Abstract
This article reviews and classifies epigenetic alterations driven by the induction of hypoxia, which are involved with genomic instability. We explore the biological relationship of key targets involved in genomic instability and heterogeneity in cancer. Subsequently, in order to reverse the key translational biologic events in tumorogenesis and metastasis, we propose a method which induces cell differentiation, by epigenetic modification. We correlate important genomic mutations, or methylations, with subsequent epimutational events that cause instability in cancer. We hypothesize that inhibition of these epigenetic aberrancies should be selective in order to overcome genetic instability. Further, we discuss our findings in preclinical and clinical settings related to our hypothesis and conclude that such an approach may overcome the current therapeutic challenges in refractory solid tumors with genomic instability.
Keywords
Epimutation, epigenetic therapy
Background
Recent research has opened scientists’ eyes to look deeper into biology of cancer cell and describe epigenetic networks, which drive tumor cells. Recent work connects epigenetics to stem cell plasticity and carcinogenesis, beyond what the genomics of a cell could explain. Although exciting, this new perspective of medicine brings complexity into clinical approaches, as it challenges the traditional concepts of cancer biology by discovering the nature of evolutionary epigenetics, which still remains to be fully understood.
One of the main therapeutic challenges in identifying actionable targets to treat in cancer is the tumor genomic instability in concert with its stem cell plasticity and epigenomic instability. Literature regarding epimutations in cancer identifies common epigenomic signatures, such as CpG island methylator phenotype (CIMP) and its genomic equivalent, microsatellite instabilities (MSI). Many of the epigenetic mutations (epimutations) can be the cause of this pattern. For example, IDH1, (Tet2 mutations), are epigenetic mutations in the case of CpG island mutated phenotype (CIMP), and inactivating Set Domain Containing 2 (SETD2) mutations are reported in the case of MSI [1-11]. Also, some evidence has suggested that MLH1 mutations and epimutations can reciprocally influence each other and suggest that an altered structure of the MLH1 locus results in epigenetic alteration [5]. For example, extensive promoter methylation is associated with MLH1 inactivation, MSI, and BRAF mutation in colon cancer [3]. Somatic MLH1 hypermethylation has been detected in 91.9% of cases in some studies, with MSI BRAF-mutated CRC [4].
In general, epigenomic mutations occur with higher incidence than genomic errors. Although reversible through generations, their penetration is sometimes enough to cause cancer by itself, without further epigenetic dysregulation. For example, IDH1 mutations are sometimes enough to cause sarcomas [2]. The investigational drugs that inhibit the epigenetic drivers such as Tet2, IDH1, and DNMT have widely failed in solid tumors. The challenge in therapeutic approaches are not only the fact that each epigenetic target needs to be verified, and inhibited if mutated, but also that the downstream epigenetic targets would have to be selectively targeted to accomplish response. This cannot be achieved through the use of available demethylators such as decitabine or azacytidine, as they are non-selective in their targets.
Hypothesis
To identify the key epigenetic targets in inducing genomic instability we selected SETD2, a common mutated epigene in colorectal cancer, acting as an H3K36 methyltransferase. This is one of the key epigenetic targets that is inactivated by mutation. This occurs in variety of solid tumors and correlates with microsatellite instability. SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability [6]. Histone modifications establish the chromatin states that coordinate the DNA damage response. It is also suggested that SETD2, is required for ATM activation upon DNA double-strand breaks (DSBs). In ccRCC, loss of SETD2 may afford an alternative mechanism for the inactivation of the p53-mediated checkpoint without the need for additional genetic mutations in TP53 [8]. The DNA double-strand break (DSB), arising from exposure to ionizing radiation or various chemotherapeutic agents or from replication fork collapse, is among the most dangerous of chromosomal lesions. Mutations in SETD2 can actually cause a novel overgrowth condition [6,7,9]. Besides the reported H3K36-trimethylation pathway, it is reported that SETD2 mutation also mediates mismatch repair gene (MMR) via AKT-induced PMS2 decrease and co-loss of MLH1 loss in ccRCC. Renal clear cell carcinoma (ccRCC) is characterized by frequent mutation in SETD2, which has recently been shown to regulate mismatch repair (MMR). In the presence of a genetic lesion, downregulation of SETD2 contributes to both initiation and progression during leukemia development by promoting the self-renewal potential of leukemia stem cells [10].
This target is of key interest, as our investigation allowed us to correlate this as the most likely genomic mutational event in relation to carcinogenesis through this epigenetic pathway. We have identified that greater than 88% of mutations in this gene are accompanied by VHL inactivation, and PHD overexpression [1]. VHL inactivation appears to be an actionable target through inhibition of HIF-1, as we showed significant inhibition in our invitro studies using our epigenetic protocol (MTET).
VHL inhibition potentiates the HIF response element interaction and EMT transition through Proryl hydroxylases (PHD). It also increases stem cell plasticity through HIF2 alpha activation and Oct4 [1]. We also know that VHL inactivation can induce other epimutations such as EZH-2 which enhances the methylation of H3K29 and overexpression of DNMT3B and Methyl CpG island protein domain 1 (Mbd1). This correlates with, and interacts with, (if not causes) histone demethylation at H3k4, 36 and 79. Certainly the methylation of H3k9, 27 and 20 have the opposite effect, and so is the deacetylation of H4k16 and H3k9, commonly seen in cancer [1]. Inactivation of VHL also can cause changes in micro RNAs. Since VHL plays such a key regulator in inducing a panepigenomic and genomic effect, through HIF-1, we believe that a targeted therapy that inhibits the methylation of this target, would significantly reduce the presence of genomic instability, stem cell plasticity and heterogeneity in cancer.
Methods
We used in vitro application of our compounds consisting of one histone deacetylase inhibitor and one demethylator (Quercetin a polyphenol and Sodium Phenyl butyrate, a known HDACI) and we showed synergistic HIF inhibition in variety of cancer cell lines (Figure 1).
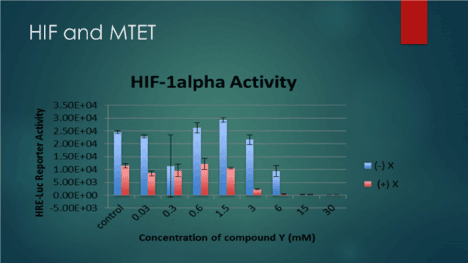
Figure 1. HIF inhibition by application of MTET
In vivo, we studied several cases of advanced solid tumor through their tumor liquid biopsy signature and we correlated their circulating tumor DNA (ctDNA) as a marker for heterogeneity and genomic instability with their response to therapy. Here we present these cases in detail and elaborate on the findings.
Case Series
Case 1
A 90-year old female with history of pancreatic cancer was referred to our clinic for evaluation and management. She was diagnosed in September 2015 and had not yet seen an oncologist due to insurance issues. She had not been staged by scans or had extensive blood workup. She had mild abdominal pain but was generally asymptomatic. She had been under the care of a gastroenterologist, and had a biliary stent placed. We referred her to an Oncologist, we drew her labs and ordered a PET scan. Her labs showed significantly elevated liver enzymes, bilirubin as well as ALK-P. Her PET scan showed an SUV of activity of 5.5 around the biliary stent. There were also retroperitoneal nodes with activity, as well as a solitary nodule in the liver that was likely metastatic. We also measured her cDNA using Guardant 360 labs, which was positive for KRAS as well as NF-1. Generally, when KRAS is mutated, we do not use erlotinib, but in her case, a combination of capecitabine and erlotinib was considered. She received IV epigenetic therapy daily for 10 treatments and tolerated it without side effects. She noted improved quality of life and energy. After 10 treatments, cDNA was repeated and showed that KRAS was no longer detected (Figure 2).

Figure 2. Case 1
Case 2
A 37-year-old-female with history of breast cancer diagnosed in January 2014, Stage II A, ER/PR positive/Her 2 negative, presented to our clinic after trying and failing multiple alternative treatment modalities. She had previously refused all conventional therapies, including hormonal blockade. She had opted for alternative treatments in Mexico, and had exhausted Insulin Potentiated Chemotherapy (IPT), Vitamin C IV drip, and other alternative treatments, including autologous immune cell therapy and hyperthermia. She was referred to us by her PMD. She was status post mastectomy done in January 2014 and had recurrence of the tumor in September 2015 at the surgical implant replacement. She was being treated at the Cleveland clinic. Imaging done in November 2015 had confirmed presence of innumerable bony metastases as well as thoracic and neck lymph involvements, extensive chest and bone disease. Her right breast mass had an SUV of 14.8, and there were several axillary, mediastinal and hilar lymphadenopathies ranging in SUV of 7-9.6. Many sclerotic bony lesions in her glenoid, sternum and sacrum, with SUVs ranging between 7.8 to 9.2 were noted.
Upon her evaluation, we found significantly elevated tumor markers, as well as an elevated IGF-1. Her circulatory DNA by liquid biopsy was positive and showed with a mutated P53 as well as ERBB2. Her serum HER-2 was also elevated (Figure 3).

Figure 3. Case 2
Her treatment plan included IV epigenetic therapies, which she received on daily basis. Her IGF-1 was repeated after two weeks of therapy and it reduced from 240 to 193 ng/mL (normal 69 – 227 ng/mL) measured on 11/9/15 and 11/23/15, respectively. She reported that her quality of life improved, and she had not changed her diet or supplements.
She was started trastuzumab based on tumor molecular profiling showing overexpression of HER-2 as well as elevated serum HER-2. She concurrently received IV epigenetic therapies, based on the molecular profiling of her tumor showing overexpression of TET2.
Her CTC was initially positive for ERBB2, cMYC, and CK19, and was repeated by BioFocus on 12/17/15 and it showed complete eradication of CTC with CK20 and ERBB2. The cMYC virtually unchanged. Her LDH dropped from 303 to 147 UI/L, after just three treatments, measured on 12/25 and 2/28/15 respectively.
Her circulatory DNA was measured through Guardant360, and it was positive for PI3K and TP53 alterations with elevated MAF, measured on 11/9/15, 12/1/15 and negative on 12/30/15. She received trastuzumab along with IV epigenetic therapies, which could explain the disappearance of ERBB2 from her CTC and/or cDNA, however there are no expected positive effects from trastuzumab on P53, or PI3K, nor the cMYC present at her initial CTC. We contribute these results to the IV epigenetic therapies she received in the interim, as this therapy in fact targets cMYC, P53 and PI3K simultaneously.
On 1/20/2016, her CTC was repeated, and it showed complete resolution of CTC post therapy.
She stopped the IV epigenetic therapies on in January 2016, as she moved back home to Cleveland, and stayed on trastuzumab every three weeks, along with monthly leuprolide and denosumab injections, and oral tamoxifen. Her restaging PET scan in August 2016 showed significant progression of her disease with increased sizes and metabolic activities in all her lesions in thoracic and bones. There was a new L1 lesion with SUV activity of 16.9. Her sternal lesion enlarged and had an SUV activity that increased to 16.9 from 5.6, as well as a sacral lesion that enlarged with SUV activity of 16.9 from 5.6.
She immediately returned to our clinic. At this point the CTC was repeated, and it showed the presence of HER-2 positive cells again. She was started back on the IV therapies again on 8/25/16, administered on daily basis. Her restaging scan was ordered on 9/15/16 and showed partial response to therapy with significant reduction in all her lesions sizes and SUV metabolic activities. Her cervical nodes improved, right breast implant with SUV of 8.2 from 12.2, axillary nodes 4.2 down from 6.0, pulmonary hilar nodules 4.3 from 9.2, and bony lesions in sternum 10.2 from 16, L1 from 16.3 down to 11.9, right scapula from 13.9 down to 8.4, and sacrum from 7.4 to 6.6.
Case 3
A 50-year-old female with a suspicious left breast mass presented to us for evaluation and management in November 2015. She had not yet had a biopsy when she referred to us, and initially refused to do so. Her breast cancer tumor markers were elevated and had been drawn by her chiropractor after she found a mass in her left breast growing for over a year. A thermogram was performed which was suspicious. Upon her arrival to our clinic, we ordered biomarkers as well as a staging PET scan, which showed a significantly metabolically active tumor in her left breast as well as axilla, and SUV activity in her iliac bones as well as thoracic and lumbar spine. The most active lesion had an SUV activity of 9.9. Her tumor markers were extremely elevated and had positive cDNA on her liquid biopsy done by Guardant360, which was positive for PI3K as well as cKIT alterations, measured on 11/19/15. Her transforming growth factor (TGF), was also elevated, as expected from her cKIT overexpression. Her CTC came back positive for cMYC and for ERBB2 (HER-2). She eventually agreed to biopsy, which showed both ductal and lobular carcinoma that was ER positive and HER-2 negative.
She was started on combination of MTET and trastuzumab. Her TGF also dropped from 24,000 measured on 1/19/16 down to 1258 on 2/22/16. Her cancer was restaged on 4/1/16, through a whole-body PET, which showed a near complete resolution of thoracic lymph nodes by FDG activity, as well as complete resolution of FDG activity in T3 and L4 bony mets (the SUV dropped to 2.0 compared to 9.1). There were pulmonary nodules with remaining activity.
She was further started on everolimus in December 2016. Her cDNA was repeated, and it showed stable KIT, and resolution of PI3K to non-detectable. (Please see below). Further cDNA showed reduction of MAF to 0.5%. She had another restaging PET scan on 1/3/17, which showed improvement in both size and activity of all metastatic lesions in the lungs, and bony pelvis. No new lesion was seen. The left upper lobe lung lesion decreased in size from 1.4 to 1.2 cm, and SUV from 12.6 to 4.9, left upper lobe lesion 1 cm from 1.6 cm, activity down from 11.7 to 1.1, right upper lobe lung lesion 0.9 cm from 1.3 cm, activity down from 10 to 3.7, axillary nodes SUV of 0.9. Her left iliac wing lesion decreased SUV activity from 8.3 to 4.7. The left ilium decreased from 9.9 to 4.5. The inferior pubic ramus decreased from 5.3 to 3. The L4 lesion stable. Further she stopped the epigenetic therapies and continued with her conventional treatments with afinitor, her c DNA showed significant increase on May 22,2017 (Figure 4 and 5).

Figure 4. Case 3

Figure 5. Case 4
Case 4
An 80-year-old male with history of multiorgan failure, and locally advanced left posterior bladder cancer was referred to us for evaluation and management in June 2015. He had been suffering from partial obstruction of the rectum, secondary to inflammation versus malignancy in rectal region, confirmed by the pelvic CT. He also had renal failure that is also possibly secondary to bladder mass causing obstruction of urine flow, with hydronephrosis in both sides, right more than left, seen in his imaging. He also reported bad odor and mouth dryness, which I contribute to the uremia he has with majorly impaired kidney function. We ordered laboratory studies for him, including Guardant 360 for cDNA. His labs showed elevated CEA and CA19-9. His cDNA revealed many alterations including BRCA2, PI3K and P53. He was started on IV epigenetic therapies and anti-inflammatory therapies aiming at reducing his pain and improving his creatinine. After 10 IV treatments, his cDNA was repeated, and showed reduced heterogeneity by disappearance of several alterations, as well as decreased MAF (Figure 6).
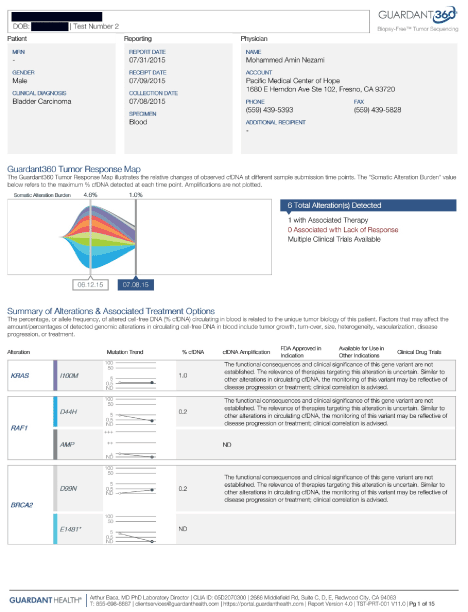
Figure 6. Case 4
Case 5
A 54-year-old female with history of EGFR mutated lung adenocarcinoma was referred to us in February 2018. She had been on afatinib, which was started in 2016, and resulted in decreased CEA levels for several months, but now has progression of disease. Her oncologist ordered lab tests by Travogene, which were negative for any mutation in T790M. She is complaining of back pain at this time which is perhaps due to increased tumor growth at thoracic spine (T4), she also has noticed an enlarged Left axillary node for about 2 months. Her brain MRI was done in December 2017 and was normal. Our plan of care included IV epigenetic therapies, which we hoped would increase the response to the afatinib. After 10 treatments, her cDNA ws repeated, which showed improvement, with many alterations no longer detected, and her EGFR down to 5.5 MAF from 30.4 (Figure 7).
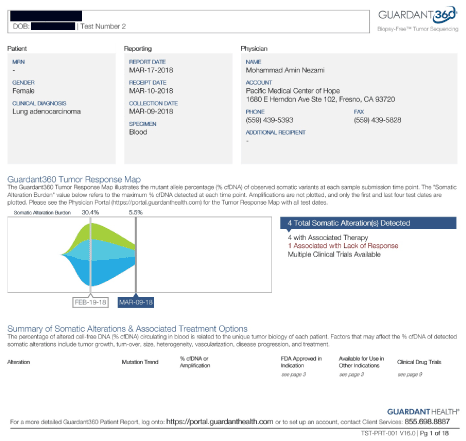
Figure 7. Case 5
Case 6
A 57-year-old female with a history of invasive ductal breast carcinoma, presented for evaluation and management. She was first diagnosed in October 2000. She was treated with alternative treatments in Mexico and remained in remission until 2008, when she had right breast pain. She went back to Mexico and had right mastectomy for recurrent breast cancer. Her pathology showed ER/PR/HER2 triple positive with a Ki67 of 60%. After that, she received trastuzumab and hormonal blockade with anastrozole. Her PET scan done at that time confirmed metastatic disease with bone involvement. She then was treated at St. Jude’s where she continued hormonal blockade, trastuzumab, and zoledronic acid. She progressed slowly and was under the care of her local oncologist. She also tried and failed denosumab. She was started on tamoxifen and offered radiation. She had been switched to letrozole near the time of presentation, but review of her PET scan on 6/8/16 again showed progression of disease with complete destruction of the sternum. There were multiple lung metastases, as well as extensive disease in the peritoneum. There were no liver lesions noted. She has never had a paracentesis, although she appeared to have ascites. She referred to us seeking alternative and integrative treatments. Upon her arrival she was evaluated, and her labs drawn, which showed presence of CTCs in the blood, along with expression of ERBB2 and cMYC. These CTCs were ER negative.
2021 Copyright OAT. All rights reserv
Her molecular profiling was performed which showed presence of several mutations in her tissue biopsied from her humerus. First, they were HER-2 negative, second, they were ER positive. Third they were MSH-6 positive, making them responsive to the IV epigenetic therapies which started immediately on daily basis.
Her labs reported decreased tumor markers, CA 27.29 at 685 from 765, CA 15.3 at 533 from 574, serum HER-2 at 75 from 98, measured on 6/28/16 after two weeks of therapy. Her cDNA showed a marked reduction in MAF, from 7.4 to 5.0 at the level of PI3K. It was repeated after continued treatment and showed more reduction from 5 to 1.9, measured on 8/2/16.
She received trastuzumab after two weeks of MTET. Her initial response to MTET was reduction of her serum HER-2 level. She did not respond to either lapatinib or trastuzumab, as her serum HER-2 increased again. Since this seemed to be a case of inherent resistance to trastuzumab, she was started on epigenetic therapies again, which resulted in decreased serum HER-2. It was decided to use afatinib (a pan-EGFR blocker) in conjunction with trastuzumab, in her case. Her tumor markers dropped, and she continued to respond to therapy with improved markers and her c DNA continued to improve with reduced PI3k and CCND1, measured on August 2016 (Figure 8).
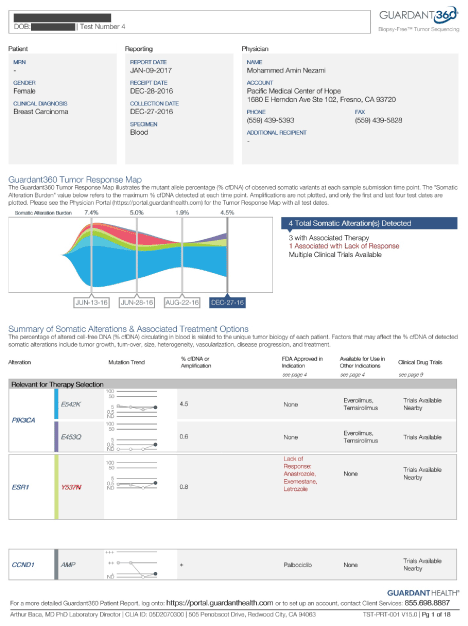
Figure 8. Case 6
Conclusion
We propose further trials based on our promising findings in our samples using MTET protocol to target VHL/ HIF pathway. We also recommend further generation of hypothesis based on the clinical model of response which includes application of companion diagnostic tools such as circulating DNA.
References
- Thirlwell C, Schulz L, Dibra H, Beck S (2011) Suffocating cancer: Hypoxia-associated epimutations as targets for cancer therapy. Clinical Epigenetics 3: 9 [Crossref]
- Roy DM, Walsh LA, Chan TA (2014) Driver mutations of cancer epigenomes. Protein & Cell 5: 265-296 [Crossref]
- Vilkin A, Niv Y, Nagasaka T, Morgenstern S, Levi Z, et al. (2008) T2093 Microsatellite Instability (MSI), Hmlh1 Promoter Hypermethylation and BRAF Mutation Analysis in Sporadic Colorectal Cancers (Crcs) of Different Ethnic Groups in Israel. Gastroenterology 134 [Crossref]
- Moreira L, Muñoz J, Cuatrecasas M, Quintanilla I, Leoz ML (2014) Prevalence of somatic mutl homolog 1 promoter hypermethylation in Lynch syndrome colorectal cancer. Cancer 121: 1395-1404. [Crossref]
- Cini G, Carnevali I, Quaia M, Chiaravalli AM, Sala P (2015) Concomitant mutation and epimutation of the MLH1 gene in a Lynch syndrome family. Carcinogenesis 36: 452-458 [Crossref]
- Pfister S, Ahrabi S, Zalmas L, Sarkar S, Aymard F (2014) SETD2-Dependent Histone H3K36 Trimethylation Is Required for Homologous Recombination Repair and Genome Stability. Cell Reports 7: 2006-2018. [Crossref]
- Clark A, Low K (1988) Pathways and Systems of Homologous Recombination in Escherichia coli. The Recombination of Genetic Material 155-215
- Carvalho S, Vítor A, Sridhara S, Martins F, Raposo A (2018) SETD2 is required for DNA double-strand break repair and activation of the p53-mediated checkpoint. [Crossref]
- Luscan A (2018) Mutations in SETD2 cause a novel overgrowth condition. [Crossref]
- Zhu X, He F, Zeng H, Ling S, Chen A, et al. (2018) Identification of functional cooperative mutations of SETD2 in human acute leukemia. [Crossref]
- Feng C, Wen H (2015) MP39-03 LOSS OF MLH1 CONFERS RESISTANCE TO PI3Kβ INHIBITORS IN RENAL CLEAR CELL CARCINOMA WITH SETD2 MUTATION. The Journal of Urology 193