Abstract
Infiltrative cardiomyopathy (ICM) refers to a heterogeneous group of myocardial disorders mainly cardiac amyloidosis, sarcoidosis, and hemochromatosis with characteristic deposition of abnormal substances within cellular and intercellular spaces, which leads to the development of ventricular diastolic dysfunction, systolic dysfunction or both. ICM results from a broader spectrum of inherited and acquired cardiac or systemics disorders. Despite being relatively rare, ICM is often misdiagnosed due to variable physiologic and morphologic characteristics. Early diagnosis of ICM can lead to potentially curative treatment. This paper reviews published studies on ICM, with a particular focus on its definitions, epidemiology, risk factors, clinical presentation, prognosis, etiopathogenesis, diagnosis, as well as clinical management. The ultimate aim of the review is to provide an in-depth understanding of ICM as well as improve its diagnostic and clinical management.
Introduction
Cardiomyopathies define abnormalities in the structure and functions of the ventricular myocardium in the absence of coronary artery diseases (CAD) or abnormal loading conditions [1-3]. The European Society of Cardiology (ESC) [4] classifies cardiomyopathies into primary or secondary myocardial disorders. Primary cardiomyopathies involves the heart as the only organ, while secondary cardiomyopathies manifests as a cardiac or systemic disorder [3]. The ESC’s classification system defines cardiomyopathies using specific morphological and functional criteria and then classifying them into genetic and non-genetic subtypes, regardless of the existence of extra-cardiac disease [3]. In this regard, ICM has been defined as a form of cardiac muscle disease where the ventricular walls become progressively and excessively rigid, disrupting ventricular filling [2]. It is characterized by diastolic dysfunction (a reduction in diastolic volume of the ventricles) and systolic dysfunction in some cases. ICM can be idiopathic or associated with other diseases such as fibrosis, endomyocardial, or amyloidosis, resulting to interstitial fibrosis [2].
Infiltrative cardiomyopathies, generally refers to a group of diseases, which are characterized by restrictive cardiac physiology [5]. Sarcoidosis, haemochromatosis, and cardiac amyloidosis are the three most prevalent forms of ICM [6]. Other forms of ICM include Fabry disease, Friedreich ataxia, Danon disease and Mucopoly saccharidoses [1].
Despite the preservation of EF, ICM patients exhibit severe limitations in exercise tolerance [5]. ICM are rare and thus often underdiagnosed [6]. In addition, their prevalence are incompletely documented and understood [2]. However, several clinical studies have been carried out to investigate the incidence and prevalence of various forms of ICM. Crooks [7] investigated 501 people with haemochromatosis and found the incidence of diagnosis of the condition increased by 2-fold between 1987 and 2002. Clinical evidence of heart-related sarcoidosis has been reported in at least 2-7% of patients diagnosed with sarcoidosis [8]. The prevalence of 2-7% have been reported in a number of studies [9-11]. Sarcoidosis can affect any part of the heart, with protein manifestations [8]. While most cardiac sarcoidosis-related deaths are because of arrhythmias or conduction defects, Lynch [8] contend that granulomatous infiltration of the myocardium can prove lethal. Antemortem recognition of cardiac sarcoidosis is rare due to high likelihood of sudden death as the presenting feature [12, 13]. Hamzeh [14] underscored the lack of a universally agreed diagnosis criteria of cardiac sarcoidosis. In their 2002 seminal paper, Deng [15] undertook an in-depth review of cardiac sarcoidosis, during which they discussed novel prevalence and epidemiological insights of cardiac sarcoidosis that have developed over the years.
Incidence of cardiac involvement has been reported in 19.5 to 28% of patients diagnosed with sarcoidosis in the U.S. and UK [12, 16, 17]. However, necropsy studies have reported a higher incidence of cardiac sarcoidosis (50-78%) among Japanese patients [18]. Cardiac involvement is the leading cause of death (accounting for 77-85% of total deaths) among patients diagnosed with sarcoidosis [19,20]. However, in the U.S., myocardial involvement is responsible for 13-50% of sarcoidosis-related deaths [12,21]. Cardiac sarcoidosis can manifest itself in various forms, including as a total heart block, congestive HF, pulmonary hypertension, ventricular arrhythmias, and ventricular aneurysms [22]. In Figure 1, cardiac amyloid is characterized by an increased LV wall thickness.
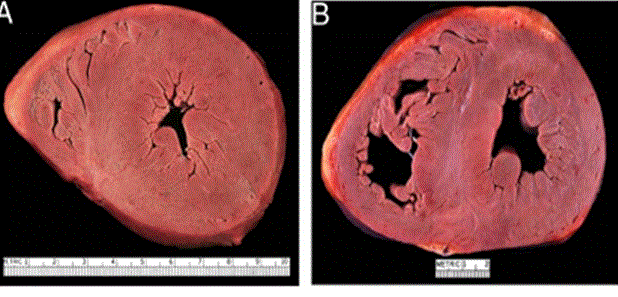
Figure 1: Cardiac Amyloidosis in ICM
Two Hearts Exhibiting Significantly Thickened LV Wall. (A) Amyloidosis (B) Hypertrophic Cardiomyopathy. Adopted from: Seward and Casaclang-Verzosa, (2010), p.1773, [2].
Given its underdeveloped diagnostic framework, the risk factors for the development of ICM is quite underreported. However, ICM is more associated with advanced age, female gender, and co-morbidities such as diabetes, hypertension, chronic kidney disease, and obesity [23,24]. ICM can result from a broader spectrum of both hereditary and acquired conditions with varying systemic manifestations [25]. Other forms of ICM like the Fabry disease caused by lack of lysosomal enzyme, α-galactosidase A, plays an essential role in breaking down neutral glycosphingolipids [26].
ICM presents itself with an occasional dynamic LV outflow, which appears in a similar manner to conditions with true myocyte hypertrophy such as hypertensive heart disease and hypertrophic cardiomyopathy [2]. In addition, some forms of ICM leads to increased wall thickness and small ventricular volume [2]. While some ICM lead to increased ventricular wall thickness, others result into chamber enlargement with secondary wall thinning [2].
There is no correlation between increased wall thickness and an increase in QRS complex amplitude. QRS complex amplitude may decrease with the increase in wall thickness is a phenomenon that is often witnessed with accumulation within the interstitium as opposed to within myocytes [2]. The development of certain forms of ICM such as cardiac amyloid and Friedreich ataxia could lead to the absence of increased voltage on ECG, notwithstanding the appearance of hypertrophy. [2]. However, research, as summarized in Table 1, has shown that low-voltage QRS complex phenomenon is not uniform across the various forms of ICM. For instance, the findings presented in Table 1 suggest that some forms of ICM such as Danon disease and Fabry disease are characterized by increased-voltage QRS complex, which is often evident in circumstances of increased size of cardiac myocytes, normal intraventricular conditions, as well as synchronous activation within the myocardium.
Table 1: Infiltrative Cardiomyopathies with Thick Ventricular Walls and Increased LV Mass.
|
Condition
|
Age at Presentation (Years)
|
History & Clinical Presentation
|
Echocardiography
|
ECG Profile
|
CMR LGE
|
Biopsy
|
Ref. #
|
|
Cardiac amyloid
|
> 30
|
HF symptoms, nephrotic syndrome, hepatomegaly, idiopathic peripheral neuropathy.
|
Symmetrical increase in wall thickness of the LV & RV, dilated RA & LA, decreased EF in advanced cases, granular appearance of myocardium.
|
Decreased/normal QRS complex voltage and pseudo-infarction in inferolateral leads.
|
Global, diffuse, pronounced in subendocardium (RV & LV walls)
|
Myocyte atrophy, normal cardiac tissue is replaced by amyloid.
|
[27-28]
|
|
Danon Disease
|
< 20
|
HF, skeletal myopathy, mental retardation.
|
A thicker LV (20-60 mm), thick RV in certain cases, decreased EF.
|
Increased to normal QRS complex voltage, short PR interval.
|
Sub-endocardial, lack of correspondence to perfusion territory.
|
Sarcoplasmic vacuolization, focal storage of PAS-positive material, myofibrillar disarray.
|
[30-32]
|
|
Cardiac oxalosis
|
> 20
|
Nephrocalcinosis and juvenile urolithiasis.
|
Symmetrical increase in wall thickness of LV & RV, patchy, normal EF, and echo dense speckled reflection.
|
Increased to normal QRS complex voltage, total heart block.
|
Increased myocardium attenuation on CT.
|
Intra and extracellular deposition of oxalate crystals without concomitant inflammation and necrosis.
|
[33-35]
|
|
Fabry disease
|
Male: 11+/- 7
Female: 23+/-16
|
Neuropathic pain, skin rashes, impaired sweating.
|
Symmetrical increase in wall thickness of LV and RV, and normal EF.
|
Increased to normal QRS complex voltage, short/prolonged PR interval.
|
Focal, midwall, inferolateral wall.
|
Enlarged myocytes with clusters of concentric glycolipid within lysosomes.
|
[26, 27, 36, 37]
|
|
Friedreich ataxia
|
25 (range 2-51)
|
Gait abnormality
|
Increased wall thickness in LV and posterior, normal EF, patchy, echo dense speckled.
|
Normal QRS complex voltage, ventricular tachycardia.
|
-
|
Not specific
|
[38-41]
|
|
Mucopoly saccharidoses
|
1-24 (10 is the median year)
|
Variable depending on condition subtype, delayed mental development, corneal clouding, coarse facial features, hepatosplenomegaly, skeletal deformities.
|
Asymmetrical septal hypertrophy, normal EF, mitral/aortic valve stenosis/insufficiency.
|
Increased/decreased QRS complex voltage, malignant arrhythmia.
|
-
|
Swollen myocytes with clear cytoplasm due to accumulation of muco-polysaccharides within lysosomes.
|
[42-45]
|
Adopted from: Seward and Casaclang-Verzosa, (2010), p.1771, [2]
Table 2: The AHA Diagnostic Strategies for ICM.
|
No.
|
AHA Recommendations for Cardiac Amyloidosis
|
-
|
In patients suspected with cardiac and systematic amyloidosis, detection of amyloid proteins in tissues such as abdominal fat pad, gingiva or rectum or affected organs such as heart, liver and kidney are recommended for amyloidosis diagnosis.
|
-
|
Upon detection of transthyretin (TTR) from biopsy specimen, DNA mutational analysis should distinguish between senile and hereditary amyloidosis.
|
-
|
After the confirmation of cardiac amyloidosis, cardiac imaging and laboratory examination should be considered to identify cardiac involvement.
|
-
|
Echocardiography or cardiac MRI is recommended to patients with suspected cardiac amyloidosis to determine cardiac restrictive filling and heart failure.
|
-
|
Endomyocardial diagnosis is recommended to diagnose cardiac amyloidosis in suspected patients when there is no evidence of non-cardiac tissue amyloidosis
|
-
|
Natriuretic peptide (BNP of NT-pro BNP) is recommended to detect pre-clinical cardiac involvement in patients diagnosed with amyloidosis.
|
-
|
ECG abnormalities such as low QRS voltage in the presence of ventricular thickening may suggest cardiac amyloidosis.
|
-
|
Nuclear imaging may assist in differentiating AL from TTR amyloidosis.
|
|
No.
|
AHA Recommendations for Cardiac Amyloidosis
|
-
|
Echocardiogram to assess LV function in patients with signs and symptoms of HF
|
-
|
Endomyocardial biopsies is useful to confirm cardiac sarcoidosis when pathology reveals non-caseating granulomas.
|
Infiltrative cardiomyopathy generally refers to a group of diseases that are characterized by restrictive cardiac physiology [5]. ICM is among the severest forms of preserved ejection fraction. There is no standard prognosis for ICM, as a condition, given that each form of ICM has its own prognosis. This section therefore reviews the prognosis for various forms of ICM [5].
Cardiac amyloid characteristic echocardiographic appearance involves increased thickening of the LV and right ventricular walls, normal LV cavity size, as well as a nonspecific granular appearance of the myocardium, as shown in Figure 2. The cardiac amyloid is also characterized by atrial enlargement, thickened papillary muscles, small to moderate pericardial effusion, as well as valve leaflets [2].
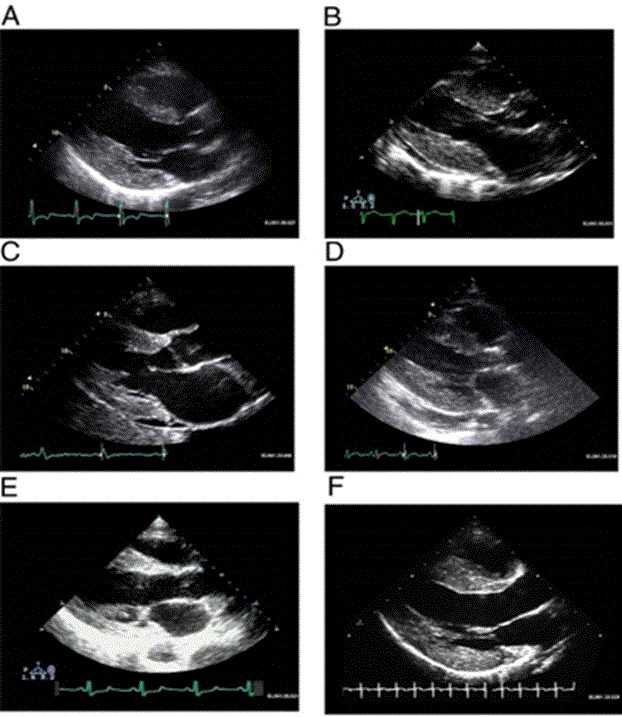
Figure 2: Increased LV Mass and Thickening of Ventricular Walls in Cardiac Amyloidosis
(A) Hypertrophic non-obstructive cardiomyopathy; (B) Hypertensive heart disease with secondary renal failure; (C) Cardiac amyloid; (D) Mucopolysaccharidosis; (E) Cardiac oxalosis; (F) Friedreich ataxia. Adopted From: Seward and Casaclang-Verzosa, (2010), p.1774, [2]
Kyle [46] study to establish long-term survival (≥ 10 years) among 30 patients diagnosed with primary cardiac amyloid found patients who remain untreated have a median survival of less than six months after the outset of heart failure. Whereas post chemotherapy stem cell transplantation yields impressive results [47], the mortality rate associated with transplantation ranges between 11% and 40% [48].
Cardiac involvement in Fabry disease, which is mostly associated with men in their thirties, may mimic the morphological and clinical features associated with HCM, including extremely thick walls. This condition accounts for 3% of cases that have been diagnosed as HCM [49]. A study conducted by Goldman [26] found that the thickening of the LV and LV mass increase as the patient’s age advances. Similarly, the severity of disease also increases with advancement in the patient’s age [26]. Progressive diastolic dysfunction is evident in patients diagnosed with Fabry disease as well as preservation of both EF and fractional shortening [26].
Danon disease mostly affect men in their teens and women in their twenties [2]. Affected men present with HF, mental retardation, and skeletal myopathy [31]. The affected women on the other hand present with affected cardiac myocytes [50]. Whereas cardiac symptoms become evident during adolescence, patients diagnosed with the disease are likely to die of HF in their thirties [31]. The echocardiographic characteristics associated with the Danon disease include LV wall thickness (20 mm to 60 mm) [30] or a significant increase in right ventricular wall thickness (≥ 10 mm) in the absence of pulmonary disease [31]. A severely impaired LV systolic dysfunction has also been reported among patients diagnosed with this condition [30, 31]. Danon disease may also present as sub-endocardial LGE [32] contrary to HCM in which LGE appears mid-epicardial and patchy [30]. Short PR interval and delta waves (Wolff-Parkinson-White syndrome) is a common feature in Danon disease, and consequently resulting into syncope in most patients diagnosed with the disease [31].
This condition results from expanded Guanine-Adenine-Adenine repeats (120-1,700 times, instead of the usual 8-22) in the frataxin gene on chromosome 9 [51]. The symptoms of the condition begin to show at the age of between 2 and 51 years [51]. However, the cardiac manifestations are likely to occur at the age of between 4 and 5 years, following the onset of the neurologic disorders [2]. Eventually, the development of cardiac disease is expected in 90% to 100% of the patients [39]. As shown in Figure 2, studies have revealed that cardiac involvement is characterized by an increase in wall thickening of the interventricular septum or posterior wall [38,39,52]. Severe HF might develop in some patients with mild LV dilation and systolic dysfunction [38]. Morvan [39] found that if present, valvular insufficiency is often mild in patients diagnosed with this condition. Non-cardiac dyspnea and frequent respiratory infections associated with Friedreich ataxia are because of severe scoliosis and neuromuscular impairment of respiratory muscles [38]. Leone [53] analyzed survival among Italian patients diagnosed with Friedreich’s disease found the 10-year, 20-year, and 30-year survival rates among were 96%, 80%, and 61% respectively. Inter-current pulmonary infection and cardiac dysfunction are the causes of death among patients diagnosed with Friedreich’s disease [38,42].
This condition is characterized by an enhanced generation of oxalic acid, caused by the deposition oxalate crystals in various organs, including the kidney and the heart [33, 54]. Palka Schulze [33] and Palka [34] identified biventricular symmetrically thickened wall (Figure 2), considered the echocardiographic characteristic of myocardial oxalosis. The early stages of this disease may be characterized by a normal EF [34]. However, mild biventricular dilation in advanced cases of the disease [34]. Computed tomography may demonstrate oxalate deposits as increased attenuation of the myocardium during the very late stages of the disease (Figure 3). Diastolic function in patients diagnosed with the condition maybe severely impaired, with restrictive filling and elevated filling pattern [33,34].
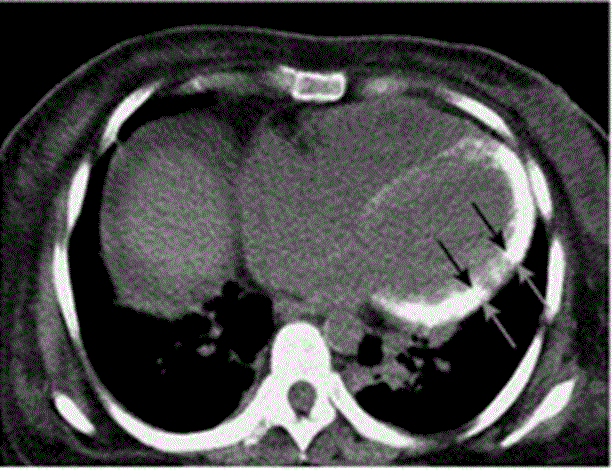
Figure 3: A Computed Tomographic Scan of an Unenhanced Chest.
Increased attenuation of myocardium is evident (arrow), compatible with advanced oxalate deposition. Adopted From: Seward and Casaclang-Verzosa, (2010), p.1775, [2].
This condition is a representation of inborn errors of metabolism due to deficiencies in lysosomal enzymes that are responsible for the breaking down of glycosaminoglycan [55]. The proper functioning of the cell is impaired by the accumulation of partially degraded mucopolysaccharides resulting into a range of clinical manifestations. These disorders are hereditary in nature [2]. The inheritance of the disorders occur in an autosomal recessive manner, and have an equal effect on both men and women [56]. Patients diagnosed with the condition may exhibit normal intellect or mental retardation, as well as experience developmental delay or severe behavioral problems [57]. Whereas the occurrence of cardiac disease is nearly 100% in all subtypes of Mucopolysaccharidosis, Type I Mucopolysaccharidosis (Hurler-Scheie syndrome) presented the most severe cardiac involvement [58]. Common findings associated with Mucopolysaccharidosis include significant valvular thickening and secondary pulmonary hypertension [42, 58]. Rigante [58] reports that asymmetrical septal hypertrophy precedes thickening of the valves. According to Rigante [58], the mitral and the aortic valves are more often involved, and hence producing insufficiency and/or stenosis. LV systolic function is normal in most cases [58]. However, ECG evidence of LV hypertrophy is uncommon [55, 58]. The characteristic small QRS complex voltages to poor conductance of glycosaminoglycan and rare intraventricular conduction delay or malignant arrhythmias are common among patients diagnosed with Mucopolysaccharidosis [58].
The etiopathogenesis for ICM is incompletely understood. However, a number of studies have reported etiologic or pathologic processes for the different etiopathogenic mechanisms of ICM. These mechanisms includes cardiac amyloidosis and cardiac sarcoidosis as discussed in the subsequent sections.
The extracellular deposition of amyloid fibrils within the heart result in abnormalities in three vital processes for the normal cardiac function namely: conduction, contractility, and coronary blood flow [1]. The thickening of the myocardial wall, and its consequent firm, rubbery, and noncompliant state is due to amyloid accumulation [59]. The thickening of the myocardial wall leads to a rise in intra-cardiac pressures, impending progressive biventricular diastolic dysfunction [60,61]. Cumulative myocyte damage and local fibrosis could lead to systolic dysfunction as the disease progresses [1]. However, systolic dysfunction is typically evident in extremely advanced disease [2]. Ridolfi [62] analyzed the conduction system in cardiac amyloid and established the involvement of the sinoatrial node, bundle branches, atrioventricular (AV) note could manifest at various degrees of heart block, and complex ventricular arrhythmias can be evident.
Sarcoidosis needs at least three key events to develop: exposure to antigen; acquired cellular immunity directed against the antigen mediated through antigen presenting cell and antigen specific T lymphocytes; as well as the appearance of immune effector cells necessary for promoting a more no-specific inflammatory response [22]. Granulomatous infiltration leading to the disruption of normal organ function is the major pathologic feature of sarcoidosis [1]. Figure 4 illustrates the pathogenesis of cardiac sarcoidosis.
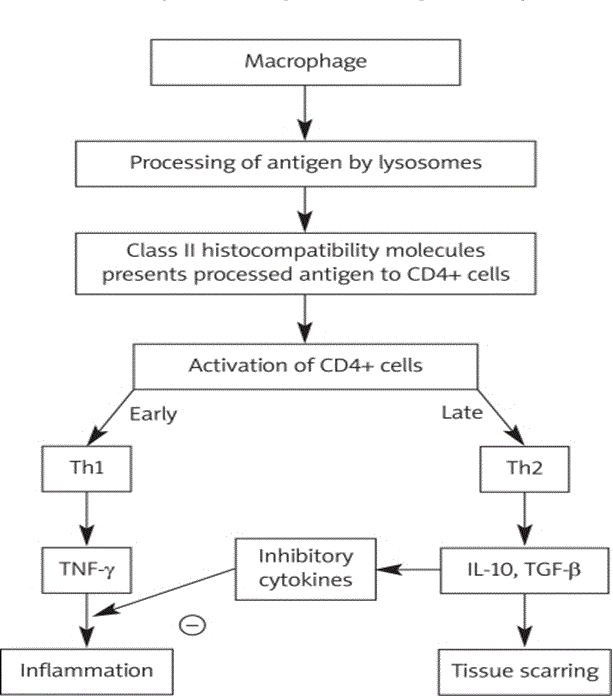
Figure 4: An Illustration of Pathogenesis of Cardiac Sarcoidosis.
Adopted From: Sekhri et al. (2011), p.547 [22].
In Figure 4, cytokine profile shifts to that of T helper type 2 response at a later stage of lesion evolution [22]. This shift exerts anti-inflammatory effects, and leads to tissue scarring. Cardiac sarcoidosis is characterized by three main successive stages namely: edema; granuloma formation; and fibrosis, which results in scar formation [22]. Sarcoid can involve any part of the heart, including the myocardium, endocardium, and pericardium [1]. However, Roberts [63] established that the disease’s involvement is typical of the LV free wall and the basal aspect of the interventricular septum. In Figure 5, Iannuzzi [9] illustrates the presence of CD4+ T cells, interacting with antigen-presenting cells to initiate the formation and maintenance of granulomas, is a cardinal feature of the sarcoidosis.
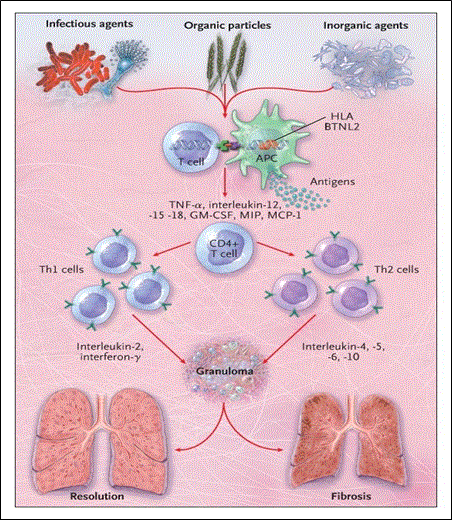
Figure 5: A Hypothesis for the Immunopathogenesis of Sarcoidosis.
Adopted From: Iannuzzi, Rybicki, and Teirstein (2007), p.2156 [9]
The CD4+ T cells interacting with antigen-presenting cells causes the myocardium to thicken and predominate leading to diastolic dysfunction in the initial stages of the disease [1]. However, in the later stages of the disease dilation of the ventricles, global or segmental hypokinesia, and predomination of systolic dysfunction occurs. The patchy nature of myocardial involvement can lead to uneven wall motion abnormalities that fail to match any given coronary distribution [64]. Despite the various genetic or acquired etiologies, the hallmark etiopathogenesis process of the ICM is the infiltration of the heart by abnormal substances [1]. Given the systemic nature of the underlying disease, ICM occurs in various age groups presenting as extra-cardiac clinical manifestations. Whereas conduction abnormalities and diastolic HF with restrictive physiology predominate the initial stages of the disease, the advanced stages of the disease on the other hand are characterized by systolic dysfunction.
The diagnosis for ICM requires a high degree of clinical suspicion [1]. The 2016 Scientific Statement from American Heart Association on current diagnostic and treatment strategies for specific dilated cardiomyopathies recommends the basis of ICM diagnosis is the detection of its two primary underlying infiltrative disorders – amyloidosis or sarcoidosis [64]. The most commonly adopted diagnostic methods for obtaining confirmatory evidence for ICM are cardiac imaging methods – endomyocardial biopsy, echocardiography, and CMR [1]. Other methods providing important clues for differential diagnosis include electrocardiography [66], tissue diagnosis [67-69], nuclear imaging [70, 71]
The AHA recommends the diagnosis of ICM require multiple diagnostic methods to detect cardiac amyloidosis and cardiac sarcoidosis as summarized in Table 4 [64].
Endomyocardial biopsy is the gold standard for the diagnosis cardiac amyloidosis [22]. Endomyocardial biopsy has a high diagnostic accuracy for cardiac sarcoidosis. However, an evaluation of endomyocardial biopsies by found that the sensitivity of endomyocardial biopsy for non-caseating granulomas was low (< 20%) [65]. The low sensitivity associated with endomyocardial biopsy is due to patchy myocardial involvement, which frequently involves the intraventricular septum and the LV [22]. Despite the low sensitivity, endomyocardial biopsy remains the gold standard for diagnosing cardiac sarcoidosis [1].
Echocardiography has been adopted for most forms of ICM, including cardiac amyloid [61, 72-76], cardiac sarcoidosis [77], Fabry disease [78], Danon disease [79], as well as Friedreich’s disease [39, 80]. Echocardiography is useful for the assessment of thickening of the LV wall, while Doppler echocardiography for the assessment of abnormal alterations in cardiac hemodynamics such as basal strain rate, peak early velocity, peak late velocity, and isovolumic relaxation time [72-76].
The use of CMR as a diagnostic method has for ICM has been investigated in various studies involving various forms of ICM such as cardiac amyloid [81], cardiac sarcoidosis [82, 83], Fabry disease [78], and Danon disease [84]. However, whereas the accuracy of cardiac MRI has not been established in assessment of myocardium in ICM patients and is not routinely employed for diagnosing the Friedreich’s disease Weidemann [85], the modality is helpful in determining the extent of cardiac involvement.
Differentiating various forms of ICM such as cardiac amyloidosis and Fabry disease from hypertrophic cardiomyopathy (HCM) is challenging since these condition share a number of clinical and hemodynamic features such as LV hypertrophy and diastolic dysfunction [75,78,118,119]. Accurate diagnosis is therefore critical when handing patients suspected of ICM [120]. A number of studies have attempted to differentiate various forms of ICM from HCM. Kubo [117], through a case control study comprising 46 consecutive patients with ICM (11 patients) or HCM (35 patients), sought to investigate the potential of high-sensitivity cardiac troponin T (hs-cTnT) in differentiating ICM from HCM. The study found that serum hs-cTnT was significantly higher in ICM patients than in their HCM counterparts (0.083 ± 0.057 ng/ml versus 0.027 ± 0.034 ng/ml, p < 0.001). The findings led Kubo [117] to conclude that serum hs-cTnT is an effective and accurate diagnostic indicator for differentiating ICM from HCM.
Baccouche [75] differentiated cardiac amyloidosis (CA) from HCM using 3-dimensional (3D) speckle tracking-derived functional parameters. Using a cohort of 12 patients with CA and another cohort of 12 patients with HCM, diagnosed using CMR, Baccouche [75] performed a 3D speckle tracking echocardiographic with wall motion analysis at radial strain (RS) longitudinal, and circumferential. The results indicated that 3D speckle-tracking function parameters were relatively lower in HCM patients and minimal in CA patients. In addition, basal RS was significantly reduced in CA patients (7.5 ± 19.7 vs. 22.3 ± 22.7; P < 0.0001) [75]. The stud conclude that the 3D speckle tracking echocardiography was a useful predictor for the accurate differentiation of CA from HCM. De Cobelli [78] performed a comparison of the myocardial location and distribution patterns of delayed enhancement between patients with Fabry disease and those with HCM. The study found that patients with Fabry disease exhibited LV delayed enhancement with a typical and consistently established pattern, characterized by the involvement of the inferolateral basal or mid basal segments and a mesocardial distribution, spreading to the subendocardium, and thus suggestive of Fabry disease.
The present meta-analysis sought to combine current research findings on studies investigating the diagnosis of ICM to advance the knowledge and understanding of diagnostic methods and features. Studies were included if they (a) recruited patients either prospectively and retrospectively; (b) investigated at least one type of ICM diagnostic method; (c) provided clinical outcomes of the diagnostic methods. The relevant studies were searched from online databases, both indexed and non-indexed, as well as from screening of bibliographies of included studies and review studies. Broad-based search terms were used including diagnosis” OR “diagnostic methods” AND “cardiac sarcoidosis” OR “Friedreich’s disease” OR “Fabry disease” OR “cardiac amyloidosis”. Titles and/or abstracts of the retrieved studies believed to satisfy the inclusion criteria were included for further review pending inclusion. Additional studies were obtained from screening bibliographies of the included studies and review articles. There was no restriction on publication date or language. Case reports (investigate few patients) or studies with data that was not readily extracted were excluded. Each included study was reviewed against the inclusion criteria, data extracted and recorded on Microsoft Excel Worksheet. The extracted data was name of the first author, period of study; type of ICM investigated; sample size of patients recruited; diagnostic method used; diagnostic feature targeted and a summary of the main findings. Table 3 summarizes the results of the meta-analysis of these different diagnostic methods for various forms of ICM.
Table 3: Summary of Data from Studies Investigating Diagnosis of ICM.
|
1st Author
[Ref. #]
|
Period of Study
|
Form of ICM
|
Sample Size
|
Diagnostic Method(s)
|
Diagnostic Feature
|
Summary of Main Findings
|
|
Murtagh [66]
|
2000-2005
|
Cardiac amyloidosis
|
127
|
Electrocardiography
|
systemic amyloidosis and biopsy-proved cardiac involvement
|
-Low-voltage QRS witnessed in 50% of cases of AL amyloidosis, suggestive of cardiac amyloidosis.
-A pseudoinfarct pattern (poor R-wave progression QS waves) in the precordial leads observed in 50% of cases, suggestive of cardiac amyloidosis.
|
|
Lee [73]
|
1995-2012
|
Cardiac amyloidosis
|
117
|
Echocardiography
|
Left ventricular wall thickness (LVWT)
|
-A third of patients diagnosed as having an LVWT of ≤ 12 mm.
-There is need for early detection using sensitive diagnostic methods even when LVWT is not increased.
|
|
Falk [74]
|
1982-1987
|
Cardiac amyloidosis
|
31
|
Echocardiography
|
Ventricular myocardium
|
-Increased myocardial echogenicity observed in 16 patients (sensitivity of 63%; specificity of 74% for diagnosis of amyloidosis).
-Increased atrial septal thickness observed in 60% of amyloid patients.
-Combination of increased myocardial echogenicity and increased atrial thickness was 60% sensitive and 100% specific for the diagnosis of amyloidosis.
|
|
Klein [61]
|
1987-1989
|
Cardiac amyloidosis
|
64
|
Doppler echocardiography
|
Left ventricular diastolic function.
|
-53 patients (with cardiac involvement) exhibited prominent abnormalities of left ventricular diastolic filling (LVWT: early > 12 mm but < 15 mm; advanced ≥ 15 mm).
-Relaxation appeared normal in early amyloidosis, with decreased peak early velocity (75 ± 20 versus 86 ± 16 cm/s; p < 0.01), increased late velocity (71 ± 22 versus 56 ± 13 cm/s; p < 0.01), decreased early to late velocity ratio (1.2 ± 0.6 versus 1.6 ± 0.5; p < 0.01) and prolonged isovolumic relaxation time (87 ± 15 versus 73 ± 13 ms; p < 0.01) compared with normal values.
|
|
Baccouche [75]
|
2007-2012
|
Cardiac amyloidosis
|
12
|
Echocardiography
|
Basal, midventricular, and apical.
|
-Patients with amyloidosis exhibited a significantly reduced basal radial strain (RS) (7.5 ± 19.7 vs. 22.3 ± 22.7; P < 0.0001).
|
|
Koyama [76]
|
2001-2002
|
Cardiac amyloidosis
|
119
|
Echocardiography
|
Tissue velocity, strain, and strain rate at the basal, mid, and apical LV multiple walls.
|
-70 patients exhibited cardiac involvement defined as the mean value of LVWT > 12 mm.
-32 patients died during a mean follow-up period of 285 ± 136 days.
-The mean basal strain, a measure of longitudinal LV function, proven an effective predictor of clinical outcome among patients with AL amyloidosis.
|
|
Syed [81]
|
2006-2007
|
Cardiac amyloidosis
|
120
|
CMR
|
Late gadolinium enhancement (LGE)
|
-Abnormal LGE found in 34 (97%) of the 35 patients with histologically verified cardiac amyloidosis (CA), and increased echocardiographic LVWT in 32 (91%) of the 35 patients.
-LGE is common in CA and detects interstitial expansion from amyloid deposition.
|
|
Perugini [70]
|
2004-2005
|
Cardiac amyloidosis
|
25
|
Radionuclide Imaging
|
Left ventricular mass and renal function.
|
- Heart and heart/whole-body tracer retention were significantly higher (p < 0.05) in patients with cardiac involvement of TTR-related.
- Cardiac 99mTc-DPD uptake was present in all patients with cardiac involvement of TTR-related.
-Through the use of genotyping/immunohistochemistry as the reference technique, the accuracy of 99mTc-DPDscintigraphy in distinguishing TTR-related and AL etiology proved to be 100%, and thus making 99mTc-DPDscintigraphy a useful step in the work-up of the differential diagnosis of TTR versus AL etiology in patients with documented CA.
|
|
Bokhari [71]
|
2005-2008
|
Cardiac amyloidosis
|
45
|
Radionuclide Imaging
|
Cardiac retention.
|
-Patients with TTR CA had a significantly higher semi-quantitative cardiac visual score than the AL cohort (2.9±0.06 vs. 0.8±0.27, p<0.0001) as well as a higher quantitative score (1.80±0.04 vs.1.21±0.04, p<0.0001).
- 99mTc-PYP cardiac imaging distinguishes AL from ATTR CA and may be a simple, widely available method for identifying subjects with ATTR CA.
|
|
Ardehali [67]
|
1982-1997
|
Cardiac amyloidosis
|
845
|
Tissue Diagnosis
|
Endomyocardial biopsy (EMBx)
|
-The final diagnosis differed from the initial clinical diagnosis in 264 (31%) of the 845 patients, with EMBx constitution 75% (evident in 196 patients) of the 264 patients.
EMBx establishes the final diagnosis in 75% of the 845 patients, with a high degree of specificity.
|
|
Burstow [77]
|
1983-1987
|
Cardiac sarcoidosis
|
88
|
Echocardiography
|
LV systolic dysfunction
|
-Echocardiographic evidence of LV systolic dysfunction recorded in 14% (in 12 patients) of the 88 patients, suggestive of cardiac sarcoidosis.
-The 12 patients exhibited a high incidence of congestive HF (9 of 12) and conduction system disease (7 of 12).
- The remaining 76 patients had normal echocardiograms or abnormalities attributed to non-sarcoid sources.
|
|
Smedema [82]
|
1998-2004
|
Cardiac sarcoidosis
|
58
|
CMR
|
Late gadolinium enhancement (LGE)
|
-Diagnosis of cardiac sarcoidosis (CS) established in 21% (in 12 patients) of the 58 patients.
-CMR revealed LGE, mostly involving basal and lateral segments (73%), in 19 patients.
-Scintigraphy appeared normal in 8 of the 19 patients, despite the presence of patchy LGE.
-CMR was characterized by a sensitivity and specificity of 100% (95% confidence interval, 78% to 100%) and 78% (95% confidence interval, 64% to 89%), thus making it a useful diagnostic tool for determining cardiac involvement in patients with CS.
|
|
Greulich [113]
|
2002-2011
|
Cardiac sarcoidosis
|
155
|
CMR
|
Presence of LGE
|
-The presence of LGE established in 25.5% (in 39 patients) of the 115 patients.
-The presence of LGE yielded a Cox hazard ratio (HR) of 31.6 for death, aborted sudden cardiac death, or appropriate ICD discharge, and of 33.9 for any event.
-The presence of myocardial scar indicated by LGE in patients with sarcoidosis provides the best independent predictor of potentially lethal events, as well as other adverse events, yielding a Cox HR of 31.6 and of 33.9, respectively.
|
|
Ohira [114]
|
2000-2006
|
Cardiac sarcoidosis
|
21
|
18F-fluoro-2-deoxyglucose positron emission tomography (18F-FDG PET) and magnetic resonance imaging (MRI)
|
Cardiac involvement
|
-Both 18F-FDG PET and MRI provided high sensitivity for diagnosing CS in patients with suspected cardiac involvement, but the 18F-FDG PET’s specificity was not as high as previously reported.
- The 18F-FDG PET appeared more sensitive but less specific than CMI, at may be appropriate in patients with contradictions to CMI or in circumstances where MRI is unavailable.
|
|
Namdar [115]
|
201-2010
|
Fabry Disease
|
30
|
Electrocardiography
|
Manifestation of cardiac involvement as left ventricular hypertrophy (LVH)
|
-Significantly shorter PQ-interval (131±18 vs 155±20 ms, p<0.000001) and the QRS width (83±11 vs 90±9 ms, p<0.05).
-Repolarization more pronounced in patients with FD (QTc dispersion: 66±32 vs 40±24 ms, p<0.001, Tpeak−Tend dispersion: 56±20 vs 37±16 ms, p<0.0005), and hence making electrocardiography a useful tool for early detection of the Fabry disease.
|
|
De Cobelli [78]
|
2002-2007
|
Fabry Disease
|
23
|
CMR
|
Myocardial location and distribution patterns of delayed enhancement
|
-Patients with Fabry disease – related hypertrophy exhibited LV delayed enhancement, with a typical and consistently found pattern characterized by the involvement of the inferolateral basal or mid basal segments and a mesocardial distribution that spared the subendocardium, and hence suggestive of the Fabry disease.
|
|
Morvan [39]
|
1989-1992
|
Friedreich's ataxia (FA)
|
10
|
Doppler-echocardiography
|
Cardiac involvement.
|
-Presence of FA-associated cardiomyopathy with left ventricular hypertrophy, suggestive of the presence of diastolic function abnormalities.
|
|
Delatycki [80]
|
1990-1996
|
Friedreich's ataxia
|
83
|
Echocardiography
|
Interventricular septal thickness, cardiac wall thickness.
|
-65% of patients exhibited increased interventricular septal thickness and cardiac wall thickness.
|
|
Maron [79]
|
2008-2009
|
Danon disease
|
7
|
Doppler-echocardiography
|
lysosome-associated membrane protein gene (LAMP2; Danon disease)
|
-Patients developed LV systolic dysfunction (mean [SD] ejection fraction, 25% [7%]) and cavity enlargement.
-Echocardiographic abnormalities such as marked LV hypertrophy, accompanied by outflow tract obstruction observed.
|
Nineteen (19) studies, 10 prospective and 9 retrospective meeting the inclusion criteria were included in the present meta-analysis. The 19 studies covered a period of thirty years, published between 1982 and 2012. The long period enables the assessment of the development and accuracy of diagnostic methods for ICM over time. The 19 studies recruited a combined population of 104 patients diagnosed with ICM or its main underlying cardiac etiologies. Cardiac amyloidosis was the most frequently investigated and diagnosed etiologic cause of ICM by 53% of the included studies [61,66,67,70,71,73,74,75,76,81] followed by cardiac sarcoidosis by 21% of the included studies [77,82,113,114]. The other less common cardiac conditions investigated were Fabry disease (FD) [78,115] and Friedreich ataxia (FA) [39,80] at 11% each, and finally Danon disease at 5% [79]. The most studied diagnostic method was echocardiography imaging studied by 47% followed cardiac MRI by 16%, electrocardiography by 11%, radionuclide imaging by 11%, and tissue diagnosis by 5% of the included studies.
Diagnosis of ICM depends on detection of the primary etiology. The most common cardiac condition leading to ICM is cardiac amyloidosis followed by cardiac sarcoidosis and Fabry disease. The mainstay of the diagnosis of cardiac amyloidosis was cardiac imaging by echocardiography, which could may be complemented by cardiac MRI and radionuclide imaging techniques. Echocardiographic imaging was used for characterizing LV dysfunction defined as increased LV wall thickness ≤ 12 mm and increased atrial septal thickness [61,73,76], and ventricular myocardium in infants defined as myocardial hyper-echogenicity [74]. Doppler Tissue echocardiography was also useful to detect changes in hemodynamics defined as significantly reduced basal strain rate, peak early velocity and increased late velocity and prolonged isovolumic relaxation time [61,75]. Cardiac MRI reveals abnormal LGE in cardiac amyloidosis as well as detects interstitial expansion due to amyloid deposition but its use was not common [81]. In the reviewed studies, cardiac LGE-MRI was the most common imaging method for diagnosis of cardiac sarcoidosis. Radionuclide imaging is an important imaging technique for detecting abnormal LV mass and renal function [70,71]. Non-imaging methods such as ECG abnormalities – low voltage QRS and pseudo-infarct pattern (poor R-wave progression, QS waves in precordial leads) suggests cardiac amyloidosis [66].
Infiltrative cardiomyopathies refer to a heterogeneous group of cardiac conditions characterized by restrictive cardiac physiology. As a result, the basis of diagnosis is the detection of the causative cardiac condition, mainly cardiac amyloidosis, cardiac sarcoidosis, Fabry disease and Friedreich's ataxia. Diagnosis of cardiac amyloidosis requires a combination of clinical examination and cardiac imaging. Echocardiography-defined increased LV wall thickness ≥ 2 mm and atrial dilatation may suggest cardiac amyloidosis. Cardiac LGE-MRI may be useful in the detection of the extent of cardiac involvement while radionuclide imaging, although insensitive, is useful in the differentiation of AL amyloidosis (immunoglobulin light chain amyloidosis) and transthyretin (TTR) amyloidosis. The present findings are consistent with AHA recommendation on the diagnosis of cardiac amyloidosis in patients suspected with infiltrative cardiomyopathies [64]. The AHA recommends echocardiography imaging-modality in the diagnosis of cardiac amyloidosis because of the uncertainty of the accuracy of cardiac LGE-MRI and low sensitivity of nuclear imaging. Echocardiographic features such as thickened ventricular walls with normal ventricular size, bi-atrial dilation, presence of pericardial effusion and valvular thickening without significant dysfunction. Although insensitive, myocardial hyper-echogenicity increases the suspicion of cardiac amyloidosis. ECG abnormalities – reduced QRS voltage despite thickened ventricular walls, pseudo-infarct pattern in pre-cordial leads, atrial fibrillation and atrioventricular conduction abnormalities suggest cardiac amyloidosis [64].
Cardiac sarcoidosis is the second most common cardiac condition, whose diagnosis suggests ICM. However, the diagnosis of cardiac sarcoidosis is challenging because non-caseating granuloma, which is characteristic pathological lesion, is not specific to sarcoidosis but is could result from various infectious and non-infectious etiologies [71,75,76]. Consistent with the present findings, the AHA guidelines recommend echocardiography as essential in the diagnosis of cardiac sarcoidosis by characterizing abnormal alterations in ventricular systolic and diastolic dysfunction, wall motion abnormalities, increased septal thickness and abnormal Doppler fillings. In addition, ECG abnormalities including bundle branch block (BBB), atrioventricular (AV) block, frequent premature ventricular complexes and pathological Q-waves and ST-T changes suggest cardiac sarcoidosis. If echocardiography is non-conclusive, the AHA guidelines recommend cardiac LGE-MRI as a valuable imaging method for the diagnosis of cardiac sarcoidosis. Early enhancement of in T2-weighted gadolinium images reveals fibrotic changes and scarring [64]. Finally, although other cardiac conditions such as Fabry disease and Friedreich ataxia and Danon disease may suggest diagnosis of ICM, these conditions are rare and not included in the AHA guidelines for clinical diagnosis of ICM [64].
Clinical management methods for ICM varies depending on the form of ICM. According to the AHA clinical guidelines, clinical management t should focus on treating the underlying cardiac etiologies.
Clinical management of cardiac amyloidosis targets treatment of HF caused by restrictive cardiomyopathy and therapy targeting the underlying protein disorder [1]. The use of loop diuretics helps in attaining euvolemia [1]. However, beta-blockers and angiotensin converting enzyme (ACE) inhibitors are typically avoided in the clinical management of cardiac amyloidosis [1]. Beta-blockers are avoided because cardiac output is largely dependent on heart rate in the setting of a fixed stroke volume while ACE-inhibitors increases the likelihood of neurohormonal blockade resulting in significant hypotension in the setting of the underlying automatic neuropathy [86, 87]. Device therapy – implantable cardioverter defibrillators (ICD) may be considered for preventing sudden cardiac death but its protective value has not been established in patients with cardiac amyloidosis [1].
For the treatment of AL amyloidosis, a melphalan-based cytotoxic chemotherapy regimen with or without autologous stem-cell transplantation is often recommended [88]. However, a recent study by Mikhael [89] reported that cyclophosphamide-bortezomib-dexamethasone (CyBorD) generates rapid and complete hematologic response in patients diagnosed with AL amyloidosis. Even though rare, cardiac transplantation has been found to be a viable clinical management option for patients with AL amyloidosis, with isolated cardiac disease that makes them inappropriate candidates for neoadjuvant chemotherapy [90]. Liver transplantation has been found to be a potential curative clinical management method for ATTR amyloidosis, particularly among patients without existing cardiac involvement [100]. Finally, A number of pharmacotherapies such as the nonsteroidal anti-inflammatory drugs (NSAIDs) diflunisal and its non-NSAID analog tafamidis [101, 102], as well as RNAs [103], antisense oligonucleotides [104], and green tea extracts [105] have been used for purposes of reducing, stabilizing, or silencing TTR activity or production.
Glucocorticoids are the mainstay in clinical management of cardiac sarcoidosis initiated promptly following diagnosis [1]. Due to paucity of randomized controlled trials confirming the efficacy of glucocorticoids, clinical management strategies involving glucocorticoids rely on observational studies [106, 107]. The ultimate duration of treatment is determined by clinical response and can be guided by monitored improvement in LGE on CMR [108]. For patients indicated for pacemaker and ICD, careful consideration is necessary due to the high incidence of SCD [1]. The existence of CHB or high-grade AV block is a demonstration of permanent pacemaker implantation, even in if the AV block reverses transiently [109]. Cardiac implantation is a potential option for managing cardiac sarcoidosis in young patients with end-stage, New York Heart Association (NYHA) Class IV HF, despite optimal medical therapy, and for patients diagnosed with intractable ventricular arrhythmias [1].
Enzyme replacement is the mainstay method of clinical management of the Fabry disease, which should be initiated prior to the development of tissue injury and fibrosis [110].
The performance of cardiac transplantation is a potentially lifesaving clinical management procedure for patients diagnosed with the Danon disease [111]. However, patients with the disease often die early in life [1].
The clinical management approach to the Friedreich’s disease is largely supportive, and limited to conventional HF medication, device implantation, and antiarrhythmic drugs [112].
The present meta-analysis combines findings from previous research articles on clinical management of ICM. The intention is to advance the knowledge and understanding on the most common clinical management approaches in use today and their clinical effectiveness.
Study selection
Studies investigating clinical management methods of ICM were searched on online electronic search engines - PubMed, EMBASE, Scopus and Google Scholar. Relevant medical subject headings (MESH) terms and a combination of key words to ensure no relevant study was left out. The key search words used were infiltrative cardiomyopathy” OR “Cardiac Amyloidosis” OR “Cardiac Sarcoidosis” AND “Clinical Management” OR “Treatment” OR “Implantation”, “Chemotherapy” OR “Medical Therapy”. The search was not limited to publication year, language or the age of the recruited patients. Study selection began with screening of titles and abstracts useful for excluding case studies (involving one or a few ICM patients) animal studies and conference papers (still subject to revision) and compared abstracts to the inclusion criteria. The inclusion criteria were the study (a) recruited patients diagnosed with ICM or one of the underlying cardiac conditions; (b) included at least one clinical management methods; and (c) reported outcomes on clinical management. After selection, relevant data was extracted from all the included studies. The extracted data was recorded in Microsoft Excel Worksheet. The extracted data included first author, study period, sample, mean age, type of ICM, treatment, follow-up period, and the main outcomes. Table 4 provides a summary of data extracted from the included studies.
Table 4: Summary of Results of Studies on Clinical Management of ICM.
|
1st Author
[Ref #]
|
Study Period
|
Sample Size
|
Mean Age (Yrs.)
|
ICM Form
|
Treatment
|
Follow-Up Period (Months)
|
Outcome(s)
|
|
Varr [116]
|
2009-2012
|
31
|
68
|
Cardiac amyloidosis
|
Implantable cardioverter-defibrillator (ICD)
|
23
|
-ICD successful in most patients, with therapy resulting in the termination of the arrhythmia in 80% of the patients, and hence demonstrating its potential for primary prevention of sudden cardiac death (SCD).
|
|
Mikhael [89]
|
2008-2010
|
17
|
59
|
Confirmed and symptomatic AL
|
Cyclophosphamide, bortezomib, and dexamethasone (CyBorD)
|
21
|
-16 of 17 (94%) of patients responded to CyBorD, with 71% achieving complete hematologic response and 24% a partial response.
- CyBorD produced rapid and complete hematologic responses in most of the patients.
|
|
Maurer, [90]
|
1997-2004
|
25
|
63
|
Systemic amyloidosis and HF
|
Cardiac transplantation followed by stem cell or liver transplantation.
|
6
|
-Patients who received cardiac transplantation followed by stem-cell or liver transplantation showed improved survival, and hence a novel therapeutic option for amyloid patients.
|
|
Dubrey [100]
|
1990-1995
|
11
|
63
|
Familial amyloidosis (FAP)
|
Liver transplantation
|
102
|
-5 patients showed progression of LVWT, with increased LV mass after liver transplantation. Three of the five patients exhibited reduction in electrocardiographic voltage despite abolition of the mutant protein from the serum.
Despite 9 of the 11 patients showing symptomatic improvement in neurologic symptoms, 1 patient developed HF and a second patient suffered SCD.
|
|
Berk [101]
|
2006-2012
|
130
|
59.7
|
Familial amyloid polyneuropathy
|
Pharmacotherapy (diflunisal)
|
24
|
-Reduced rate of progression of neurologic endpoints observed.
|
|
Coelho [102]
|
2010-2012
|
125
|
61
|
Transthyretin familial amyloid polyneuropathy (TTR-FAP).
|
Pharmacotherapy(Tafamidis treatment)
|
18
|
-A higher-than-anticipated liver transplantation dropout rate was evident.
-Reduced rate of progression of neurologic endpoints observed.
|
|
Kristen [105]
|
2011-2012
|
9
|
65
|
amyloidosis transthyretin (ATTR) 7 of 9 cardiomyopathy
|
Green tea (GT) and/or green tea extracts (GTE)
|
12
|
-No increase of LVWT and LV myocardial mass was observed by echocardiography after 12 months of follow-up.
Observations suggestive of inhibitory effect of GT and/or GTE on the progression of cardiac amyloidosis.
|
|
Vignaux [108]
|
1999-2000
|
12
|
40
|
histologically proven sarcoidosis
|
Monitoring
|
12
|
-Monitoring the improvement in LGE on CMR guides clinical response, which determines the ultimate length before LV systolic function declines.
|
|
Echaniz-Laguna [111]
|
2001-2005
|
8
|
30
|
Danon disease
|
Heart transplantation
|
60
|
-1 patient successfully treated through heart transplantation, suggestive of the potential of heart transplantation in the treatment of Danon disease.
|
|
Total/ Average
|
|
368
|
57
|
|
|
31
|
|
After all the potentially relevant studies were screened against the inclusion criteria, nine (9) studies were included in this meta-analysis. The studies included six (6) retrospective and three (3) prospective studies. The nine studies recruited a combined population of 368 patients diagnosed with various ICM-related disorders with a mean age of 57 years followed up for a mean period of 31 months, range 6 to 102 months. A greater majority of the studies (78%) investigated cardiac amyloidosis, while the remaining studies (11% each) investigated treatment option for cardiac sarcoidosis and Danon Disease. The most common clinical management method was medical therapy assessed by a third of the studies (45%), followed by cardiac/liver transplantation (33%), ICD (11%) and clinical monitoring (11%).
Medical therapy was the most common treatment for cardiac amyloidosis. A combination of cyclophosphamide, bortezomib, and dexamethasone (CyBorD) had 94% positive response with a rapid and complete hematologic response in 71% of the patients and a partial response in 24% of the patients [89]. Nonsteroidal anti-inflammatory drug (NSAID) – diflunisal – reduced the rate of progression [101]. Tafamidis, a specific transthyretin (TTR) stabilizer or dissociation inhibitor, was affective in the treatment of TTR amyloidosis. The drug preserved LV wall thickness and LV myocardial mass after 12 months [102]. Green tea extracts (GTE) was also effective in inhibiting the progression of cardiac TTR amyloidosis [105]. Besides medication, cardiac or liver transplantation was a common clinical management approach in ICM patients. In patients with systemic amyloidosis and heart failure, heart transplantation followed by liver transplantation significantly improve survival [90]. Liver transplantation alone is less effective in cardiac amyloidosis since about half of patients with cardiac amyloidosis exhibit progression of LV wall thickness. Cardiac transplantation also showed great improvement in clinical outcomes of patients with Danon Disease [111]. For patients with cardiac sarcoidosis, routine monitoring reveal improvement in LV systolic function imaged using cardiac LGE-MRI [108].
Discussion
The principal target of clinical management approaches for patients diagnosed with ICM is treating the primary underlying etiology. In the present findings, clinical management of ICM focused on treating cardiac amyloidosis, sarcoidosis and Danon disease, which represent the main primary etiologies of ICM. The target of therapy was relieving restrictive filling (reducing LV wall thickness), positive hematologic response and the treatment of heart failure. The primary therapy was medication to achieve hematologic response and reverse LV wall thickening. Cardiac transplantation followed by or liver transplantation conveyed a protective effect improving survival in patients with systemic amyloidosis and symptomatic heart failure. Consistent with these findings, the current recommendations by AHA propose medication, cardiac transplantation and liver transplantation as effective therapy for TTR amyloidosis without significant cardiac involvement. However, the recommendations caution that conventional HF medication such as ACE-inhibitors and beta-blockers should be avoided because of the risk or neurohormonal blockade and preservation of heart rate [64]. Although the present analysis finds ICD is important in protecting against sudden cardiac death, the AHA recommendations suggests its clinical value on preventing sudden cardiac death has not been conclusively demonstrated [64].
The treatment of cardiac sarcoidosis-induced ICM has limited clinical trials. Whereas the present findings suggest monitoring of cardiac function by cardiac LGE-MRI to predict subsequent risk of developing systolic dysfunction, the AHA guidelines recommend corticosteroids are the mainstay of cardiac sarcoidosis therapy in symptomatic patients. However, randomized clinical trials on the efficacy of corticosteroids are lacking but retrospective studies suggest a clinically beneficial effect in improving LV function. Furthermore, treatment of cardiac sarcoidosis in asymptomatic or minimally symptomatic patients remains controversial [64]. Standard HF therapy should be considered for patients with cardiac sarcoidosis and significantly reduced LV ejection fraction [106, 107]. Finally, clinical guidance for under underlying etiologies of ICM such as Fabry Disease and Danon Disease are lacking, with management varying in between studies. In summary, the mainstay clinical management of ICM is the identification and treatment of the underlying etiologies, principally, cardiac amyloidosis and sarcoidosis. The primary treatment is medical therapy. Additional therapies are cardiac and/or liver transplantation for cardiac amyloidosis, and corticosteroids and conventional HF treatment for cardiac sarcoidosis.
Infiltrative cardiomyopathy define a heterogeneous group of myocardial disorders characterized by the deposition of abnormal substances within the intercellular spaces leading to diastolic and/or systolic dysfunction. The cardiac condition manifests as increased rigidity of ventricular walls disrupting ventricular filling. It can be idiopathic or secondary to cardiac or systemic disorders. It is a rare form of cardiomyopathy with a prevalence of 2 to 7% of reported cardiomyopathies. Risk factors include advanced age, female gender and the presence of diabetes, hypertension, chronic kidney disease, and obesity. Clinically, ICM mainly manifests as cardiac amyloidosis and cardiac sarcoidosis and in fewer cases, Fabry disease, Danon Disease and Friedreich's ataxia. Prognosis of ICM is challenging since it varies considerably based on the primary underlying etiology. The etiopathogenesis of ICM remains incompletely understood but cardiac amyloidosis and sarcoidosis have been implicated as the key etiopathogenic factors. The heterogeneity of underlying etiologies complicates diagnosis of ICM. Although endomyocardial biopsy is the gold standard for diagnosis of cardiac amyloidosis, echocardiography is the most common imaging method for diagnosis. Cardiac LGE-MRI is valuable in the diagnosis of cardiac sarcoidosis. The two cardiac imaging methods diagnose ICM by characterizing increasing LV wall thickness and restrictive ventricular filling. Finally, clinical management approaches target to relieve hemodynamics (reduce LV wall thickness), achieve complete hematologic response and relieve symptoms of heart failure. The common treatment options include medical therapy, implantable cardioverter defibrillator and cardiac/liver transplantation.
References
- Bejar D, Colombo PC, Latif F, Yuzefpolskaya M (2015) Infiltrative cardiomyopathies. Clinical Medicine Insights: Cardiology 9: 29-38.
- Seward JB, Casaclang VG (2010) Infiltrative cardiomyopathy diseases –Cardiomyopathies that look alike. Journal of the American College of Cardiology 55: 1769-1779.
- Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, et al. (2008) Classification of the cardiomyopathies: A position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. European Heart Journal 29: 210-276.
- European Society of Cardiology (2014) 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy. European Heart Journal 35: 2733-2779.
- Smart FW (2015) Unusual cardiomyopathies: Some may be more usual than previously thought and simply underdiagnosed. Cardiovascular Innovations and Applications 1: 29-36.
- Moinuddin MJ, Figueredo V, Amanullah AM (2010) Infiltrative diseases of the heart. Rev Cardiovasc Med 11: 218-227. [Crossref]
- Crooks CJ, West J, Solaymani DM (2009) The epidemiology of haemochromatosis: A population-based study. Alimentary Pharmacology & Therapeutics 29: 183-92.
- Lynch JP, Hwang J, Bradfield J, Fishbein M, Shivkumar K, et al. (2014) Cardiac involvement in sarcoidosis: Evolving concepts in diagnosis and treatment. Seminars in Respiratory and Critical Care Medicine 35: 372-390.
- Iannuzzi MC, Rybicki BA, Teirstein AS (2007) Sarcoidosis. N Engl J Med 357: 2153-2165. [Crossref]
- Ayyala US, Nair AP, Padilla ML (2008) Cardiac sarcoidosis. Clin Chest Med 29: 493-508. [Crossref]
- Baughman RP, Teirstein AS, Judson M (2001) Case Control Etiologic Study of Sarcoidosis (ACCESS) research group. Clinical characteristics of patients in a case control study of sarcoidosis. Am J Respir Crit Care Med 164: 1885-1889.
- Perry A, Vuitch F (1995) Causes of death in patients with sarcoidosis. A morphologic study of 38 autopsies with clinicopathologic correlations. Arch Pathol Lab Med 119: 167-172. [Crossref]
- Bagwan IN, Hooper LV, Sheppard MN (2011) Cardiac sarcoidosis and sudden death. The heart may look normal or mimic other cardiomyopathies. Virchows Arch 458: 671-678.
- Hamzeh NY, Wamboldt FS, Weinberger HD (2012) Management of cardiac sarcoidosis in the United States: a Delphi study. Chest 141: 154-162. [Crossref]
- Deng JC, Baughman RP, Lynch JP (2002) Cardiac involvement in sarcoidosis. Semin Respir Crit Care Med 23: 513-527. [Crossref]
- Fleming HA (1974) Sarcoid heart disease. Br Heart J 36: 54-68. [Crossref]
- Sharma OP, Maheshwari A, Thaker K (1993) Myocardial sarcoidosis. Chest 103: 253-258. [Crossref]
- Yazaki Y, Isobe M, Hiramitsu S (1998) Comparison of clinical features and prognosis of cardiac sarcoidosis and idiopathic dilated cardiomyopathy. Am J Cardiol 82: 537-540. [Crossref]
- Tachibana T, Ohmori F, Ueda E (1986) Clinical study on cardiac sarcoidosis. Ann N Y Acad Sci 465: 530-542. [Crossref]
- Matsui Y, Iwai K, Tachibana T, Fruie T, Shigematsu N, et al. (1976) Clinicopathological study of fatal myocardial sarcoidosis. Ann N Y Acad Sci 278: 455-469. [Crossref]
- Gideon NM, Mannino DM (1996) Sarcoidosis mortality in the United States 1979-1991: an analysis of multiple-cause mortality data. Am J Med 100: 423-427. [Crossref]
- Sekhri V, Sanal S, Delorenzo LJ, Aronow WS, Maguire GP (2011) Cardiac sarcoidosis: a comprehensive review. Arch Med Sci 7: 546-554. [Crossref]
- Mesquita ET, Jorge AJL, Junior CVS, de Andrade TR (2017) Cardiac amyloidosis and its new clinical phenotype: Heart failure with preserved ejection fraction. Review Article 1: 71-80.
- Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, et al. (2006) Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med 355: 251-259. [Crossref]
- Selvanayagam JB, Joseph M, Karamitsos T, Neubauer S (2015) Infiltrative cardiomyopathy. Oxford Medicine Online 1: 1-6.
- Goldman ME, Cantor R, Schwartz MF, Baker M, Desnick RJ (1986) Echocardiographic abnormalities and disease severity in Fabry's disease. J Am Coll Cardiol 7: 1157-1161. [Crossref]
- Vohringer M, Mahrholdt H, Yilmaz A, Sechtem U (2007) Significance of late gadolinium enhancement in cardiovascular magnetic resonance imaging (CMR). Herz 32: 129-37.
- Gertz MA, Lacy MQ, Dispenzieri A (1999) Amyloidosis: recognition, confirmation, prognosis, and therapy. Mayo Clin Proc 74: 490-494. [Crossref]
- Carroll JD, Gaasch WH, McAdam KP (1982) Amyloid cardiomyopathy: Characterization by a distinctive voltage/mass relation. Am J Cardiol 49: 9-13.
- Arad M, Maron BJ, Gorham JM, Johnson WH Jr, Saul JP, et al. (2005) Glycogen storage diseases presenting as hypertrophic cardiomyopathy. N Engl J Med 352: 362-372. [Crossref]
- Charron P, Villard E, Sébillon P, Laforêt P, Maisonobe T, et al. (2004) Danon's disease as a cause of hypertrophic cardiomyopathy: a systematic survey. Heart 90: 842-846. [Crossref]
- Piotrowska KD, Kownacki L, Kuch M (2009) Cardiovascular magnetic resonance findings in a case of Danon disease. J Cardiovasc Magn Reson 11: 1-12.
- Schulze MR, Wachter R, Schmeisser A, Fischer R, Strasser RH (2006) Restrictive cardiomyopathy in a patient with primary hyperoxaluria type II. Clin Res Cardiol 95: 235-240. [Crossref]
- Palka P, Duhig E, Carey L, Galbraith A (2001) Primary oxalosis with cardiac involvement: echocardiographic features of an unusual form of cardio- myopathy. Circulation 103: 122-133.
- Vélez RS, Depierreux M, Nortier J, Unger P (2006) Cardiac oxalosis: a rare cause of diastolic dysfunction. Eur Heart J 27: 2496. [Crossref]
- De Cobelli F, Esposito A, Belloni E (2009) Delayed-enhanced cardiac MRI for differentiation of Fabry’s disease from symmetric hypertrophic cardiomyopathy. Am J Roentgenol 192: 97-102.
- Pochis WT, Litzow JT, King BG, Kenny D (1994) Electrophysiologic findings in Fabry's disease with a short PR interval. Am J Cardiol 74: 203-204. [Crossref]
- Child JS, Perloff JK, Bach PM, Wolfe AD, Perlman S, et al. (1986) Cardiac involvement in Friedreich's ataxia: a clinical study of 75 patients. J Am Coll Cardiol 7: 1370-1378. [Crossref]
- Morvan D, Komajda M, Doan LD, Brice A, Isnard R, et al. (1992) Cardiomyopathy in Friedreich's ataxia: a Doppler-echocardiographic study. Eur Heart J 13: 1393-1398. [Crossref]
- Alboliras ET, Shub C, Gomez MR (1986) Spectrum of cardiac involvement in Friedreich’s ataxia: clinical, electrocardiographic and echocardiographic observations. Am J Cardiol 58: 518-524.
- Isnard R, Kalotka H, Durr A (1997) Correlation between left ventricular hypertrophy and GAA trinucleotide repeat length in Friedreich’s ataxia. Circulation 95: 2247-2249.
- Kumar V, Abbas AK, Fausto N, Mitchell RN (2007) Robbins Basic Pathology. Philadelphia, PA: Saunders Elsevier.
- Schieken RM, Kerber RE, Ionasescu VV, Zellweger H (1975) Cardiac manifestations of the mucopolysaccharidoses. Circulation 52: 700-705. [Crossref]
- Wraith JE (1995) The mucopolysaccharidoses: a clinical review and guide to management. Arch Dis Child 72: 263-267. [Crossref]
- Rigante D, Segni G (2002) Cardiac structural involvement in mucopolysaccharidoses. Cardiology 98: 18-20. [Crossref]
- Kyle RA, Gertz MA, Greipp PR, Witzig TE, Lust JA, et al. (1999) Long-term survival (10 years or more) in 30 patients with primary amyloidosis. Blood 93: 1062-1066. [Crossref]
- Comenzo RL, Gertz MA (2002) Autologous stem cell transplantation for primary systemic amyloidosis. Blood 99: 4276-4282. [Crossref]
- Gertz MA, Lacy MQ, Gastineau DA, Inwards DJ, Chen MG, et al. (2000) Blood stem cell transplantation as therapy for primary systemic amyloidosis (AL). Bone Marrow Transplant 26: 963-969. [Crossref]
- Colucci WS, Lorell BH, Schoen FJ, Warhol MJ, Grossman W (1982) Hypertrophic obstructive cardiomyopathy due to Fabry's disease. N Engl J Med 307: 926-928. [Crossref]
- Sugie K, Yamamoto A, Murayama K (2002) Clinicopathological features of genetically confirmed Danon disease. Neurology 58: 1773-1778.
- Dürr A, Cossee M, Agid Y, Campuzano V, Mignard C, et al. (1996) Clinical and genetic abnormalities in patients with Friedreich's ataxia. N Engl J Med 335: 1169-1175. [Crossref]
- Alboliras ET, Shub C, Gomez MR (1986) Spectrum of cardiac involvement in Friedreich’s ataxia: clinical, electrocardiographic and echocardiographic observations. Am J Cardiol 58: 518-524.
- Leone M, Rocca WA, Rosso MG, Mantel N, Schoenberg BS, et al. (1988) Friedreich’s disease: Survival analysis in an Italian population. Neurology 38: 1433-1438.
- Gilbert EF (1987) The effects of metabolic diseases on the cardiovascular system. Am J Cardiovasc Pathol 1: 189-213. [Crossref]
- Schieken RM, Kerber RE, Ionasescu VV, Zellweger H (1975) Cardiac manifestations of the mucopolysaccharidoses. Circulation 52: 700-705.
- Hamosh A (2004) Online Mendelian Inheritance in Men. Baltimore, MD: Johns Hopkins University.
- Wraith JE (1995) The mucopolysaccharidoses: a clinical review and guide to management. Arch Dis Child 72: 263-267. [Crossref]
- Rigante D, Segni G (2002) Cardiac structural involvement in mucopolysaccharidoses. Cardiology 98: 18-20.
- Kushwaha SS, Fallon JT, Fuster V (1997) Restrictive cardiomyopathy. N Engl J Med 336: 267-276. [Crossref]
- Chew C, Ziady GM, Raphael MJ, Oakley CM (1975) The functional defect in amyloid heart disease. The "stiff heart" syndrome. Am J Cardiol 36: 438-444. [Crossref]
- Klein AL, Hatle LK, Burstow DJ (1989) Doppler characterization of left ventricular diastolic function in cardiac amyloidosis. J Am Coll Cardiol 13: 1017-1026.
- Ridolfi RL, Bulkley BH, Hutchins GM (1977) The conduction system in cardiac amyloidosis. Clinical and pathologic features of 23 patients. Am J Med 62: 677-686.
- Roberts WC, McAllister HA, Ferrans VJ (1977) Sarcoidosis of the heart. A clinicopathologic study of 35 necropsy patients (group 1) and review of 78 previously described necropsy patients. Am J Med 63: 86-108.
- Bozkurt B, Colvin M, Cook J, Cooper LT, Deswal A, et al. (2016) Current diagnostic and treatment strategies for specific dilated cardiomyopathies: a scientific statement from the American Heart Association. Circulation 134: e579-e646.
- Uemura A, Morimoto S, Hiramitsu S (1999) Histologic diagnostic rate of cardiac sarcoidosis: Evaluation of endomyocardial biopsies Am Heart J 138: 299-302.
- Murtagh B, Hammill SC, Gertz MA, Kyle RA, Tajik AJ, et al. (2005) Electrocardiographic findings in primary systemic amyloidosis and biopsy-proven cardiac involvement. Am J Cardiol 95: 535-537.
- Ardehali H, Qasim A, Cappola T, Howard D, Hruban R, et al. (2004) Endomyocardial biopsy plays a role in diagnosing patients with unexplained cardiomyopathy. Am Heart J 147: 919-923. [Crossref]
- Falk RH (2005) Diagnosis and management of the cardiac amyloidoses. Circulation 112: 2047-2060. [Crossref]
- Ansari Lari MA, Ali SZ (2004) Fine-needle aspiration of abdominal fat pad for amyloid detection: a clinically useful test? Diagn Cytopathol 30: 178-181. [Crossref]
- Perugini E, Guidalotti PL, Salvi F (2005) Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J Am Coll Cardiol 46: 1076-84.
- Bokhari S, Castano A, Pozniakoff T, Deslisle S, Latif F, et al. (2013) Tc-pyrophosphate scintigraphy for differentiating light-chain cardiac amyloidosis from the transthyretin-related familial and senile cardiac amyloidoses. Circ Cardiovasc Imaging 6: 195-201.
- Tsang W, Lang RM (2010) Echocardiographic evaluation of cardiac amyloid. Curr Cardiol Rep 12: 272-276. [Crossref]
- Lee GY, Kim K, Choi JO, Kim SJ, Kim JS, et al. (2014) Cardiac amyloidosis without increased left ventricular wall thickness. Mayo Clin Proc 89: 781-789. [Crossref]
- Falk RH, Plehn JF, Deering T, Schick EC Jr, Boinay P, et al. (1987) Sensitivity and specificity of the echocardiographic features of cardiac amyloidosis. Am J Cardiol 59: 418-422. [Crossref]
- Baccouche H, Maunz M, Beck T (2012) Differentiating cardiac amyloidosis and hypertrophic cardiomyopathy by use of three-dimensional speckle tracking echocardiography. Echocardiography 29: 668-677.
- Koyama J, Falk RH (2010) Prognostic significance of strain Doppler imaging in light- chain amyloidosis. JACC Cardiovasc Imaging 3: 333-342.
- Burstow DJ, Tajik AJ, Bailey KR, DeRemee RA, Taliercio CP (1989) Two-dimensional echocardiographic findings in systemic sarcoidosis. Am J Cardiol 63: 478-482.
- De Cobelli F, Esposito A, Belloni E (2009) Delayed-enhanced cardiac MRI for differentiation of Fabry’s disease from symmetric hypertrophic cardiomyopathy. Am J Roentgenol 192: 97-102.
- Maron BJ, Roberts WC, Arad M, Haas TS, Spirito P, et al. (2009) Clinical outcome and phenotypic expression in LAMP2 cardiomyopathy. JAMA 301: 1253-1259. [Crossref]
- Delatycki MB, Paris DB, Gardner RJ, Nicholson GA, Nassif N, et al. (1999) Clinical and genetic study of Friedreich ataxia in an Australian population. Am J Med Genet 87: 168-174. [Crossref]
- Syed IS, Glockner JF, Feng D (2010) Role of cardiac magnetic resonance imaging in the detection of cardiac amyloidosis. JACC Cardiovasc Imaging 3: 155-164.
- Smedema JP, Snoep G, van Kroonenburgh MP (2005) Evaluation of the accuracy of gadolinium-enhanced cardiovascular magnetic resonance in the diagnosis of cardiac sarcoidosis. J Am Coll Cardiol 45: 1683-1690.
- Youssef G, Beanlands RSB, Birnie DH, Nery PB (2011) Cardiac sarcoidosis: applications of imaging in diagnosis and directing treatment. Heart 97: 2078-2087.
- Nucifora G, Miani D, Piccoli G, Proclemer A (2012) Cardiac magnetic resonance imaging in Danon disease. Cardiology 121: 27-30.
- Weidemann F, Störk S, Liu D, Hu K, Herrmann S, et al. (2013) Cardiomyopathy of Friedreich ataxia. J Neurochem 126: 88-93. [Crossref]
- Banypersad SM, Moon JC, Whelan C, Hawkins PN, Wechalekar AD (2012) Updates in cardiac amyloidosis: a review. J Am Heart Assoc 1: e000364. [Crossref]
- Gertz MA, Dispenzieri A, Sher T (2015) Pathophysiology and treatment of cardiac amyloidosis. Nat Rev Cardiol 12: 91-102. [Crossref]
- Merlini G, Seldin DC, Gertz MA (2011) Amyloidosis: pathogenesis and new therapeutic options. J Clin Oncol 29: 1924-1933. [Crossref]
- Mikhael JR, Schuster SR, Jimenez ZH (2012) Cyclophosphamide- bortezomib-dexamethasone (CyBorD) produces rapid and complete hematologic response in patients with AL amyloidosis. Blood 119: 4391-4394.
- Maurer MS, Raina A, Hesdorffer C (2007) Cardiac transplantation using extended-donor criteria organs for systemic amyloidosis complicated by heart failure. Transplantation 83: 539-545.
- Dubrey SW, Davidoff R, Skinner M, Bergethon P, Lewis D, et al. (1997) Progression of ventricular wall thickening after liver transplantation for familial amyloidosis. Transplantation 64: 74-80.
- Berk JL, Suhr OB, Obici L (2013) Diflunisal Trial Consortium. Repurposing diflunisal for familial amyloid polyneuropathy: Am randomized clinical trial. JAMA 310: 2658-2667.
- Coelho T, Maia LF, Martins da Silva A (2012) Tafamidis for transthyretin familial amyloid polyneuropathy: A randomized, controlled trial. Neurology 79: 785- 792.
- Coelho T, Adams D, Silva A, Lozeron P, Hawkins PN, et al. (2013) Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med 369: 819-829. [Crossref]
- Ackermann EJ, Guo S, Booten S (2012) Clinical development of an antisense therapy for the treatment of transthyretin-associated polyneuropathy. Amyloid 19: 43- 44.
- Kristen AV, Lehrke S, Buss S (2012) Green tea halts progression of cardiac transthyretin amyloidosis: An observational report. Clin Res Cardiol 101: 805-813.
- Hamzeh NY, Wamboldt FS, Weinberger HD (2012) Management of cardiac sarcoidosis in the United States: a Delphi study. Chest 141: 154-162. [Crossref]
- Yazaki Y, Isobe M, Hiroe M (2001) Central Japan Heart Study Group. Prognostic determinants of long-term survival in Japanese patients with cardiac sarcoidosis treated with prednisone. Am J Cardiol 88: 1006-1010.
- Vignaux O, Dhote R, Duboc D (2002) Clinical significance of myocardial magnetic resonance abnormalities in patients with sarcoidosis: A 1-year follow-up study. Chest 122: 1895-1901.
- Birnie DH, Sauer WH, Bogun F (2014) HRS expert consensus statement on the diagnosis and management of arrhythmias associated with cardiac sarcoidosis. Heart Rhythm 11: 1304-23.
- Weidemann F, Sanchez N, Politei J (2013) Fibrosis: A key feature of Fabrym disease with potential therapeutic implications. Orphanet J Rare Dis 8: 100-116.
- Echaniz LA, Mohr M, Epailly E (2006) Novel LA MP-2 gene mutation and successful treatment with heart transplantation in a large family with Danon disease. Muscle Nerve, 33: 393-397.
- Weidemann F, Störk S, Liu D, Hu K, Herrmann S, et al. (2013) Cardiomyopathy of Friedreich ataxia. J Neurochem 126: 88-93. [Crossref]
- Greulich S, Deluigi CC, Gloekler S (2013) CMR imaging predicts death and other adverse events in suspected cardiac sarcoidosis. JACC Cardiovasc Imaging 6: 501-511.
- Ohira H, Tsujino I, Ishimaru S (2008) Myocardial imaging with 18F-fluoro-2- deoxyglucose positron emission tomography and magnetic resonance imaging in sarcoidosis. Eur J Nucl Med Mol Imaging 35: 933-941.
- Namdar M, Steffel J, Vidovic M, Brunckhorst CB, Holzmeister J, et al. (2011) Electrocardiographic changes in early recognition of Fabry disease. Heart 97: 485-490. [Crossref]
- Varr BC, Zarafshar S, Coakley T (2014) Implantable cardioverter-defibrillator placement in patients with cardiac amyloidosis. Heart Rhythm 11: 158-162.
- Kubo T, Baba1 Y, Hirota T, Tanioka K, Yamasaki N, et al. (2015) Differentiation of infiltrative cardiomyopathy from hypertrophic cardiomyopathy using high- sensitivity cardiac troponin T: A case-control study. Cardiovascular Disorders 15: 1-7.
- Palka P, Lange A, Donnelly JE, Scalia G, Burstow DJ, et al. (2002) Doppler tissue echocardiographic features of cardiac amyloidosis. J Am Soc Echocardiogr 15: 1353-1360.
- Elliott P, Baker R, Pasquale F, Quarta G, Ebrahim H, et al. (2011) Hughes DA, and ACES study group: Prevalence of Anderson-Fabry disease in patients with hypertrophic cardiomyopathy: the European Anderson-Fabry Disease Survey. Heart 97: 1957-60.
- Weidemann F, Niemann M, Breunig F, Herrmann S, Beer M, et al. (2009) Long- term effects of enzyme replacement therapy on Fabry cardiomyopathy: Evidence for a better outcome with early treatment. Circulation 119: 524-529.