Extensive wall thinning may result in a parchment appearance (Uhl anomaly) [3,75]. Clinically, progressive disease is characterized by systolic impairment and bi-ventricular dilation in the absence or presence of ventricular aneurysm, and clinical features of HF. Single or multiple aneurysms of the RV free wall occur in 50% of the cases. LV involvement affects up to 76% of the cases [76]. In advanced disease, distinguishing AVC from DCM becomes difficult. The prevalence of AVC is approximately 1 in 5,000 but higher rates of 4.4 in 1,000 individuals have been reported in Northern Italy [77,78]. AVC is familial in at least 30% of the cases with a predominant autosomal dominant pattern of inheritance with reduced penetrance and variable expressivity [78]. In many parts of the world, phenotypic expression is more common in men than in women (2-3:1) [79]. AVC often manifests during late childhood or adolescence but may also present in the elderly and has a prevalence of 1 in 2,000 [80]. AVC is a leading cause of SCD in young adults (≤35 years of age) and may account up to 10% of cardiovascular deaths in individuals younger than 65 years old.
AVC is a disorder of the desmosome, a multi-protein complex that forms cell-to-cell junctions and links intermediate filaments of adjacent cells to establish a functional intercellular network. Desmosomes are especially prevalent in tissues exposed to mechanical stress such as the myocardium or the skin, which explains why the phenotypic spectrum of AVC involves cardiac and skin manifestations [35. Molecular mechanism of AVC includes impaired cell-to-cell adhesion and defective transmission of the contractile force. Recent evidence associates fibroadiposis with impaired WNT signalling leading to a re-direction of the cardiomyocyte fate to adipocyte fate [81].
Clinical evaluation: A confirmatory diagnosis of AVC is complicated by variations between and within families of the age of onset, the degree of penetrance, and clinical features. Typical clinical features include ventricular and supraventricular arrhythmias that may be well tolerated or result in syncope or SCD, which is frequently precipitated by exercise. It is a major cause of death in the young, occurring at a rate of 2.5% per year [78]. In 1994, an international task force established criteria for clinical diagnosis of AVC based on ECG findings (repolarization, depolarization or conduction abnormalities), the presence of arrhythmias, structural defects, histopathologic features and familial features. The criteria was revised in 2010, divided into six categories with major and minor features (Table 1). An individual satisfies the taskforce criteria for AVC by fulfilling two major, one major and two minor, or four minor criteria [82]. HF may occur with disease progression in up to 20% cases.
Table 1. Task force criteria for AVC diagnosis
Criteria |
Major |
Minor |
Global/regional dysfunction & structural alterations* |
Severe dilation & reduction or RVEF with no/mild LV impairment |
Mild global RV dilation or EF reduction with normal LV. |
Localized RV aneurysms |
Regional RV hypokinesia |
Severe segmental RV dilation |
Mild segmental RV dilation |
Tissue characterization of walls |
Fibrofatty replacement of myocardium on endomyocardial biopsy. |
-- |
Repolarization abnormalities |
-- |
Inverted T waves in right precordial leads (V1 and V3) – people aged >12 years in absence of RBBB |
Depolarization/ Conduction Abnormalities |
Epsilon T waves or localized prolongation > 110 ms of the QRS complex in right precordial leads (V1 to V3) |
Late potentials (signal average ECG) |
Arrhythmias
|
-- |
LBBB type VT (sustained/ non sustained) – ECG, Holter, exercise testing |
-- |
Frequent ventricular extrasystoles (>1,000/24 h) (Holter) |
Family History |
Familial disease confirmed at necropsy or surgery |
Familial history of premature sudden death <35 years due to suspected RV dysplasia. |
-- |
Familial history – clinical diagnosis based on present criteria |
RVEF: Right Ventricular Ejection Fraction; EF: Ejection Fraction; RBBB: Right Bundle Branch Block; LBBB: Left Bundle Branch Block; VT: Ventricular Tachycardia; ECG: Electrocardiography
* Detected by echocardiography, angiography, magnetic resonance imaging or radionuclide scintigraphy
Genetics of AVC: Analyses of first- and second-degree relatives of AVC patients suggest up to half of the reported AVC patients have inherited familial form of the disease. Its most common pattern of inheritance is autosomal dominant with incomplete penetrance but there are a few cases of an autosomal recessive pattern of transmission [3,36,75,83,84]. At present, 15 genes have been described to cause AVC. Towbin and colleagues [85,86] demonstrated the involvement of compound and digenic heterozygous in the pathogenesis of AVC in up to 20 cases in association with palmoplantar keratoderma and woolly hair (Naxos disease) causing a more severe disease. Due to incomplete penetrance, genotype positive relatives express variable and mild (or in some cases none) phenotypes and thus the prevalence of inherited disease may be underestimated in clinical practice [36,79].
The first gene associated with AVC was junctional plakoglobin (JUP) caused by a homozygous 2-nucleotide deletion in JUP in patients with Naxos disease. The JUP encodes plakoglobin protein, which is a key protein of the desmosome in the intercalated disk. In autosomal dominant AVC, the desmosomal protein encoding DSP gene was identified as disease causing [75,73,79]. Today, most pathogenic AVC variants are present in five genes encoding desmosomal proteins – plakoglobin, desmoplakin (DSP), desmocollin-2 (DSC-2), desmoglein-2 (DSG2) and plakophilin (PKP2) [73]. In about 80% of genotype-positive AVC patients, alterations occur in PKP2, DSP and DSG2 genes [80]. Homozygous or compound heterozygous DSP and JUP variants have been described in DCM and AVC patients, woolly/kinky hair, and palmoplantar hyperkeratosis (Naxos and Carvajal syndrome).
A few mutations in non-desmosomal genes encoding proteins that interact with desmosomal proteins mat also cause AVC. These proteins include: (i) transforming growth factor β3 that conveys cytokine stimulating fibrosis and modulates cell adhesion and growth. (ii) Transmembrane protein 43 (TMEM-43), which is a response element for PPAR-γ and am adipogenic transcription factor. (iii) DES, which binds desmoplakin. (iv) TTN, which bridges the sarcomere along its longitudinal axis and forms a continuous filament along the myofibril [3,35,75,79,87,88].
The key mechanism underlying the contribution of genetic mutation in the pathogenesis of AVC and SCD is the disruption of the integrity of the intercalated disk. Recent evidence indicates that the loss of desmosomal integrity may considerably affect gap junctions, sodium channel function and electrical propagation, which induces ventricular arrhythmias in the absence of overt structural myocardial damage [89,90]. These mechanisms provide an overlapping phenotype (CM coexisting with arrhythmias) because of the disruption of two final common pathways – desmosome and ion channel [88,91] (Table 2).
Table 2. Summary of the included studies
1st Author [Ref#]
|
Year
|
Country
|
Patients No. & CM Type
|
Male (%)
|
Age (yrs.)
|
Genes Studied
|
Methods
|
Affected Patients
|
Summary of Findings
|
Kamisago [8] |
2000 |
The U.S |
21 DCM family members |
NR |
24 |
MYH, TNNT2, TNNI3, TPM1 |
Oligonucleotide-selective sequencing |
4 |
Sarcomeric gene mutations accounts for ~10% of cases for familial DCM more prevalent in families with early onset ventricular dilatation and dysfunction. |
Millat [127] |
2010 |
France |
192 unrelated patients with HCM |
70.3 |
31.6 |
MYBPC3, MYH7, TNNT2 and TNNI3 |
Denaturing liquid chromatography/ sequencing analysis |
92 |
MYBPC3 (25%) and MYH7 (12%) are the most common cause of inherited HCM. MYBPC3 has early onset |
Meder [128] |
2011 |
Germany |
10 patients with inherited DCM (5) or HCM (5) |
50 |
42.3 |
MYH7, MYBPC3, LMNA |
Micro-array-based subgenomic enrichment followed by NGS |
6 |
Detects CM-causing mutations with high accuracy, is fast and cost-efficient and suitable for routine clinical practice of genetic testing |
Millat [129] |
2011 |
France |
105 unrelated patients with DCM |
65 |
35.8 |
MYH7, TNNT2, TNNI3 and LMNA |
High Resolution Melting (HRM)/sequencing |
20 |
Detects 19% of 105 unrelated DCM patients. LMNA and TNNT2 are the most frequent mutations |
Brito [130] |
2012 |
Portugal |
77 unrelated probands with familial or sporadic HCM |
52 |
57 |
MYBPC3, MYH7, TNNT2, TNNI3 and MYL2 |
PCR and sequencing |
41 (27 with HCM) |
Disease-associated mutations were more prevalent in familial than sporadic HCM. Mutations in MYBPC3 and MYH7 accounted for the most cases of sarcomere-related HCM |
Haas [131] |
2014 |
Multi-national |
639 familial or sporadic DCM |
66 |
|
Plakophilin-2, MYBPC3, and desmoplakin |
Ultra-high coverage next-generation sequencing of 84 genes |
294 |
High analytical quality and feasibility of Next-Generation Sequencing in clinical genetic diagnostics and provide a sound database of the genetic causes of DCM |
Akinrinade [132] |
2015 |
Finland |
145 unrelated familial or sporadic DCM patients |
|
44.3 |
Sarcomere and Z-disk, Desmosomal |
Oligonucleotide-selective sequencing |
119 |
NGS has high diagnostic yield especially in familial DCM. Bioinformatics variant filtering is a reliable step in the process of interpretation of genomic data in a clinical setting. |
Zhao [133] |
2015 |
China |
21 unrelated DCM patients |
71 |
48.7 |
Sarcomeric, Cytoskeleton and others |
Next-generation sequencing |
12 |
Genetic testing is useful in testing for pathogenic mutations to guide clinical management of familial DCM and may assist in predicting disease risk for family members before symptom onset |
Forleo [134] |
2017 |
Italy |
38 unrelated patients with DCM (16), HCM (14) or AVC (8) |
|
38 |
Sarcomeric, Cytoskeleton and others |
Next-generation sequencing |
12 |
Data obtained using targeted NGS could contribute to the molecular diagnosis of CM, early identification of patients at risk for arrhythmia, and better management of CM |
Viswanathan [135] |
2017 |
The U.S. |
80, symptomatic/ asymptomatic HCM |
62.5 |
42.4 |
Sarcomeric, Cytoskeleton and others |
Next-generation sequencing |
|
MYBPC3 mutations are a prominent cause of HCM and are phenotypically indistinguishable from HCM caused by MYH7 mutations |
AVC: Arrhythmogenic Ventricular Cardiomyopathy; CM: Cardiomyopathy; DCM: Dilated Cardiomyopathy; FCM: Hypertrophic Cardiomyopathy; NGS: Next Generation Sequencing
Left ventricular non-compaction cardiomyopathy
Clinical description: The typical characteristics of isolated LVNC is heavily trabeculated or spongy appearance of the LV myocardium. The main pathogenic mechanism is the arrest of myocardial compaction during the first trimester of embryonic development. The disorder can also be acquired based on case observations of LVNC after previous normal echocardiographic findings [92]. A recent literature review associates LVNC with mitochondrial disorders followed by Barth syndrome, an X-linked condition characterized by early onset CM (often DCM sometimes LVNC), neutropenia, muscle weakness and growth delay. The true prevalence of LVNC is unknown but reports approximate a range between 0.014% and 1.3% [92].
The LVNC CM has an early onset with variable clinical expression, ranging from asymptomatic to progressively poor cardiac function, ventricular hypertrophy, frequent thromboembolic events and SCD [93]. The disorder typically involves the LV but about half of the patients exhibit RV involvement [94,95]. Clinical manifestations and radiographic findings of LVNC resemble those found in DCM patients and can co-exist with DCM or HCM in the same individual patient or family [92]. Whether LVNC is a distinct cardiac entity is an ongoing controversy. The WHO [4] and the ESC [7] lists LVNC as an unclassified CM, whereas the AHA classifies LVNC as a primary genetic CM [3].
Clinical evaluation: Cardiac non-invasive imaging by echocardiography remains the most commonly used diagnostic test for LVNC. Three diagnostic criteria have been proposed for LVNC. The criteria constitutes the detection of thickened myocardium, and a ratio of a two-layered myocardial structure composed of thin compacted epicardial layer and thick non-compacted endocardial layer, or trabecular meshwork with deep endomyocardial spaces, in the absence of coexisting cardiac abnormalities. The ratio of non-compacted to compacted layers is ≥ 2.0 measured at the end of systole [96-99].
Genetic aetiology: The two dominant transmission patterns for AVC in childhood are autosomal dominant (70%) and X-linked (30%) [100-102]. Autosomal recessive and mitochondrial patterns of inheritance also occurs [100]. In LVNC due to congenital heart disease (CHD), the congenital cardiac defect is heterogeneous in families and the transmission pattern of the LVNC is autosomal dominant along with the CHD. The affected relatives may have no CHD at initial evaluation because cardiac defect includes minor forms of CHD such as small ventricular septal defects (VSD), atrial septal defect (ASD), and patent ductus arteriosus, which may have normalized or reduced penetrance, whereas others may have severe forms of CHD [100,103]. Since LVNC is a rare disorder, its genetic aetiology remained incompletely understood. However, available data suggests the disease results from variants in known DCM and HCM genes encoding sarcomeric proteins (ACTC1, MYH7, MYBPC3 and TNNT2) [104-106]; the Z-disk (LBD3) [107,108]; nuclear lamina (LMNA) [109]; and dystrophin-associated glycoprotein complex (DTNA) [110]. Notably, the NKX2.5 gene that encodes a cardiac specific transcription factor is involved in the aetiology of LVNC [111] but no supporting data on its pathogenic variants in individuals with isolated LVNC.
The 2011 Heart Rhythm Society of America and the European Heart Rhythm Association consensus statement on genetic testing for the channelopathies and cardiomyopathies gave the lowest grading for the role of genetic testing in LVNC patients [112]. The statement recommends only variant specific genetic testing for at-risk first-degree relatives following the identification of a pathogenic LVNC variant in the proband. Genetic testing is available only because all genes associated with LVNC are also involved in other CMs, for which testing has been widely adopted. Detection rates remain incompletely defined and currently restricted to isolated reports. Unpublished research by the Laboratory for Molecular Medicine diagnostic testing of a large referral population detected a clinically significant variant in 24% of 108 LVNC patients. Variants occurred in MYH7 (13.6%), MYBPC3 (4.0%), TNNI3 (2.0%), VCL (2.8%), TAZ (1.1%) and TNNT2 (1.0%) [35]. Splice variants in the MYH7 gene that are very rare in HCM and DCM, appear more prevalent in LVNC patients.
Restrictive cardiomyopathy
Clinical description: Restrictive CM is a heart muscle disorder characterized by impaired LV diastolic filling with rapid early filling and slow late filling but with normal or decreased diastolic volumes in one or both ventricles. It occurs secondary to pathological conditions that stiffen the myocardium by infiltration or fibrosis such as eosinophilic endomyocardial disease, hemochromatosis, amyloidosis, sarcoidosis, scleroderma, carcinoid, Gaucher’s disease, Fabry disease, glycogen storage disease, metastatic malignancies, anthracycline cardiotoxicity or radiation injury. These pathophysiological conditions may be local to the heart or systemic (affect multiple organs). Several infiltrative diseases are familial such as amyloidosis, hemochromatosis, Gaucher’s disease and glycogen storage disease [3-5]. In some individuals, RCM occurs without a precipitating condition, referred to as idiopathic RCM. This form of RCM generally does not exhibit a familial pre-disposition, but several small families have presented with the disease. The phenotypic expression in these families is variable – isolated RCM, RCM with AV conduction block and skeletal myopathy. Familial RCM expressed autosomal dominant and autosomal recessive patterns of inheritance [9-12].
Natural history: The Prognostication of RCM varies based on the causative agent or pathologic condition. However, a disproportional percentage experience progressive disease in the setting of congestive HF with a high incidence of premature mortality. Diuretic or vasodilator therapy may improve prognosis (improve symptoms) in RCM. Other specific therapies such as iron chelation and immunosuppression therapy may improve clinical outcomes in patients with hemochromatosis and primary amyloidosis respectively. Heart transplantation may be beneficial to selected individual with disease pathologies local to the heart [9,11].
Clinical evaluation: Typical presentation of RCM includes dyspnoea, fatigue, exercise intolerance associated with cardiac inability to increase cardiac output by tachycardia without further reducing ventricular filling. In severe cases, signs of tight HF may manifest including hepatomegaly, ascites and peripheral oedema. Some patients may exhibit elevated the jugular venous pressure with an inspiratory increase in height. Non-invasive cardiac imaging, particularly, transthoracic echocardiography may detect thickening of the LV and/or RV walls with variable effects on chamber diameter and contractile function. The cardinal feature of cardiac amyloidosis is the presence of sparkling echodense spots in the myocardial walls. Thickening of the mitral and aortic valve leaflets may occur. Transmitral interrogation reveals increased peak E-wave velocity, producing an increase in the E/A wave ratio and shortened E-wave deceleration time although these findings are not specific to RCM and may manifest in other cardiac disorders that increase LV diastolic stiffness such as HCM and CAD. Typical haemodynamic feature of RCM is the dip and plateau sign consisting of rapid decline in ventricular pressure at the onset of a diastole (dip), followed by a rapid rise to a plateau in early diastole (plateau) [9-11].
Genetic aetiology: Idiopathic RCM is exceedingly rare and its genetic aetiology is only beginning to be defined. Recent data suggests variants in sarcomere or Z-disk protein genes including TNNI3, TNNT2, MYH7, ACTC1, TPM1, MYL3, and MYL [85-88]. Missense variants in the desmin gene (DES) has also been described in several families with desmin-related myopathy, which may present with RCM [89]. Same to LVNC, the current Heart Rhythm Society European Heart Rhythm Association guidelines recommend variant-specific testing for at-risk relative after the identification of a pathogenic variant in the proband [112]. Data from the Laboratory for Molecular Medicine detected a clinically significant variant in 35% of 50% individual with reported RCM. Variants were present in TNNI3 (18%), MYH7 (14%) and MYBPC3 (2%) [35].
The diversity of the CM is a consequence of genetic, allelic, epigenetic, and environmental heterogeneity contribute to variable phenotypic expression. Molecular genetic familial studies have considerably improved the current clinical understanding of monogenic conditions and their polygenic counterparts.
Incomplete and age-related penetrance
Like other disorders with an autosomal dominant transmission pattern, inherited CMs express marked phenotypic variability even within families. Penetrance (the proportion of individuals with mutations with clinically detectable disease) increases with age but does not reach 100% In HCM disease, hypertrophy manifests in adolescence, while the age of onset in patients with sarcomeric DCM is bimodal, peaking at childhood and mid-adulthood [113]. DCM is progressive due to LMNA mutation [114]. AVC has a low-level penetrance, such that it is uncommon to find many people with clinically apparent disease.
Variable expressivity
Variable expressivity is the range of clinical signs and symptoms that can manifest in different individuals with the same genetic disorder. Earlier studies described severe form of each of the CMs. Recent studies reveal that most of the affected individuals have mild disease, sometime atypical disease; as a result, the proportion of familial disease is higher than originally indicated. In the general population, individuals with subtle features of inherited CMs are difficult to detect. Thus, population screening is generally ineffective. Instead, cascade screening (sequential identification of related family members guided by genetic testing) is key to diagnosis [115].
Genetic heterogeneity and allelic disorders
HCM and DCM can be allelic, each cause by specific missense mutation in the same genes encoding sarcomeric proteins. However, no reliable documentation of families are available, in which a single sarcomeric mutation has caused HCM in some members and DCM in others. However, other aspects of CM phenotype can vary within families suggesting the absence of a precise relationship between mutation and biophysical consequences. For instance, apical HCM mostly occurs in families affected primarily by typical HCM and in only a minority of cases does apical HCM have a consistent relationship with specific mutations such as ACTC1 [116].
Phenocopies
Phenocopies refer to similar disorders but with different causes. Distinguishing phenocopies is clinically relevant because although they have a similar cardiac structure, they may exhibit different inheritance patterns, natural histories or responses to therapy. Certain autosomal dominant CMs (such as those caused by PRKAG2 mutations) and X-linked CM (such as Fabry and Danon’s disease) share similar clinical features with sarcomeric HCM, although they are clinically distinct disorders [117-119]. Phenocopies may also inform our understanding of disease mechanisms. For instance, in HCM due to PRKAG2 mutations (often attributed to glycogen deposition), a simple bulk effect cannot explain the increase in cardiac mass. The glycogen probably initiated signalling mechanisms involved in sarcomeric HCM [120,121].
Genotype-phenotype correlations
In certain circumstances, knowledge of the gene underlying CMs can alter patient care. For instance, phenocopies of HCM with different inheritance patterns and natural histories, and the susceptibility to conduction disease of DCM patients due to LMNA mutations that is sufficient to warrant pacemaker insertion, the use of implantable cardioverter-defibrillator should be considered [122,123]. However, for most CMs, correlations between disease gene and the phenotype are of limited utility for management of care of individual patients because of substantial overlap between disease gene-groups, allelic heterogeneity and insufficient clinical data [124-126]. Long-term efforts are warranted to accumulate reliable evidence on genotype-phenotype correlations.
The clinical relevance of genetic mutations as aetiological agents of CM is becoming evident. Since CMs contribute to the high morbidity and mortality of HF, understanding the contribution of genetics in the pathogenesis of CM is vital to improve the management of CM and CM-associated HF. Genetic involvement in CMs exhibit a wide heterogeneity and complexity. Novel sequencing technologies, in particular, the next generation sequencing (NGS) have considerably improved the availability of molecular testing, the efficiency of genetic analyses and the affordability of genetic testing. This development has increased the accessibility of genetic testing and the use of NGS-based sequencing in routine clinical diagnostics. Nearly 100 disease-associated genes can cause CMs [9].
The knowledge of pathogenic mutations in CMs is vital to support genetic counselling, risk stratification and prognostication, therapy guidance, and thus, increased therapeutic effectiveness. Family cascade screening for known familial pathogenic mutation cam lead to early diagnosis in affected family members and prophylactic interventions instituted to avoid, or delay disease onset or progression. Understanding the cellular basis of genetic CMs may provide new insights into the molecular biology of impaired cardiac cell function. Increased understanding of the molecular and genetic pathophysiology of CM will improve the identification of novel therapeutic targets and lead to the development of novel and specific treatment options. The present meta-analysis explores the genetics of CMs based on findings of genetic tests reported in published clinical trials.
The search for studies was performed on PubMed (http://www.ncbi.nlm.nih.gov/pubmed) and Embase (http://www.elsevier.com/online-tools/Embase) to identify articles that consider or suspect CM to be a genetic disorder. Key search terms used were inherited cardiomyopathies, genetic cardiomyopathies, idiopathic cardiomyopathies or sarcomeric cardiomyopathies, and genetic testing or next-generation sequencing. The excluded articles were case reports, conference papers, review articles and editorials, or did not provide raw data on genetics for a pooled analysis. Data was presented as frequency and percentage (categorical data), mean and standard deviation (continuous data) and event rate and 95% confidence interval (dichotomous data). Inconsistency index (I2) estimated for heterogeneity across studies and p < 0.05 indicated statistical significance.
Findings
After subjecting articles yielded by the online search to the inclusion criteria, the final dataset consisted of 10 primary studies that evaluated genetic testing in individuals diagnosed with various morphological and structural forms of familial or sporadic CMs [8,127-135]. The publication dates of the 10 studies ranged from 2000 to 2017 conducted in different countries: the U.S. [8,135], France [127,129], German [128], Portugal [130], Finland [132], China [133], and Italy [134]. One study [131] was multinational involving cardiomyopathic patients from eight countries – Denmark, England, France, Germany, Italy, Netherlands, Spain and Sweden. Six studies enrolled unrelated patients with CM [127,129,130,132-134] and the remaining four included family members diagnosed with CM [8,128,131,135]. Half of the studies enrolled DCM patients [8,129,131-133], three enrolled HCM patients [127,130,135] and the remaining two enrolled HCM and DCM patients [128] and HCM, DCM and AVC patients [129]. The main genes studies in all studies were those encoding sarcomeric proteins and more recent studies [132-135] studied sarcomeric, Z-disk, desmosomal and cytoskeleton genes. Methods of genetic testing employed included oligonucleotide-selective sequencing [8,132], denaturing liquid chromatography/sequencing analysis [127], high resolution melting (HRM)/sequencing [129], and the rest used NGS [128,131-135].
In total, the 10 studies enrolled 1,328 CM patients. Nearly two-thirds were males (62.4%) and the mean age was 40.5%. In nine (9) studies [8,127-134] enrolling 1,248 patients, 600 of them tested positive for pathogenic mutations for various forms of CM translating into 46.2% (95% CI: 34.1 to 58.9: Figure 3). In three studies [127,129,130], 153 patients identified to have pathogenic genetic mutations had 128 different mutations. Of 112 patients with pathogenic mutations [127,129], there were 94 different mutations, out of which 43 were novel mutations. In four studies, [127,129-131] with 447 patients with proven pathogenic gene mutations, 94 patients had multiple gene mutations. The most prevalent cause of CM is mutations in the sarcomere proteins – MYBPC3 mutations in 95 of 344 patients, event rate (ER) 31.35 (95% CI: 10.2% to 64.6%) Figure 4 [127,130,132,133,135] followed by MYH7 mutations in 48 out of 364 patients (ER: 15.6%; 95% CI: 6.4% to 33.4%) Figure 5 [127,129,130,132,133,135]. Other common sarcomeric mutations were TNNT2 (ER: 9.9%; 95% CI: 4.1% to 22.2%) Figure 6 [127,129,130,132,135] and TNNI3 (ER: 5.3%; 95% CI: 2.8% to 9.6%) Figure 7 [127,129,130,135]. Four studies [132-135] using NGS tested for mutations in genes sarcomeric, cytoskeletal, desmosomal and Z-disk but the studies did not quantify the prevalence for each of the four mutation groups for comparison. However, sarcomeric mutations were the most common type of mutations for familial forms of HCM and DCM.
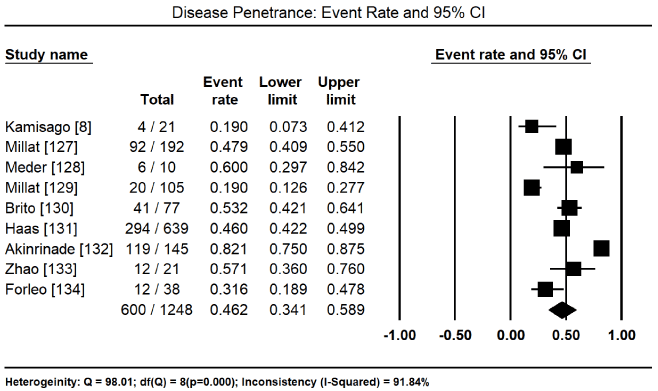
Figure 3. Event rate for disease penetrance and 95% CI
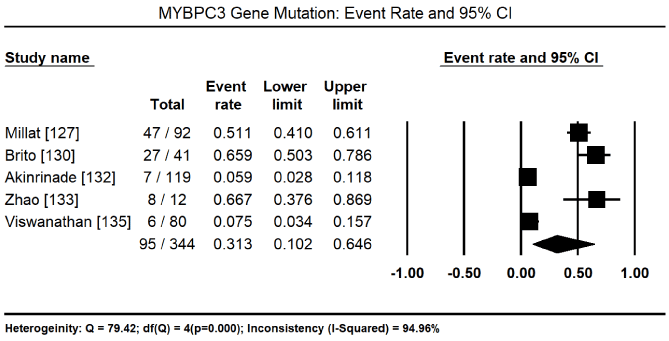
Figure 4. Event rate for MYBPC3 and 95% CI
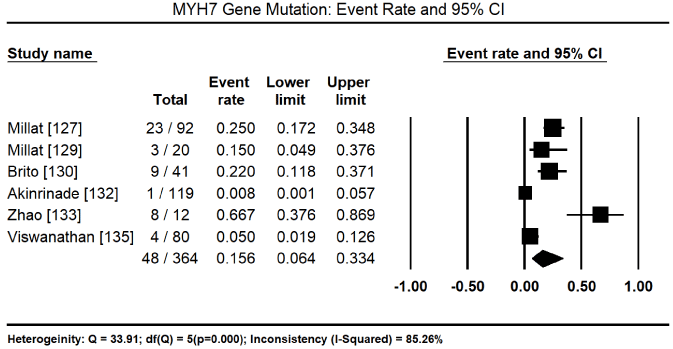
Figure 5. Event Rate for MYH7 and 95% CI

Figure 6. Event rate for TNNI3 and 95% CI
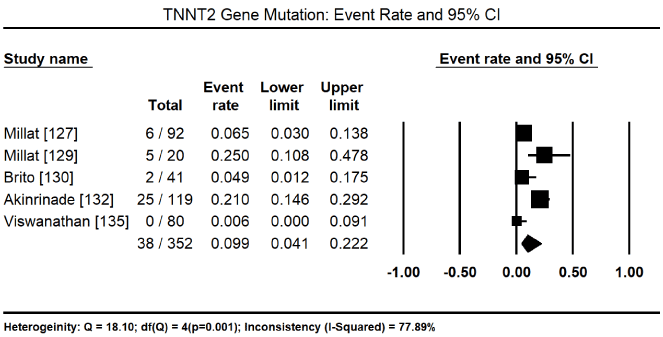
Figure 7. Event rate for TNNT2 and 95% CI
Genomics is emerging as a potential primary aetiology of Mendelian disorders. Genomic sequencing information promises to advance the current clinical knowledge of pathogenic mechanisms of hereditary diseases. A better understanding of disease manifestation is clinically relevant to support the paradigm shift in clinical practice from symptomatic to aetiologic-specific disease management. The traditional genetic testing method for mutations since the mid-1970s has been Sanger sequencing, but its slow speed, diminished accuracy and high cost significantly limited its use in routine diagnostics [136]. Four decades later (in 2005), the inception and continued development of NGS has enabled massive parallel sequencing and the generation of substantial amounts of data within a relatively shorter time. However, Sanger sequencing persists as the gold standard for validation in clinical sequencing experiments [137]. Still, NGS allows high throughput sequencing and transformed the field of molecular biology by its detection of many more mutations in addition to those identified by Sanger sequencing. The NGS advanced the understanding of molecular biology and genetics with expectation to provide personalized approach for specific diagnosis for the disease [137].
In the present findings, genetic mutations are a common cause of inherited forms of CMs, accounting for nearly a half of all report cases (46.2%). Pin particular, pathogenic mutations are more common in hereditary forms of HCM and DCM. However, because nearly all the studies included HCM or DCM patients, it may explain the low prevalence of other familial CMs such as RCM and AVC. Additional studies on genetic testing on RCM and AVC patients may clarify whether genetic mutations are the cause in a majority of these patients. Despite increased accuracy in the diagnosis of inherited CM, the use of genetic testing in the management of inherited CMs remains limited. The present findings suggest that many patients diagnosed with inherited CM have different mutations, some already identified and others are novel mutations as well as thousands of variants in the genetic mutations. In addition, about a fifth of CM patients have multiple mutations. Numerous mutations some novel and multiple mutations in CM patients limit the use of genetic testing to inform management in clinical practice. Sarcomeric mutations were the most common causes of inherited CMs although non-sarcomeric mutations such as cytoskeletal and desmosomal can also be frequent. In sarcomeric CM, MYBPC3 and MYH7 mutations are the two most common causes of HCM and DCM. TNNT2 and TNNI3 have also been described as causes of inherited sarcomeric CMs.
Utility of genetic testing
In HCM: Consistent with the present findings, previous studies support the clinical utility of genetic testing in patients diagnosed with familial forms of HCM, DCM, AVC and RCM. Genetic testing can diagnose about 60% to 70% of consecutive inherited HCM patient but the yield reduces with the inclusion of sporadic disease to ~30% [138]. Troponin T mutations (TNNT2 and TNNI3) may contributed to SCD in the absence of traditional risk factors and detection of these mutations may warrant early ICD therapy. In other mutations, SCD without the traditional risk factors is exceedingly rare limiting the utility of genetic testing in this cohort. Patients (such as those with Danon’s disease or PRKAG2 mutation) exhibiting special characteristics such as conduction abnormalities, pre-excitation or systemic disease may suggest phenocopies of sarcomere-related HCM and focused genetic testing may assist diagnosis and management [138].
Studies evaluating prognostication of mutational analysis have had inconsistent findings, but a family history of SCD remains a significant risk factor suggesting genetic background may prove useful in risk assessment [30,32,45,139]. A large cohort of unrelated HCM patients demonstrated a genetic diagnosis of any myofilament mutation has a four-times more likelihood of developing adverse outcomes including cardiac death, stroke and disease progression to advanced HF compared to genotype-negative patients [140]. However, the lack of specific changes in therapy for patients based on these findings, limits clinical utility of genetic testing in patient management. The possibility for prophylactic or curative medical therapy in pre-clinical genotype positive individuals remains unrealized, but early investigations in humans and animals show promise [141-143]. At present, clinical relevance of genetic testing in HCM patients is cascade screening to stratify family members at risk of disease development [115].
In DCM: Diagnostic yield of genetic testing in inherited DCM is low (~30%). Inherited DCM has a wide genetic heterogeneity and the majority of mutations have very low prevalence warranting the sequencing of large number of genes for effective genetic testing. A high prevalence of private mutations among individual family members and the need to assess individual for new pathogenic mutations complicates analysis of findings of genetic testing. DCM with conduction disease and/or arrhythmia is a special subset of Inherited DCM in which targeted testing for LMNA, desmosomal and SCN5A mutations may have considerable clinical impact. For genotype-positive relative with a strong family history of ventricular arrhythmias, heart block or SCD, early prophylactic ICD placement may be beneficial. Definitive detection of pathogenic mutation in the presence of clinical disease allow cascade family screening to identify at risk family members for follow-up in members testing negative and appropriate monitoring for those testing positive to prevent disease progression and adverse events. Already, there is promising evidence for prophylactic use of ACE-inhibitors in genotype-positive, phenotype-negative patients with Duchene’s muscular dystrophy for the prevention or delay of disease development, and the use of ACE-inhibitors in asymptomatic LV dysfunction [144,145].
In AVC: Several factors limit clinical utility of genetic testing for AVC patients. A desmosomal variant appears in about half of AVC patients who fulfil clinical diagnostic criteria but interpretation remains problematic. Many of these variants are single nucleotide changes occurring in up to 16% of healthy volunteers. Besides well-described founder populations, private mutations are prevalent and require individual determination of their pathogenicity, that is, sufficient to cause disease or to modify disease in about 10% to 15% of AVC cases who have more than one variant [138]. The presence of multiple variant mutations increases the risk of disease severity and a nearly five-fold increase in the risk of penetrant disease. This evidence demonstrate clinical significant of multiple variants in clinically significant AVC and the relevance of genetic testing of all five desmosomal genes when evaluating AVC proband and family [146]. The identification of pathogenic mutation enables detection of pre-clinical disease and the discharge of unaffected patients. In the case where clinical diagnosis of probands is definitive or certain, genetic testing is reasonable if cascade screening is feasible or desired. However, the evidence on genetic variants in ACM patients is insufficient to support diagnosis in borderline or clinical uncertain cases but clinical follow-up of the proband and family members is recommended [138].
In other CMs: Current classification systems sub-classify inherited RCM and LVNC as distinct myocardial entities but available evidence reveals considerable overlap between these CMs, and HCM and DCM. There is increased recognition of inherited RCM as a specific phenotype of HCM. Data shows that it can occur in patients having mutations expressed as classical HCM in other family members [147,148]. LVNC is also an imaging diagnosis with overlap with both DCM and HCM phenotypes and their pathogenic mutations [149]. However, the prevalence of pure inherited versus sporadic RCM and LVNC without HCM and/or DCM is unknown. The definition and clinical phenotype remains debatable, population prevalence varies widely depending on the patient population studies and the diagnostic criteria utilized as well as the clinical course of LVNC remains unclear, some reporting adverse events and other a relatively benign prognosis [150-153]. Genetic testing for LVNC is reserved for patients with syndromic manifestations and patients with clear familial disease. Due to overlap with DCM and HCM, active family assessment is important in evaluating these patients.
Familial RCM is the rarest of the primary myocardial disease and is increasingly recognized as familial disease associated with sarcomeric mutations [138]. The population prevalence of pure inherited RCM is unknown. Typical clinical manifestation include atrial enlargement with normal ventricles, a high burden of arrhythmias, progression to advanced HF, and HF or arrhythmia-associated death. Some RCM patient with troponin I mutations alter troponin I inhibition of actin-myosin ATPase resulting in elevated calcium sensitivity at the actin/myosin bridge leading to increased myocardial stiffness due to altered sarcomere response to calcium homeostasis. This mechanism is clinically relevant in distinguishing the development of RCM from that of HCM phenotypes [148,154]. Despite the low utility of genetic testing for inherited CMs in clinical practice, continued research and understanding of the pathogenic role of genetic mutations promises to widen the use of genetic testing in the diagnosis and clinical management of inherited CMs.
Increased recognition of the genetic basis of inherited diseases promises to create new ways to understand disease manifestation in humans. Inherited CMs are a genetically heterogeneous group of myocardial disorders. The understanding of the structure of sarcomere and intercalated disks, and mechanisms of myocardial contraction (actin-myosin interaction including troponin-tropomyosin complex and calcium ions) is essential to appreciate the genetics of CMs. The most common forms of inherited CMs are HCM and DCM, and the less prevalent ones are AVC, LVNC, and RCM. The most common type of mutation is sarcomeric mutations. The others are mutations affecting cytoskeletal, desmosomal and Z-disk proteins. The introduction of the next generation sequencing method revolutionized genetic testing and enabled the identification of a greater number of genetic mutations. However, a wide heterogeneity, multiple mutations, newer (previously unidentified) mutations, the effect of modifier genes and environmental effects, genotype-phenotype association and incomplete penetrance considerably limit diagnostic accuracy and therapeutic implications of genetic testing. At present, the only clinical relevance of genetic testing in inherited CMs is cascade screening to identify at risk family members to prevent or delay disease progression.
- Lee CS, Chien CV, Bidwell JT, Gelow JM, Denfeld QE, et al. (2014) Comorbidity profiles and inpatient outcomes during hospitalization for heart failure: An analysis of the U.S. Nationwide inpatient sample. BMC Cardiovasc Disord 14: 73. [Crossref]
- Shamszad P, Hall M, Rossano JW, Denfield SW, Knudson JD, et al. (2013) Characteristics and outcomes of heart failure-related intensive care unit admissions in children with cardiomyopathy. J Card Fail 19: 672-677. [Crossref]
- Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, et al. (2006) Contemporary definitions and classification of the cardiomyopathies: an American Heart Association scientific statement from the council on clinical cardiology, heart failure and transplantation committee; quality of care and outcomes research and functional genomics and translational biology interdisciplinary working groups; and council on epidemiology and prevention. Circulation 113: 1807-1816. [Crossref]
- Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, et al. (1996) Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation 93: 841-842. [Crossref]
- Bozkurt B, Colvin M, Cook J, Cooper LT, Deswal A, et al. (2016) Current diagnostic and treatment strategies for specific dilated cardiomyopathies: a scientific statement from the American Heart Association. Circulation 134: e579-e646. [Crossref]
- Pinto YM, Elliott PM, Arbustini E, Adler Y, Anastasakis A, et al. (2016) Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J 37: 1850-1858. [Crossref]
- Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, et al. (2007) Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 29: 270-276. [Crossref]
- Kamisago M, Sharma SD, DePalma SR, Solomon S, Sharma P, et al. (2000) Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med 343: 1688-1696. [Crossref]
- Towbin JA (2014) Inherited cardiomyopathies. Circ J 78: 2347-2356. [Crossref]
- Walker CA, Spinale FG (1999) The structure and function of the cardiac myocyte: a review of fundamental concepts. J Thorac Cardiovasc Surg 118: 375-382. [Crossref]
- Fatkin D, Graham RM (2002) Molecular mechanisms of inherited cardiomyopathies. Physiol Rev 82: 945-980. [Crossref]
- Pinnell J, Turner S, Howell S (2007) Cardiac muscle physiology. Continuing Education in Anaesthesia, Critical Care and Pain 7: 85-88.
- Bowditch HP (1871) Ueber die Eigenthuemlichkeiten der Reizbarkeit, welche die Muskelfasern des Herzens zeigen. Ber Sachs Ges (Akad) Wiss 23: 652-689.
- Janssen PM (2010) Myocardial contraction-relaxation coupling. Am J Physiol Heart Circ Physiol 299: H1741-H1749. [Crossref]
- Huxley AF (1957) Muscle structure and theories of contraction. Prog Biophys Biophys Chem 7: 255-318. [Crossref]
- Holmes KC (1997) The swinging lever-arm hypothesis of muscle contraction. Curr Biol 7: R112-R118. [Crossref]
- Spudich JA, Sivaramakrishnan S (2010) Myosin VI: an innovative motor that challenged the swinging lever arm hypothesis. Nat Rev Mol Cell Biol 11: 128. [Crossref]
- Holmes KC, Geeves MA (2000) The structural basis of muscle contraction. Philos Trans R Soc Lond B Biol Sci 355: 419-431. [Crossref]
- Rayment I, Holden HM, Whittaker M, Yohn CB, Lorenz M, et al. (1993) Structure of the actin-myosin complex and its implications for muscle contraction. Science 261: 58-65. [Crossref]
- Yanagida T, Esaki S, Iwane AH, Inoue Y, Ishijima A, et al. (2000) Single–motor mechanics and models of the myosin motor. Philos Trans R Soc Lond B Biol Sci 355: 441-447. [Crossref]
- Solaro JR, Van Eyk J (1996) Altered interactions among thin filament proteins modulate cardiac function. J Mol Cell Cardiol 28: 217-230. [Crossref]
- Poetter K, Jiang H, Hassanzadeh S, Master SR, Chang A, et al. (1996) Mutations in either the essential or regulatory light chains of myosin are associated with a rare myopathy in human heart and skeletal muscle. Nat Genet 13: 63-69. [Crossref]
- Sanbe A, Gulick J, Hayes E, Warshaw D, Osinska H, et al. (2000) Myosin light chain replacement in the heart. Am J Physiol Heart Circ Physiol 279: H1355-H1364. [Crossref]
- Gruen M, Gautel M. (1999) Mutations in the β-myosin S2 that cause familial hypertrophic cardiomyopathy abolish the interaction with the regulatory domain of myosin binding protein-C. J Mol Biol 286: 933-949. [Crossref]
- Gautel M, Zuffardi O, Freiburg A, And Labeit S (1995) Phosphorylation switches specific for the cardiac isoform of myosin binding protein-C: a modulator of cardiac contraction? EMBO J 14: 1952–1960. [Crossref]
- Weisberg A, Winegrad S (1998) Relation between crossbridge structure and actomyosin ATPase activity in rat heart. Circ Res 83: 60-72. [Crossref]
- Kunst G, Kress KR, Gruen M, Uttenweiler D, Gautel M, et al. (2000) Myosin binding protein C, a phosphorylation-dependent force regulator in muscle that controls the attachment of myosin heads by its interaction with myosin S2. Circ Res 86: 51–58. [Crossref]
- Bowles NE, Bowles KR, Towbin JA (2000) The “final common pathway” hypothesis and inherited cardiovascular disease: The role of cytoskeletal proteins in dilated cardiomyopathy. Herz 25: 168-175. [Crossref]
- Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, et al. (1995) Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA study. Circulation 92: 785-789. [Crossref]
- Niimura H, Bachinski LI, Sangwatanaroj S, Watkins H, Chudley AE., et al. (1998) Mutations in the gene for cardiac myosinbinding protein C and late-onset familial hypertrophic cardiomyopathy. N Engl J Med 338: 1248-1257. [Crossref]
- Moolman JC, Corfield VA, Posen B, Ngumbela K, Seidman CE, et al. (1997) Sudden death due to troponin T mutations. J Am Coll Cardiol 29: 549-555. [Crossref]
- Watkins H, Mckenna WJ, Thierfelder L, Suk HJ, Anan R, et al. (1995) Mutations in the genes for cardiac troponin T and α-tropomyosin in hypertrophic cardiomyopathy. N Engl J Med 332: 1058-1064. [Crossref]
- Krajinovic M, Pinamonti B, Sinagra G, Vatta M, Severini GM., et al. (1995) Linkage of familial dilated cardiomyopathy to chromosome 9. Am J Hum Genet 57: 846-852. [Crossref]
- Olson TM, Doan TP, Kishimoto NY, Whitby FG, Ackerman MJ, et al. (2000) Inherited and de novo mutations in the cardiac actin gene cause hypertrophic cardiomyopathy. J Mol Cell Cardiol 32: 1687-1694. [Crossref]
- Teekakirikul P, Kelly MA, Rehm HL, Lakdawala NK, Funke BH (2013) Inherited cardiomyopathies: molecular genetics and clinical genetic testing in the postgenomic era. J Mol Diagn 15: 158-170. [Crossref]
- Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, et al. (2001) HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies. Heart Rhythm 8: 1308-1339. [Crossref]
- Maron BJ, Maron MS (2013) Hypertrophic cardiomyopathy. Lancet 381: 242-255. [Crossref]
- Marston S, Copeland O, Jacques A, Livesey K, Tsang V, et al. (2009) Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ Res 105: 219-222. [Crossref]
- Purevjav E, Arimura T, Augustin S, Huby AC, Takagi K, Nunoda S, et al. (2012) Molecular basis for clinical heterogeneity in inherited cardiomyopathies due to myopalladin mutations. Hum Mol Genet 21: 2039-2053. [Crossref]
- Lopes LR, Zekavati A, Syrris P, Hubank M, Giambartolomei C, et al. (2013) Genetic complexity in hypertrophic cardiomyopathy revealed by high-throughput sequencing. J Med Genet 50: 228-239. [Crossref]
- Diegoli M, Grasso M, Favalli V, Serio A, Gambarin FI, et al. (2011) Diagnostic work-up and risk stratification in X-linked dilated cardiomyopathies caused by dystrophin defects. J Am Coll Cardiol 58: 925-934. [Crossref]
- Ho CY (2010) Genetics and clinical destiny: Improving care in hypertrophic cardiomyopathy. Circulation 122: 2430-2440. [Crossref]
- Bos JM, Will ML, Gersh BJ, Kruisselbrink TM, Ommen SR, et al. (2014) Characterization of a phenotype-based genetic test prediction score for unrelated patients with hypertrophic cardiomyopathy. Mayo Clin Proc 89: 727-737. [Crossref]
- Lopes LR, Rahman MS, Elliott PM (2013) A systematic review and meta-analysis of genotype-phenotype associations in patients with hypertrophic cardiomyopathy caused by sarcomeric protein mutations. Heart 2013; 99: 1800-1811. [Crossref]
- Bos JM, Towbin JA, Ackerman MJ (2009) Diagnostic, prognostic, and therapeutic implications of genetic testing for hypertrophic cardiomyopathy. J Am Coll Cardiol 54: 201-211. [Crossref]
- Theis JL, Bos JM, Bartleson VB, Will ML, Binder J, et al. (2006) Echocardiographic-determined septal morphology in Z-disc hypertrophic cardiomyopathy. Biochem Biophys Res Commun 351: 896- 902. [Crossref]
- Geske JB, Bos JM, Gersh BJ, Eidem BW, Ackerman MJ (2014) Deformation patterns in genotyped patients with hypertrophic cardiomyopathy. Eur Heart J Cardiovasc Imaging 15: 456-465. [Crossref]
- Dec GW And Fuster V (1994) Idiopathic dilated cardiomyopathy. N Engl J Med 331: 1564-1575. [Crossref]
- Towbin JA, Lowe AM, Colan SD, Sleeper LA, Orav EJ, et al. (2006) Incidence, causes and outcomes of dilated cardiomyopathy in children. JAMA 296: 1867-1876. [Crossref]
- Grunig E, Tasman JA, Kucherer H, Franz W, Kubler W, et al. (1998) Frequency and phenotypes of familial dilated cardiomyopathy. J Am Coll Cardiol 31: 186-194. [Crossref]
- Keeling PJ, Gang Y, Smith G, Seo H, Bent SE, et al. (1995) Familial dilated cardiomyopathy in the United Kingdom. Br Heart J 73: 417- 421. [Crossref]
- Michels VV, Moll PP, Miller FA, Tajik AJ, Chu JS, et al. (1992) The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N Engl J Med 326: 77-82. [Crossref]
- Mestroni L, Rocco C, Gregori D, Sinagra G, Di Lenarda A, et al. (1999) Familial dilated cardiomyopathy: evidence for genetic and phenotypic heterogeneity. Jam Coll Cardiol 34: 181-190. [Crossref]
- Cahill TJ, Ashrafian H, Watkins H (2013) Genetic cardiomyopathies causing heart failure. Circ Res 113: 660-675. [Crossref]
- Finsterer J, Stollberger C, Wahbi K (2013) Cardiomyopathy in neurological disorders. Cardiovasc Pathol 22: 389-400. [Crossref]
- Fatkin D, Members of the CSANZ Cardiac Genetic Diseases Council Writing Group (2011) Guidelines for the diagnosis and management of familial dilated cardiomyopathy. Heart Lung Circ 20: 691-693. [Crossref]
- Givertz MM, Mann DL (2013) Epidemiology and natural history of recovery of left ventricular function in recent onset dilated cardiomyopathies. Curr Heart Fail Rep 10: 321-330. [Crossref]
- Hershberger RE, Lindenfeld J, Mestroni L, Seidman CE, Taylor MR, et al. (2009) Heart Failure Society of America. Genetic evaluation of cardiomyopathy: A Heart Failure Society of America practice guideline. J Card Fail 15: 83-97. [Crossref]
- Towbin JA, Bowles NE (2002) The failing heart. Nature 415: 227-233. [Crossref]
- Towbin JA, Lowe AM, Colan SD, Sleeper LA, Orav EJ, Clunie S, et al. (2006) Incidence, causes and outcomes of dilated cardiomyopathy in children. JAMA 296: 1867-1876. [Crossref]
- Towbin JA, Hejtmancik JF, Brink P, Gelb B, Zhu XM, et al. (1993) X-linked dilated cardiomyopathy (XLCM): Molecular genetic evidence of linkage to the Duchenne muscular dystrophy gene at the Xp21 locus. Circulation 87: 1854-1865. [Crossref]
- Muntoni F, Cau M, Ganau A, Congiu R, Arvedi G, et al. (1993) Brief report: Deletion of the dystrophin muscle-specific promoter region associated with X-linked dilated cardiomyopathy. N Eng J Med 329: 921-925. [Crossref]
- Diegoli M, Grasso M, Favalli V, Serio A, Gambarin FI, Klersy C, et al. (2011) Diagnostic work-up and risk stratification in X-linked dilated cardiomyopathies caused by dystrophin defects. J Am Coll Cardiol 58: 925-934. [Crossref]
- Rahimov F, Kunkel LM (2013) The cell biology of disease: Cellular and molecular mechanisms underlying muscular dystrophy. J Cell Biol 201: 499-510. [Crossref]
- Constantin B (2014) Dystrophin complex functions as a scaffold for signalling proteins. Biochim Biophys Acta 1838: 635-642. [Crossref]
- Chelly J, Desguerre I (2013) Progressive muscular dystrophies. Handb Clin Neurol 113: 1343- 1366. [Crossref]
- Morales A, Hershberger RE (2013) Genetic evaluation of dilated cardiomyopathy. Curr Cardiol Rep 2013; 15: 375. [Crossref]
- Mestroni L, Taylor MR (2013) Genetics and genetic testing of dilated cardiomyopathy: A new perspective. Discov Med 15: 43-49. [Crossref]
- Debold EP, Schmitt JP, Patlak JB, Beck SE, Moore JR, et al. (2007) Hypertrophic and dilated cardiomyopathy mutations differentially affect the molecular force generation of mouse alpha-cardiac myosin in the laser trap assay. Am J Physiol Heart Circ Physiol 293: H284-H291. [Crossref]
- Ehler E, Perriard JC (2000) Cardiomyocyte cytoskeleton and myofibrillogenesis in healthy and diseased heart. Heart Fail Rev 5: 259-269. [Crossref]
- Basso C, Czarnowska E, Della Barbera M, Bauce B, Beffagna G, et al. (2006) Ultrastructural evidence of intercalated disc remodelling in arrhythmogenic right ventricular cardiomyopathy: An electron microscopy investigation on endomyocardial biopsies. Eur Heart J 27: 1847-1854. [Crossref]
- Yang Z, Bowles NE, Scherer SE, Taylor MD, Kearney DL, et al. (2006) Desmosomal dysfunction due to mutations in desmoplakin causes arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Res 99: 646-655. [Crossref]
- Protonotarios N, Tsatsopoulou A (2004) Naxos disease and Carvajal syndrome: Cardiocutaneous disorders that highlight the pathogenesis and broaden the spectrum of arrhythmogenic right ventricular cardiomyopathy. Cardiovasc Pathol 13: 185-194. [Crossref]
- Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, et al. (2010) Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed modification of the task force criteria. Circulation 121: 1533-1541. [Crossref]
- Ellinor PT, MacRae CA, Thierfelder L (2010) Arrhythmogenic right ventricular cardiomyopathy. Heart Fail Clin 6: 161-177. [Crossref]
- Corrado D, Basso C, Thiene G, Mckenna WJ, Davies MJ, et al. (1994) Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: a multicenter study. J Am Coll Cardiol 30: 1512-1520. [Crossref]
- Rampazzo A, Nava A, Danieli GA, Buja G, Daliento L, et al. (1994) The gene for arrhythmogenic right ventricular cardiomyopathy maps to chromosome 14q23– q24. Hum Mol Genet 3: 959-962. [Crossref]
- Thiene G, Basso C, Danieli G, Rampazzo A, Corrado D, et al. (1997) Arrhythmogenic right ventricular cardiomyopathy. Trends Cardiovasc Med 7: 84-90. [Crossref]
- Azaouagh A, Churzidse S, Konorza T, Erbel R (2011) Arrhythmogenic right ventricular cardiomyopathy/dysplasia: A review and update. Clin Res Cardiol 100: 383-394. [Crossref]
- Herren T, Gerber PA, Duru F (2009) Arrhythmogenic right ventricular cardiomyopathy/dysplasia: A not so rare “disease of the desmosome” with multiple clinical presentations. Clin Res Cardiol 98: 141-158. [Crossref]
- Lombardi R, Marian AJ (2011) Molecular genetics and pathogenesis of arrhythmogenic right ventricular cardiomyopathy: a disease of cardiac stem cells. Pediatr Cardiol 32: 360e365. [Crossref]
- Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, et al. (2010) Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J 31: 806e814. [Crossref]
- Hamid MS, Norman M, Quraishi A, Firoozi S, Thaman R, et al. (2002) Prospective evaluation of relatives for familial arrhythmogenic right ventricular cardiomyopathy/dysplasia reveals a need to broaden diagnostic criteria. J Am Coll Cardiol 40: 1445-1450. [Crossref]
- Te Rijdt WP, Jongbloed JD, de Boer RA, Thiene G, Basso C, et al. (2013) Clinical utility gene card for: Arrhythmogenic right ventricular cardiomyopathy (ARVC). Eur J Hum Genet 22. [Crossref]
- Xu T, Yang Z, Vatta M, Rampazzo A, Beffagna G, et al. (2010) Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol 55: 587-597. [Crossref]
- Bauce B, Nava A, Beffagna G, Basso C, Lorenzon A, Smaniotto G, et al. (2010) Multiple mutations in desmosomal proteins encoding genes in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart Rhythm 7: 22-29. [Crossref]
- Patel DM, Green KJ (2014) Desmosomes in the heart: A review of clinical and mechanistic analyses. Cell Commun Adhes 21: 109-128. [Crossref]
- Vatta M, Marcus F, Towbin JA (2007) Arrhythmogenic right ventricular cardiomyopathy: A ‘final common pathway’ that defines clinical phenotype. Eur Heart J 28: 529-530. [Crossref]
- Delmar M, McKenna WJ (2010) The cardiac desmosome and arrhythmogenic cardiomyopathies: From gene to disease. Circ Res 107: 700-714. [Crossref]
- Noorman M, Hakim S, Kessler E, Groeneweg JA, Cox MG, et al. (2013) Remodelling of the cardiac sodium channel, connexin43, and plakoglobin at the intercalated disk in patients with arrhythmogenic cardiomyopathy. Heart Rhythm 10: 412-419. [Crossref]
- Towbin JA, Lorts A (2011) Arrhythmias and dilated cardiomyopathy common pathogenetic pathways? J Am Coll Cardiol 57: 2169-2171. [Crossref]
- Oechslin E, Jenni R (2011) Left ventricular non-compaction revisited: a distinct phenotype with genetic heterogeneity? Eur Heart J 2011, 32: 1446-1456. [Crossref]
- Pignatelli RH, McMahon CJ, Dreyer WJ, Denfield SW, Price J, et al. (2003) Clinical characterization of left ventricular noncompaction in children: a relatively common form of cardiomyopathy. Circulation 108: 2672-2678. [Crossref]
- Ritter M, Oechslin E, Sutsch G, Attenhofer C, Schneider J, et al. (1997) Isolated noncompaction of the myocardium in adults. Mayo Clin Proc 72: 26-31. [Crossref]
- Weiford BC, Subbarao VD, Mulhern KM (2004) Non-compaction of the ventricular myocardium. Circulation 109: 2965-2971. [Crossref]
- Chin TK, Perloff JK, Williams RG, Jue K, Mohrmann R (1990) Isolated noncompaction of left ventricular myocardium. A study of eight cases. Circulation 82: 507-513. [Crossref]
- Jenni R, Oechslin E, Schneider J, Jost CA, Kaufmann PA (2001) Echocardiographic and pathoanatomical characteristics of isolated left ventricular non-compaction: a step towards classification as a distinct cardiomyopathy. Heart 86: 666-6671. [Crossref]
- Stollberger C, Finsterer J (2004) Left ventricular hypertrabeculation/noncompaction. J Am Soc Echocardiogr 17: 91-100. [Crossref]
- Stollberger C, Finsterer J, Blazek G (2002) Left ventricular hypertrabeculation or noncompaction and association with additional cardiac abnormalities and neuromuscular disorders. Am J Cardiol 90: 899-902. [Crossref]
- Towbin JA (2010) Left ventricular noncompaction: A new form of heart failure. Heart Fail Clin 6: 453-469. [Crossref]
- Ichida F, Hamamichi Y, Miyawaki T, Ono Y, Kamiya T, et al. (1999) Clinical features of isolated noncompaction of the ventricular myocardium: Long-term clinical course, hemodynamic properties, and genetic background. J Am Coll Cardiol 34: 233-240. [Crossref]
- Sasse-Klaassen S, Gerull B, Oechslin E, Jenni R, Thierfelder L (2003) Isolated noncompaction of the left ventricular myocardium in the adult is an autosomal dominant disorder in the majority of patients. Am J Med Genet A 119A: 162-167. [Crossref]
- Stahli BE, Gebhard C, Biaggi P, Klaassen S, Valsangiacomo Buechel E, et al. (2013) Left ventricular non-compaction: Prevalence in congenital heart disease. Int J Cardiol 167: 2477-2481. [Crossref]
- Finsterer J (2009) Cardiogenetics, neurogenetics, and pathogenetics of left ventricular hypertrabeculation/noncompaction. Pediatr Cardiol 30: 659-681. [Crossref]
- Hoedemaekers YM, Caliskan K, Majoor-Krakauer D, van de Laar I, Michels M, et al. (2007) Cardiac β-myosin heavy chain defects in two families with non-compaction cardiomyopathy: linking non-compaction to hypertrophic, restrictive, and dilated cardiomyopathies. Eur Heart J 28: 2732-2737. [Crossref]
- Klaassen S, Probst S, Oechslin E, Gerull B, Krings G, et al. (2008) Mutations in sarcomere protein genes in left ventricular noncompaction. Circulation 117: 2893-2901. [Crossref]
- Vatta M, Mohapatra B, Jimenez S, Sanchez X, Faulkner G, et al. (2003) Mutations in Cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non-compaction. J Am Coll Cardiol 42: 2014-2027. [Crossref]
- Xing Y, Ichida F, Matsuoka T, Isobe T, Ikemoto Y, Higaki T, et al. (2006) Genetic analysis in patients with left ventricular noncompaction and evidence for genetic heterogeneity. Mol Genet Metab 88: 71-77. [Crossref]
- Hermida-Prieto M, Monserrat L, Castro-Beiras A, Laredo R, Soler R, et al. (2004) Familial dilated cardiomyopathy and isolated left ventricular noncompaction associated with lamin A/C gene mutations. Am J Cardiol 94: 50-54. [Crossref]
- Ichida F, Tsubata S, Bowles KR, Haneda N, Uese K, et al. (2001) Novel gene mutations in patients with left ventricular non-compaction or Barth syndrome. Circulation 103: 1256-1263. [Crossref]
- Pauli RM, Scheib-Wixted S, Cripe L, Izumo S, Sekhon GS (1999) Ventricular noncompaction and distal chromosome 5q deletion. Am J Med Genet 85: 419-423. [Crossref]
- Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, et al. (2011) HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm 8: 1308-1339. [Crossref]
- Lakdawala NK, Dellefave L, Redwood CS (2010) Familial dilated cardiomyopathy caused by an alpha-tropomyosin mutation: the distinctive natural history of sarcomeric dilated cardiomyopathy. J Am Coll Cardiol 55: 320-329. [Crossref]
- Fatkin D, MacRae C, Sasaki T (1999) Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med 341: 1715-1724. [Crossref]
- Wordsworth S, Leal J, Blair E (2010) DNA testing for hypertrophic cardiomyopathy: a cost-effectiveness model. Eur Heart J 31: 926-935. [Crossref]
- Arad M, Penas-Lado M, Monserrat L (2005) Gene mutations in apical hypertrophic cardiomyopathy. Circulation 112: 2805-2811. [Crossref]
- Blair E, Redwood C, Ashrafian H (2001) Mutations in the gamma (2) subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: evidence for the central role of energy compromise in disease pathogenesis. Hum Mol Genet 10: 1215-1220. [Crossref]
- Maron BJ, Roberts WC, Arad M (2009) Clinical outcome and phenotypic expression in LAMP2 cardiomyopathy. JAMA 301: 1253-1259. [Crossref]
- Monserrat L, Gimeno-Blanes JR, Marin F (2007) Prevalence of Fabry disease in a cohort of 508 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 50: 2399-2403. [Crossref]
- Banerjee SK, McGaff in KR, Huang XN, Ahmad F (2010) Activation of cardiac hypertrophic signalling pathways in a transgenic mouse with the human PRKAG2 Thr400Asn mutation. Biochim Biophys Acta 1802: 284-291. [Crossref]
- Watkins H, Ashraf ian H, McKenna WJ (2008) The genetics of hypertrophic cardiomyopathy: Teare redux. Heart 94: 1264-1268. [Crossref]
- Pasotti M, Klersy C, Pilotto A (2008) Long-term outcome and risk stratification in dilated cardiolaminopathies. J Am Coll Cardiol 52: 1250-1260. [Crossref]
- Meune C, Van Berlo JH, Anselme F, Bonne G, Pinto YM, Duboc D (2006) Primary prevention of sudden death in patients with lamin A/C gene mutations. N Engl J Med 354: 209-210. [Crossref]
- Watkins H, McKenna WJ, Thierfelder L (1995) Mutations in the genes for cardiactroponin T and alpha-tropomyosin in hypertrophic cardiomyopathy. N Engl J Med 332: 1058-1064. [Crossref]
- Niimura H, Bachinski LL, Sangwatanaroj S (1998) Mutations in the gene for cardiac myosin-binding protein C and late onset familial hypertrophic cardiomyopathy. N Engl J Med 338: 1248-1257. [Crossref]
- Van Driest SL, Ellsworth EG, Ommen SR, Tajik AJ, Gersh BJ, Ackerman MJ (2003) Prevalence and spectrum of thin filament mutations in an outpatient referral population with hypertrophic cardiomyopathy. Circulation 108: 445-451. [Crossref]
- Millat G, Bouvagnet P, Chevalier P, Dauphin C, Jouk PS, et al. (2010) Prevalence and spectrum of mutations in a cohort of 192 unrelated patients with hypertrophic cardiomyopathy. Eur J Med Genet 53: 261-267. [Crossref]
- Millat G, Bouvagnet P, Chevalier P, Sebbag L, Dulac A, et al. (2011) Clinical and mutational spectrum in a cohort of 105 unrelated patients with dilated cardiomyopathy. Eur J Med Gene 54: e570-575. [Crossref]
- Meder B, Haas J, Keller A, Heid C, Just S, et al. (2011) Targeted next-generation sequencing for the molecular genetic diagnostics of cardiomyopathies. Circ Cardiovasc Genet 4: 110-122. [Crossref]
- Brito D, Miltenberger-Miltenyi G, Pereira SV, Silva D, Diogo AN (2012) Sarcomeric hypertrophic cardiomyopathy: genetic profile in a Portuguese population. Revista Portuguesa de Cardiologia (English Edition) 31: 577-587. [Crossref]
- Haas J, Frese KS, Peil B, Kloos W, Keller A, et al. (2014) Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J 36: 1123-1135. [Crossref]
- Akinrinade O, Ollila L, Vattulainen S, Tallila J, Gentile M, et al. (2015) Genetics and genotype–phenotype correlations in Finnish patients with dilated cardiomyopathy. Eur Heart J 36: 2327-2337. [Crossref]
- Zhao Y, Feng Y, Zhang YM, Ding XX, Song YZ, et al. (2015) Targeted next-generation sequencing of candidate genes reveals novel mutations in patients with dilated cardiomyopathy. Int J Mol Med 36: 1479-1486. [Crossref]
- Forleo C, D’Erchia AM, Sorrentino S, Manzari C, Chiara M, et al. (2017) Targeted next-generation sequencing detects novel gene–phenotype associations and expands the mutational spectrum in cardiomyopathies. PloS One 12: e0181842. [Crossref]
- Viswanathan SK, Sanders HK, McNamara JW, Jagadeesan A, Jahangir A, et al. (2017) Hypertrophic cardiomyopathy clinical phenotype is independent of gene mutation and mutation dosage. PloS One 12 : e0187948.[Crossref]
- Shendure J, Balasubramanian S, Church GM, Gilbert W, Rogers J, et al. (2017) DNA sequencing at 40: past, present and future. Nature 550: 345-353. [Crossref]
- Biswas A, Rao VR, Seth S, Maulik SK (2014) Next generation sequencing in cardiomyopathy: towards personalized genomics and medicine. Mol Biol Rep 41: 4881-4888. [Crossref]
- Jacoby D, McKenna WJ (2012) Genetics of inherited cardiomyopathy. Eur Heart J 33: 296-304. [Crossref]
- Van Driest SL, Ackerman MJ, Ommen SR, Shakur R, Will ML, et al. (2002) Prevalence and severity of ‘benign’ mutations in the beta-myosin heavy chain, cardiac troponin T, and alpha-tropomyosin genes in hypertrophic cardiomyopathy. Circulation 106: 3085-3090. [Crossref]
- Olivotto I, Girolami F, Ackerman MJ, Nistri S, Bos JM, et al. (2008) Myofilament protein gene mutation screening and outcome of patients with hypertrophic cardiomyopathy. Mayo Clin Proc 83: 630-638. [Crossref]
- Semsarian C, Ahmad I, Giewat M, Georgakopoulos D, Schmitt JP, et al. (2002) The L-type calcium channel inhibitor diltiazem prevents cardiomyopathy in a mouse model. J Clin Invest 109: 1013-1020. [Crossref]
- Abozguia K, Elliott P, McKenna W, Phan TT, Nallur-Shivu G, et al. (2010) Metabolic modulator perhexiline corrects energy deficiency and improves exercise capacity in symptomatic hypertrophic cardiomyopathy. Circulation 122: 1562-1569. [Crossref]
- Penicka M, Gregor P, Kerekes R, Marek D, Curila K, et al. (2009) The effects of candesartan on left ventricular hypertrophy and function in nonobstructive hypertrophic cardiomyopathy: a pilot, randomized study. J Mol Diagn 11: 35-41. [Crossref]
- Duboc D, Meune C, Lerebours G, Devaux JY, Vaksmann G, et al. (2005) Effect of perindopril on the onset and progression of left ventricular dysfunction in Duchenne muscular dystrophy. J Am Coll Cardiol 45: 855-857. [Crossref]
- Yusuf S, Pitt B, Davis CE, Hood WB, Cohn JN (1992) Effect of enalapril on mortality and the development of heart failure in asymptomatic patients with reduced left ventricular ejection fractions. N Engl J Med 327: 685-691. [Crossref]
- Quarta G, Muir A, Pantazis A, Syrris P, Gehmlich K, et al. (2011) Familial evaluation in arrhythmogenic right ventricular cardiomyopathy: impact of genetics and revised Task Force Criteria. Circulation 123: 2701-2709. [Crossref]
- Sen-Chowdhry S, Syrris P, McKenna WJ (2010) Genetics of restrictive cardiomyopathy. Heart Fail Clin 6: 179-186. [Crossref]
- Gomes AV (2005) Mutations in human cardiac troponin i that are associated with restrictive cardiomyopathy affect basal ATPase activity and the calcium sensitivity of force development. J Biol Chem 280: 30909-30915. [Crossref]
- Pantazis AA, Elliott PM (2009) Left ventricular non-compaction. Curr Opin Cardiol 24: 209-213. [Crossref]
- Kohli SK, Pantazis AA, Shah JS, Adeyemi B, Jackson G, et al. (2008) Diagnosis of left-ventricular non-compaction in patients with leftventricular systolic dysfunction: time for a reappraisal of diagnostic criteria? Eur Heart J 29: 89-95. [Crossref]
- Sandhu R, Finkelhor RS, Gunawardena DR, Bahler RC (2008) Prevalence and characteristics of left ventricular noncompaction in a community hospital cohort of patients with systolic dysfunction. Echocardiography 2: 8-12. [Crossref]
- Aras D, Tufekcioglu O, Ergun K, Ozeke O, Yildiz A, et al. (2006) Clinical features of isolated ventricular non-compaction in adults long-term clinical course, echocardiographic properties, and predictors of left ventricular failure. J Card Fail 12: 726-733. [Crossref]
- Oechslin EN, Attenhofer Jost CH, Rojas JR, Kaufmann PA, Jenni R (2000) Long-term follow-up of 34 adults with isolated left ventricular non-compaction: a distinct cardiomyopathy with poor prognosis. J Am Coll Cardiol 36: 493-500. [Crossref]
- Ammash NM, Seward JB, Bailey KR, Edwards WD, Tajik AJ (2000) Clinical profile and outcome of idiopathic restrictive cardiomyopathy. Circulation 101: 2490-2496. [Crossref]