Background: The prediction of the phenotypic effect of a genetic variant represents a useful tool in genetic counseling. However, in coagulation factor VII (FVII) deficiency there is no straight correlation between genotype and phenotype since the residual FVII coagulant (FVII:C) activity associated with specific genetic variants does not always account for the observed clinical signs.
Objective: to better describe the correlation between genotype and clinical phenotype of F7 gene we report a family case with a deficient FVII:C activity.
Results and discussion: The case under investigation came to our attention during a genetic counseling attended by the proband, because she wanted to know, given the familiarity for breast cancer, her carrier probability for a BRCA1/2 pathogenetic variant. Pedigree analysis showed that besides cancer predisposition both the proband and her two sons suffered from recurrent spontaneous bleeding. Their coagulation pathway analysis was indicative of a FVII:C activity reduction with a pattern mimicking an autosomal dominant inheritance.
Proband F7 sequencing showed the following heterozygous variants: c.1088C>A (p.Pro363His), c.-326_-325insCCTATATCC, c.-122T>C and c.1238G>A. The molecular analysis of her sons highlighted that c.1088C>A variant was in trans configuration. The occurrence of c.1088C>A variant alone was associated with 36% of FVII:C residual activity. Conversely, when this variant was in compound heterozygosity with c.-326_-325insCCTATATCC, c.-122T>C and c.1238G>A haplotype, the FVII:C residual activity further shrinked to 22%.
c.1088C>A variant alone determined the most significant FVII:C activity reduction, however, when found in combination with c.-326_-325insCCTATATCC, c.-122T>C and c.1238G>A haplotype an additive effect on the FVII:C activity phenotype was observed.
blood coagulation factor, factor vii activity, haplotype, partial thromboplastin time, prothrombin time
Essentials
Atypical clinical phenotypic manifestation in FVII deficiency.
Intra-familial analysis for F7 genetic alterations and related coagulation activity defects.
Combination of two F7 alleles and their additive impact on FVII:C activity reduction.
Importance of intra-familial screening to highlight the novel c.1088C>A F7 genetic variant and its weight on the phenotypic output.
Congenital factor VII deficiency is a rare bleeding disorder with an estimated prevalence of 1 person out of 500.000 [1]. The clinical expression of this condition is extremely variable and can be characterized by either asymptomaticity or signs such as epistaxis, gum bleeding, menorrhagia, hematomas and hematuria. The most serious clinical features are due to recurrent hemarthrosis, cerebral and gastro-intestinal hemorrhages and bleeding during surgical procedures.
Prolonged prothrombin time (PT) with a normal activated partial thromboplastin time (aPTT) and FVII coagulant (FVII:C) activity below 70% are considered indicative of this condition [2,3]. The severity of hemorrhagic manifestations is however only partially related to the residual plasmatic FVII:C activity, besides, a wide and non-harmonic operative range of FVII:C activity has been proposed to evaluate the bleeding risk. A FVII:C activity above 8% has been generally associated with a low spontaneous bleeding risk [4]. Values ranging from 15 to 20%, instead, may favor bleeding in case of trauma, surgery and pregnancy [5], whereas an overall FVII:C activity above 20% should guarantee safety even in course of surgery [6].
Individuals carrying an heterozygous variant in the F7 gene are generally asymptomatic as the condition shows an autosomal recessive inheritance pattern. Indeed, only about the 20% of the carriers presents signs such as slight epistaxis, gum bleeding and menorrhagia [7], even though they are not likely to develop a significant clinical symptomatology during their life course.
In this report, we present a family where the proband and her two sons both show a clinical symptomatology correlated with a low FVII:C activity mimicking an autosomal dominant inheritance pattern. In order to establish the contributory effect of these variants on the activity of the FVII:C we described the correlation between the F7 genotypes and both laboratory and clinical investigations.
Sample collection
Written informed consent was signed by all screened subjects. Blood samples of the proband and her two sons were collected by standard atraumatic venepuncture technique using 0.1 mol/L trisodium citrate buffer as anticoagulant.
Coagulation tests
The FVII:C activity of all family members was determined using a human recombinant and an ox thromboplastin. PT, aPTT and FVII:C activity were measured by a one-stage semi-automated bioassay from plasma specimens in a ST4 coagulometer (Diagnostica Stago, Asnieres, France). Results have been expressed as a percentage of activity of the standard plasma supplied by the manufacturer. According to the manufacturer the normal ranges of PT and aPTT were 70-100% and 30-40 sec, whereas, those of FVII:C were 80.0-120.0% for both the human recombinant and the ox thromboplastin test, and 70.0 – 120.0% for the coagulometric test.
DNA analyses
Genomic DNA of all patients was extracted from whole blood using Qiagen DNA extraction Kit (QIAGEN, Dusseldorf, Germany). Next-Generation (NGS)/Massively Parallel Sequencing (MPS) of the coding regions of F7 gene was performed using an Illumina Custom Panel (Illumina, San Diego, CA 92122). Either large deletions or duplications in F7 gene were investigated by Multiplex Ligase-Dependent Probe Amplification scan (MLPA) (SALSA P207-F9 probemix–MRC-Holland). The variants placed in the 5’ untranslated region (5’UTR) of the F7 gene were directly sequenced as previously described [8]. Both sons were screened only for the variants identified in the proband. In order to investigate the potential phenotypic role that the F7 gene exonic variants may possess, we employed the following informatic prediction tools: SIFT (v6.2.0): (http://sift.jcvi.org), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and PROVEAN (http://provean.jcvi.org).
Besides breast cancer predisposition, the proband had previously complained epistaxis, menorrhagia, gum bleeding and spontaneous hematomas, as well as her two sons, who both suffered from recurrent spontaneous bleeding.
PT analysis of the proband resulted significantly lower as compared to normal range. Moreover, human recombinant and ox thromboplastin test showed that the FVII:C activity was 33.7% and 11.9% respectively, whereas it resulted 22% when determined by coagulometric method. Therefore, assuming that the FVII:C activity trend could follow an autosomal dominant pattern of inheritance. We decided to extend the laboratory and the genetic investigation to the patient’s offspring as well.
The first-born showed a phenotype characterized by only occasional spontaneous bleeding with PT and aPTT in the normal range. The FVII:C activity resulted 88.7% and 63.5% by human and ox thromboplastin test respectively, while the coagulometric test signaled an activity of 61%. Conversely, the second-born displayed a more severe symptomatology characterized by epistaxis, gum bleeding and spontaneous hematomas together with a significant dampening of FVII:C activity, which resulted to be 47.9% and 20.6% by human and ox thromboplastin test respectively. The coagulometric techinique, instead, showed an activity reduction up to around 36% (Table 1).
Table 1. Genotype and laboratory features revealed in the family members
|
PT % |
aPTT sec |
Allelic 1 Variant |
Allelic 2 Variants |
|
FVII:C % |
|
|
|
|
|
|
human thromboplastin |
ox brain thromboplastin |
coagulometric method |
Proband |
56.9 (70.0-100.0) |
30.7 (30.0-40.0) |
c.1088C>A |
c.-122T>C; c.326_325insCCTATATCC; 1238A>G |
33.7 (80.0-120.0) |
11.9 (80.0-120.0) |
22 (70.0-120.0) |
First born |
94.0 (70.0-100.0) |
35.4 (30.0-40.0) |
|
c.-122T>C; c.326_325insCCTATATCC; 1238A>G |
88.7 (80.0-120.0) |
63.5 (80.0-120.0) |
61 (70.0-120.0) |
Second born |
67.7 (70.0-100.0) |
28.9 (30.0-40.0) |
c.1088C>A |
|
47.9 (80.0-120.0) |
20.6 (80.0-120.0) |
36 (70.0-120.0) |
PT: prolonged prothrombin time; aPTT: activated partial thromboplastin time; FVII:C: FVII coagulation activity.
The F7 gene sequencing analysis of the proband detected the presence of four heterozygous variants: c.1088C>A (p.Pro363His), c.-326_-325insCCTATATCC, c.-122T>C and c.1238G>A whereas the MLPA analysis showed a normal amplification of all F7 gene exons. The molecular analysis of both promoter and exon 9 of the F7 gene revealed the heterozygous c.1088C>A (p.Pro363His) variant in one son whereas the other one resulted to be heterozygous for both the c.-326_-325insCCTATATCC, c.-122T> C and the c.1238G>A haplotype. The c.1238G>A variant is predicted to be benign/neutral by SIFT (score: 0.13), PolyPhen-2 (probability: 0.01), and PROVEAN (score: -1.16) in silico predictive tools. By contrast, the c.1088C>A variant is predicted to be damaging by SIFT (score: 0.00), PolyPhen-2 (probability: 1) and PROVEAN (score: -8.342). The correlation between genotype and phenotype in FVII deficiency is poorly understood since residual plasmatic FVII activity does not seem to account for the severity of the bleeding diathesis [9]. Generally, the disorder shows an autosomal recessive inheritance, although the 20% of the heterozygous carriers display slight bleeding [8].
In our case, the proband and her two sons both showed clinic and laboratory signs indicative of FVII deficiency, thus mimicking an autosomal dominant inheritance pattern.
Molecular analysis of the F7 gene performed in the proband and in her offspring allowed us to establish that the c.1088C>A variant was associated with c.-326_-325insCCTATATCC, c.-122T>C and c.1238G>A haplotype in trans configuration (Figure 1).
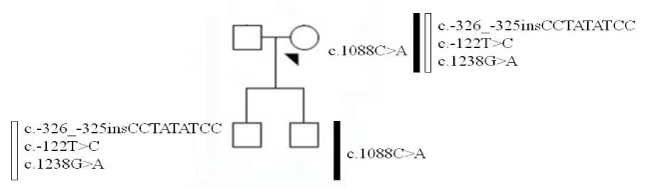
Figure 1. Schematic representation of the variant segregation among the proband and her offspring.
The c.1088C>A represents a new variant not yet reported in both international reference database of disease and Ensembl population database. This nucleotide codon (CCC) is phylogenetically highly conserved suggesting its potential involvement in the maintenance of FVII activity. Specifically, it leads to the replacement of Proline 363 (CCC) by a Histidine (CAC) residue in the protease domain of F7 gene and it is predicted to be harmful by the main in silico tools. Proline and Histidine may be considered to have only moderate physicochemical differences (Grantham dist.:77 (0-215) however, it is worth to consider that Proline has an exceptional constrained conformation linked to its alpha-N-rigid-ring structure, and its presence in the protein chain may strongly affect the protein secondary structure. As a consequence, the substitution of the Proline with a less constrained Histidine residue could increase the grade of freedom of the protein chain influencing in this way the FVII protease activity.
The c.1238G>A variant leads to the change of an Arginine with a Glycine residue in position 413 (p.Arg413Gln) and is predicted to be benign/neutral by the main in silico tools. Previous studies indicate that such genetic variant, when in an heterozygous state accounts for about 25-30% decrease of plasma FVII:C activity [10-15]. However, its net contribution is still unclear since it is in linkage disequilibrium with the c.-326_-325insCCTATATCC. The genotypic effect on FVII:C activity may be further intricated by the complete association of c-122T>C with the c.-326_-325insCCTATATCC variant, which might account for the strong reduction of the promoter activity of the F7 gene [16].
The intra-familial comparison between the phenotype of the proband and the one of her two sons allowed us to establish that the c.1088C>A variant, in heterozygous state, is the most important genetic feature for the reduction of the FVII:C activity, being it associated with 36% of residual activity evaluated by coagulometric assay. Indeed, said residual activity further decreases to 22% when this genetic variant is in compound heterozygosity with the c.-326_-325insCCTATATCC, c.-122T>C and c.1238G>A haplotype.
Therefore, the variant c.1088C>A showed a different expression compared with the c.-326_-325insCCTATATCC, c-122T>C and the c.1238G>A haplotype, mainly contributing into FVII:C activity reduction. However, the observed decrease of FVII:C activity indicates that the combination of two F7 gene alleles provides an additive effect on the dampening of FVII:C activity, even if of no relevant clinical significance [17,18]. Interestingly, the net effect on the FVII:C activity drop due to the c.-326_-325insCCTATATCC, c.-122T>C and the c.1238G>A haplotype was greater than expected (39% vs 25-30%), making difficult the prediction of their combined effect.
Despite its inter-individual variability, the might of this study is represented by the simultaneous analysis of all the family members performed by homogeneous analytical protocols in order to achieve a better establishment of the variant contributory effects on FVII:C activity. In conclusion, we report for the first time the combination of the c.1088C>A variant with the most common c.-326_-325insCCTATATCC, c.-122T>C, c.1238G>A haplotype and describe their, never analyzed before, impact on FVII:C activity through an additive model that may contribute to better classify the observed clinical phenotype.
The authors would like to express their gratitude to the proband and to her entire family for their availability towards the realization of this study.
The authors declare no conflicts of interest.
Study concept and design: G. Miolo; sample processing and measurements: G. Tessitori and A. Percesepe; processing and interpretation of data: G. Miolo, G. Tessitori and A. Percesepe; drafting of the manuscript: G. Miolo; critical revision of the manuscript: L. Caggiari, M. De Zorzi, M. Tedeschi, D. A. Santeufemia, A. Steffan, V. De Re and G. Corona.
- Giansily-Blaizot M, Aguilar-Martinez P, Biron-Andreani C, Jeanjean P, Igual H, et al. (2001) Analysis of the genotypes and phenotypes of 37 unrelated patients with inherited factor VII deficiency. Eur J Hum Genet 9: 105-112.
- Perry DJ (2002) Factor VII Deficiency. Br J Haematol 118: 689-700. [Crossref]
- Sevenet PO, Kaczor DA, Depasse F (2017) Factor VII Deficiency: From basics to clinical laboratory diagnosis and patient management. Off J Int Acad Clin Appl Thromb 23: 703-710.
- Giansily-Blaizot M, Verdier R, Biron-Adréani C, Schved JF, Bertrand MA, et al. (2004) Analysis of biological phenotypes from 42 patients with inherited factor VII deficiency: can biological tests predict the bleeding risk? Haematologica 89: 704-709.
- Siboni SM, Biguzzi E, Mistretta C, Garagiola I, Peyvandi F (2015) Long-term prophylaxis in severe factor VII deficiency. Haemophilia 21: 812-819. [Crossref]
- Mannucci PM, Duga S, Peyvandi F (2004) Recessively inherited coagulation disorders. Blood 104: 1243-1252.
- Herrmann FH, Wulff K, Strey R, Siegemund A, Astermark J, et al. (2008) Variability of clinical manifestation of factor VII-deficiency in homozygous and heterozygous subjects of the European F7 gene mutation A294V. Haematologica 93: 1273-1275.
- Wulff K, Herrmann FH (2000) Twenty-two novel mutations of the factor VII gene in factor VII deficiency. Hum Mutat 15: 489-496. [Crossref]
- Mariani G, Bernardi F (2009) Factor VII Deficiency. Semin Thromb Hemost 35: 400-406. [Crossref]
- Bernardi F, Marchetti G, Pinotti M, Arcieri P, Baroncini C, et al. (1996) Factor VII gene polymorphisms contribute about one third of the factor VII level variation in plasma. Arterioscler Thromb Vasc Biol 16: 72-76.
- Girelli D, Russo C, Ferraresi P, Olivieri O, Pinotti M, et al. (2000) Polymorphisms in the factor VII gene and the risk of myocardial infarction in patients with coronary artery disease. N Engl J Med 343: 774-780.
- Hunault M, Arbini AA, Lopaciuk S, Carew JA, Bauer KA (1997) The Arg353Gln polymorphism reduces the level of coagulation factor VII. In vivo and in vitro studies. Arterioscler Thromb Vasc Biol 17: 2825-2829.
- Ken-Dror G, Drenos F, Humphries SE, Talmud PJ, Hingorani AD, et al. (2010) Haplotype and genotype effects of the F7 gene on circulating factor VII, coagulation activation markers and incident coronary heart disease in UK men. J Thromb Haemost 8: 2394-2403.
- Mtiraoui N, Aboud N, Bouraoui H, Haizem S, Gris JC, et al. (2005) Reduction in coagulation factor VII plasma levels by R353Q but not the -323P0/10 promoter polymorphism in healthy Tunisians. Am J Hematol 79: 11-16.
- Rabelo FY, Jardim LL, Landau MB, Gadelha T, Corrêa MFB, et al. (2015) The molecular basis of low activity levels of coagulation factor VII: a Brazilian cohort. Off J World Fed Hemoph 21: 670-680.
- Sabater-Lleal M, Chillón M, Howard TE, Gil E, Almasy L, et al. (2007) Functional analysis of the genetic variability in the F7 gene promoter. Atherosclerosis 195: 262-268.
- Hahn LW, Ritchie MD, Moore JH (2003) Multifactor dimensionality reduction software for detecting gene-gene and gene-environment interactions. Bioinforma Oxf Engl 19: 376-382.
- Moore JH (2003) The ubiquitous nature of epistasis in determining susceptibility to common human diseases. Hum Hered 56: 73-82.