A 67-year-old female patient had been suffering for a year from such a severe pain syndrome that she could no longer walk. Although she had presented herself to several doctors from different disciplines, the cause of her illness was not identified. It was osteomalacia caused by a drug. This was not recognized, although the disease is common.
The aim of this article is to raise awareness of a common bone disease. In contrast to osteoporosis, this disease is very accessible to therapy. The differential diagnosis to osteoporosis and the different causes of osteomalacia, hypophosphatemia and Fanconi syndrome are explained in detail.
With the correct diagnosis and consequent treatment, the patient could be helped in such a way that she could even walk again without a walking aid. Overlooking phosphaturia can cause severe skeletal consequences.
osteomalacia, osteoporosis, hypophosphatemia, Fanconi, antiviral therapy
In the medical community bone disease is usually referred to as osteoporosis, especially if the bone mineral density has been measured as low. Thereafter, a therapy with a bisphosphonate is often started without reflection. However, some patients do not benefit from the therapy. Nephrologists with a deep understanding of bone and electrolyte disorders can be of tremendous help for these patients.
A 67-year-old female patient presented with a severe whole-body pain syndrome, in rest and especially in motion. Pain was dull and could not be localized to a single joint or bone or part of the body. The intensity of pain was 10/10 on the visual analogue scale despite the intake of 3 x 0.5 g Metamizol per day. The pain sensations had started about a year earlier and had increased continuously. During the last 8 months the patient was almost unable to walk.
Because of her complaints, the patient presented herself to numerous doctors from various disciplines. The diagnoses ranged from osteoporosis and arthrosis to suspected tumor disease. In the bone scintigraphy, in addition to an extreme accumulation in the area of both knees, a spontaneous fracture of the anterior pubic branch and the ribs 3 to 7 on the right, a pelvic fracture of the massae laterales on both sides was detected. As the bone density was reduced to T- 3.6, the patient was diagnosed with osteoporosis and therapy with alendronate 70 mg per week was initiated. However, an improvement of the pain was not observed within 6 months. Because of unbearable knee pain, a neurolysis therapy [1] was then carried out on the right.
The patient was diagnosed with a chronic hepatitis B infection, which had been known since 1981, and was treated with adefovir from 2004 to 2011 and then with tenofovir 245 mg once a day from 2011 to 2018 due to a temporary deterioration of the kidney values.
In the current clinical examination, a relatively good mobility of the arms compared to the painful lower extremity was noticeable. A hollow back, pain with thoracic compression and painful limitation of movement of the hip and knee joints were observed.
The Menell sign was positive on both sides, the Lasegue sign on the left. She had acquired genua vara with an intercondylar distance of 8 cm, and a knee joint contracture of 10 degrees on both sides.. Upon specific questioning, a decrease in height from 161 to 152 cm was recorded.
The laboratory values are listed in Table 1. Subsequently, a bone biopsy (Figure. 1) and a kidney biopsy (Figure. 2) were performed. These showed iatrogenic induced osteomalacia and a tubulopathy, respectively.

Figure 1. Histology of iliac crest biopsy: Mineralized bone tissue (green) and non-mineralized osteoid (red) in significantly increased and widened margins. Undecalcified preparation, plastic embedding, hard cut technique, Goldner staining. Original magnification x 100
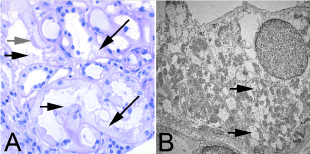
Figure 2. Histology of the kidney biopsy: A. Light microscopically potentially reversible tubule damage with defects (short black arrows) of the brush border typical for proximal tubules (grey arrow) and vacuumization of the cytoplasm of both proximal (lower black arrow) and distal tubules (upper black arrow). Periodic acid-ship paraffin histology, original magnification x 400
B. Ultrastructurally in the epithelium of distal tubules mitochondria with reduced cristae content (arrows). Transmission electron microscopy, original magnification x 6000.
Table 1. Laboratory values of the patient
Basic osteological laboratory tests |
Laboratory values of the patient |
Normal value |
Calcium |
2.23 |
2.20-2.55 (mmol/l) |
Phosphate |
0.73 |
0.84-1.45 (mmol/l) |
Alkaline phosphatase |
265 |
35-105 (U/l) |
Creatinine |
1.22 |
0.51-0.95 (mg/dl) |
Electrophoresis |
43.3 (albumin) |
40-47 (g/l) |
Blood count |
11.4 (hemoglobin) |
11.9-14.6 (g/dl) |
Erythrocyte sedimentation rate |
28/58 |
<30 (mm/h) |
TSH |
0.803 |
0.27-4.2 (mU/ml) |
Special osteological laboratory tests |
|
|
25-Cholecalciferol |
53.7 |
>40 (ng/ml) |
1,25-Cholecalciferol |
62.4 |
25-86 (pg/ml) |
FGF 23 |
30 |
26-110 (kU/l) |
Parathormone |
38.6 |
15-65 (pg/ml) |
Other laboratory tests |
|
|
Blood glucose |
98 |
82-115 (mg/dl) |
Uric acid |
1.6 |
2.4-5.7 (mg/dl) |
Immunofixation |
regular |
|
Ceruloplasmin |
regular |
|
Cystine in leucocytes |
regular |
|
Spontaneous urine |
|
|
Urine dip stic |
blood -, protein (+), glucose +++ |
|
Urine calcium |
1.2 |
2.5-10 (mmol/l) |
Urine phosphate |
8.7 |
12.9-43.9 (mmol/l) |
Urine creatinine |
31.9 |
28-217 (mg/dl) |
Urine uric acid |
19.6 |
37-92 (mg/dl) |
Collected urine (2250 ml) |
|
|
Phosphate excretion |
19.6 |
13-42 (mmol/d) |
Calcium excretion |
2.7 |
2.5-8 (mmol/d) |
Uric acid excretion |
441 |
200-1000 (mg/d) |
Hepatitis B laboratory tests |
|
|
| |
HBsAg +, HBeAg -, anti-HBs -, anti-HBe +, anti-HBc +, HBV DNA 78 IU/ml |
|
A sufficient availability of calcium and phosphate is necessary for both bone growth in adolescents and bone remodeling in adults. If one of the two substances is not available in sufficient quantities, or if their storage in the bone is disturbed, one speaks of rickets in the case of the growing skeleton (open epiphysis) and osteomalacia in the case of the adult skeleton (closed epiphysis).
In both cases, the basic bone substance osteoid is formed normally, often even excessively, but is then not sufficiently mineralized with hydroxyapatite (Ca5(OH(PO4))3). The pressure stability of the bone is reduced, it "softens" and becomes susceptible to painful bending and fractures.
Prevalence
Rickets is often remembered as a post-war disease. However, it must be assumed that both rickets [2] and osteomalacia [3] are very common even today. Post-mortem, osteomalacia has been detected in 25% of adult Europeans by iliac crest biopsy. The true prevalence could be even higher. After osteoporosis, osteomalacia is probably the second most common bone disease.
Diagnostics
Osteomalacia can be diagnosed when 4 out of 5 criteria are met [4]:
- hypophosphatemia or hypocalcemia
- elevated alkaline phosphatase
- muscle or bone pain
- low bone density (always!)
- multiple uptake in bone scan or Looser remodelling zones in X-ray.
Symptoms
Bone pain and muscle weakness are the clinically prominent features of osteomalacia. The symptoms are so unspecific that it is impossible to diagnose osteomalacia on this basis alone. But the clinical symptoms are progressive. In the absence of available building materials, healthy, mineralized bone is continuously replaced by unmineralized bone as part of the normal, permanent bone remodelling process.
Therefore, osteomalacia is not a "clear cut" diagnosis. It is rather a continuum of:
- Early stage: calcium (vitamin D) or phosphate deficiency but already histological changes.
- manifest osteomalacia: positive biochemical markers.
- Late stage: pain, bone deformation, fractures and radiological changes of the skeleton.
Differential diagnosis to osteoporosis
Similar to osteoporosis, osteomalacia has low bone density, bone pain and fractures. For this reason, osteomalacia is often misdiagnosed and treated incorrectly as osteoporosis.
In contrast to osteoporosis, for which there is no specific laboratory marker, osteomalacia (and other bone diseases) can be identified in the "bone laboratory" by the increased alkaline phosphatase. Osteoporosis, on the other hand, is a diagnosis of exclusion.
The reflexive equation of low bone density with the diagnosis of osteoporosis leads to false diagnoses. This error is so common that patients with osteomalacia have been inadvertently included even in large osteoporosis studies [5]. Since patients with osteomalacia, in contrast to osteoporosis, can be treated very well with vitamin D, frequently cited study results led to the erroneous assumption that osteoporosis could be treated with vitamin D [5,6]. Based on these data, even the authors of the earlier DVO S3 guideline of 2003 were forced to recommend vitamin D therapy for osteoporosis with evidence class 1a [7]. The revised guidelines of 2017 have recognized this and recommend vitamin D therapy for osteoporosis only in cases of vitamin D deficiency.
The efficacy of vitamin D administration in osteoporosis has recently even been completely questioned in a meta-analysis of 53,537 patients [8].
Our patient suffered from an unclear pain syndrome. The pain and spontaneous fractures, in conjunction with the significantly reduced bone density, had also led to the misdiagnosis of osteoporosis. The oral bisphosphonate therapy with alendronate caused further iatrogenic deterioration.
The patient also showed signs of a pathological "bone laboratory" (calcium, phosphate, alkaline phosphatase, creatinine, protein electrophoresis, blood count). Since both calcium and phosphate were reduced and alkaline phosphatase increased, it could not be osteoporosis.
The complaints with whole-body pain and the increased alkaline phosphatase should have pointed to a diagnosis of osteomalacia in our patient. Which was finally confirmed in the bone biopsy (Figure. 1).
Gold standard
The gold standard for diagnosing osteomalacia is a bone biopsy. The histology showed the triad of thickened osteoid border (>12.5 µm), increased osteoid volume (>10 %) and in case of a tetracycline double marking a small distance between the tetracycline bands (<0.15 µm/day). In recent years bone biopsies have been performed only rarely. However, the method has wrongly fallen out of fashion [9]. Especially since the histological changes occur before the laboratory chemical ones and since histology has a greater sensitivity for osteomalacia than the determination of calcium, phosphate and alkaline phosphatase [10].
Causes of osteomalacia
The histologically uniform picture of osteomalacia can be based on a variety of causes. For therapy, however, it is essential to classify the osteomalacia into the calcipenic or phosphopenic forms (Table 2).
Table 2. Cause of Osteomalacia
Calcipenic osteomalacia |
Phosphopenic osteomalacia |
Direct osteomalacia by altered osteoid |
- 25-Cholecalciferol deficiency |
- Mutation of the PHEX gene (phosphate regulating endopeptidase on the X chromosome)
(=X-chromosomal hypophosphatemic rickets, formerly called dominant form of Vitamin D resistant rickets). |
- Axial osteomalacia |
Dietary, malabsorption in bariatric surgery, small intestine disease or pancreatic insufficiency, inadequate sun exposure, low 25-hydroxylation in liver cirrhosis, increased breakdown by anticonvulsants, increased loss in nephrotic syndrome |
- Mutation of the Dentin Matrix Protein 1
(=autosomal recessive hypophosphatemic rickets) |
- Fibrogenesis imperfecta |
- 1,25-Cholecalciferol deficiency |
- Mutation of the Endonucleotide Pyrophosphatase / Phosphodiesterase 1 (ENPP1)
(=autosomal recessive hypophosphatemic rickets) |
- Mineralization inhibitors |
Kidney disease |
- Oncogenic osteomalacia
(increased FGF 23 production) |
Pyrophosphate (Hypophosphatasia) |
1-alpha-hydroxylase defect
(=Vitamin D resistant rickets type 1) |
- Proximal renal tubular acidosis (type 2) |
Fluorine
(Fluorosis) |
Vitamin D receptor mutation
(=Vitamin D resistant rickets type 2) |
- Disturbed phosphate reabsorption |
Aluminum |
| |
- Fanconi syndrome multiple causes |
Ethidronate |
Calcipenic osteomalacia
The majority of osteomalacia bone diseases are caused by a calcium-dependent cause with too little exposure to sunlight and/or insufficient vitamin D intake in the diet. In these cases, the 25-Cholecalciferol levels are below 15 ng/ml.
It is important to mention that a deficiency of calcium stock/supply cannot be detected by determining the calcium concentration. This can only be done with an increased detection of parathormone. However, the elevated parathormone levels should not lead to the assumption that parathormone is the cause of osteomalacia.
Since our patient's parathormone, 25-Cholecalciferol and 1,25-Cholecalciferol were within the normal range, the frequent calcipenic osteomalacia was ruled out. Due to the low serum phosphate, our patient has been diagnosed with a phosphopenic form of osteomalacia.
Phosphopenic osteomalacia
Serum phosphate is regulated by the hormone fibroblast growth factor 23 (FGF23). Physiologically, FGF23 is synthesized in the osteocyte when serum phosphate in the osteocyte is too high. This hormone causes an increased phosphate excretion in the kidney until the phosphate concentration in the serum is regulated again. In pathological low phosphate, high FGF23 values indicate an FGF23 induced cause, low FGF23 values indicate a phosphate leakage due to damage of the phosphate transporter in the kidney.
In our patient FGF23 was rather low, so that a kidney damage could be assumed (Figure 3).
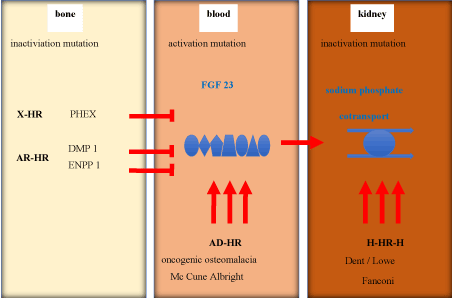
Figure 3. Differential diagnosis of Hypophosphatemia
HR: hypophosphatemic rachitis; X-HR: X chromosomal-HR; AD-HR: autosomal dominant-HR; H-HR-H: hereditary-HR-hypocalciuria; PHEX: Phosphate regulating Endopeptidase X-chromosome; DMP 1: Dentin Matrix Phosphoprotein 1; ENPP 1: Ectonucleotid Pyrophosphatase/Phosphodiesterase 1
Osteomalacia due to kidney damage
Usually the marker for kidney disease is creatinine or the glomerular filtration rate calculated from it. However, isolated tubular kidney disease may well be associated with normal or almost normal serum creatinine.
In our patient, three indications pointed to kidney disease: low uric acid, glucosuria, and phosphaturia.
A low uric acid level may indicate tubular kidney disease. Uric acid is freely filtered in the glomerulus as a small molecule with a molecular weight of 168 g/mol in the kidney and reabsorbed in the proximal tubule (URAT1 transporter). Normally only about 10% of the filtered uric acid is excreted in the urine (norm fractional uric acid excretion=7-12%). In the case of a proximal tubular disorder, this percentage increases, resulting in a decreased uric acid level in the serum.
It is important that - as in our patient - the uric acid concentration and the amount of uric acid in the urine is normal or even reduced, although there is a renal uric acid loss. A loss of uric acid via the kidney must therefore be determined on the basis of fractional uric acid excretion in spontaneous urine, which at 53 % clearly indicated tubular damage.
Glucosuria
Urine strip test can only discover tubular kidney disease if attention is paid to the discrepancy between the detection of urine glucose and normal blood glucose. Glucose, with a molecular mass of 180 g/mol, is filtered in the glomerulus and completely reabsorbed in the proximal tubule, so that normal urine is glucose-free. Glucosuria +++ with normal blood glucose levels is therefore highly suspicious for damage to the kidney, especially the proximal tubule.
Phosphaturia
A low phosphate level in serum (Table 3) combined with increased phosphate excretion is often another indicator for tubular kidney disease. However, it is rarely possible to detect a loss of phosphate via the kidneys by looking only on the measured phosphate concentration in spontaneous urine or on the phosphate amount in the collected urine. Both urine parameters are often low or normal despite renal phosphate loss. The concentration of phosphate in urine depends on this: concentration of phosphate in serum and the amount drunk.
Table 3. Causes of Hypophosphatemia
Reduced supply or reduced intestinal absorption |
Antacids, phosphate binders |
Steatorrhoea/ Diarrhea |
Vitamin D deficiency |
Vitamin D resistance |
Internal redistribution |
Insulin |
Respiratory alkalosis |
Hungry bone syndrome |
Increased renal excretion |
(a) driven by a circulating factor: |
Hyperparathyroidism |
FGF23 overproduction in oncogenic osteomalacia or intravenous iron therapy or Mc Cune Albright |
Mutation in the gene for Dentin Matrix Protein 1 (DMP1) |
Mutation in the gene for Endonucleotide Pyrophosphatase/Phosphodiesterase 1 (ENPP1) |
(b) damage to phosphate transport in the kidney: |
Congenital by mutation in the gene for: |
Sodium Phosphate Cotransporter Type 2c (SLC34A3) |
Sodium Phosphate Cotransporter Type 2a (SLC34A1) |
Sodium Proton Antiport Regulatory Factor 1 (NHERF1) |
Chloride Proton Antiport (CLCN5) Dent syndrome |
Oculo-cerebro-renal syndrome (OCRL1) Lowe syndrome |
Acquired |
Fanconi Syndrome |
Drugs: acetazolamide, cis-platinum, ifosfamide, imatinib, temsirolimus, sorafenib |
A renal phosphate loss is often only recognized by the fractional phosphate excretion in spontaneous urine (norm: 5-20 %) or better by the maximum phosphate reabsorption (norm: 0.8 to 1.35 mmol/l). These values can be easily calculated in spontaneous urine.
In our patient, the urine phosphate concentration was low and the amount of phosphate excreted in the 24-hour collected urine was normal. The fractional phosphate excretion was increased 48% and the maximum phosphate reabsorption was significantly lowered by 0.36 mmol/l. Thus, the hypophosphatemic cause of osteomalacia in our patient was proven by a renal phosphate loss. The renal tubular damage was then confirmed by the renal biopsy (Figure. 2). The latter showed strikingly swollen mitochondria in the tubule cells.
Differential diagnosis Fanconi syndrome
The swollen mitochondria - "giant mitochondria"- are an indicator of mitochondrial damage. The failure of the ATP synthesis in the mitochondria then leads to the failure of the energy supply for the active sodium pumps at the basolateral blood-side membrane (Na-K-ATPase). In a chain reaction, all passive sodium-driven transport systems on the opposite apical, urine-side membrane (sodium/phosphate, sodium/glucose, sodium/amino acid pumps) also fail. This syndrome was named after the first describer Fanconi [11].
Fanconi syndrome can have several causes (Table 4).
Table 4. Causes of Fanconi syndrome
Congenital |
Acquired |
Dent’s disease (nephrocalcinosis, kidney stone, osteomalacia) |
Monoclonal Gammopathy |
Lowe syndrome (oculo-cerebro-renal syndrome) |
Heavy metal poisoning (lead, mercury, cadmium) |
Wilson's disease (copper storage disease) |
Drugs (aminoglycosides, cisplatin, ifosfamide, retroviral medication) |
Cystinosis (cystine storage disease) |
|
Tyrosinemia |
|
Galactosemia |
|
Hereditary fructose intolerance |
|
Mitochondriopathy |
|
A congenital form of Fanconi Syndrome could almost be excluded due to the age of the patient. Late forms of copper or cystine storage disease are possible, if the toxic accumulation of copper or cystine occured late in life. However, both diseases were unlikely due to the kidney histology and could be excluded with a normal ceruloplasmin and cystine concentration in the leukocytes respectively. Since immunoelectrophoretically also a monoclonal gammopathy could be excluded as cause, a drug-induced tubular damage was obvious after exclusion of all other causes.
Drug-induced Fanconi syndrome
In fact, there are numerous reports of tubular disorder associated with tenofovir taken by the patient. Tenofovir induces ATP deficiency in the kidney via mitochondrial damage, which leads to apoptosis of human proximal tubule cells [12]. Since not only tenofovir can trigger this renal damage, this complication must be taken into account in all patients receiving antiretroviral therapy for hepatitis B or HIV [13]. The finding that tenofovir may cause Fanconi syndrome is not new. Since the first description by Verhelst and colleagues [14] even a prospective study has been performed [15]. The incidence is considered to be between 1 to 5 per 1000 treatment years depending on other co-administered drugs [16]. However, Fanconi syndrome is easily and often overlooked since serum creatinine and glomerular filtration rate may be no sensitive markers of tubular dysfunction and the specific markers, such as urine phosphate, urine glucose and aminoaciduria are rarely determined. Awareness is necessary to discover a Fanconi syndrome even among nephrologists. Furthermore, the fact that phosphaturia can cause such a severe skeletal damage is largely unknown in the medical community.
Tenofovir had been administered daily to the patient since 2014. This had resulted in a decrease in viral load, and even in the formation of an antibody against the pathogenetic (virulence-indicating) e-antigen of the hepatitis B virus, as well as normalization of liver parameters. However, the antiviral therapy was not discontinued. The AWMF guideline [17] does not mention criteria for stopping therapy.
The following diagnosis was made in the patient:
- Tenofovir induced Fanconi syndrome
- Fanconi syndrome induced hypophosphatemia
- Hypophosphatemia induced osteomalacia
- Osteomalacia induced genua valga, resulting in incorrect loading with consecutive knee joint arthrosis and fracture of the tibial plateau on both sides
- Fracture of the massae laterales on both sides
- Rib fractures
- Pubic bone branch fracture
- Hollow round back
- Pain syndrome
Therapy
In adults, calcium-dependent osteomalacia is usually treated with vitamin D (10,000 IU/d) and calcium substitution (1000 mg/d). In the rare case of 1.25 OH cholecalciferol deficiency also with active D-hormone (calcitriol 0.25 µg/d).
The therapy of phosphate-dependent osteomalacia is usually carried out with phosphate substitution (1-3 g/d). In order to increase phosphate absorption in the intestine, calcitriol (1 µg/d) can be given at the same time.
In our patient the only therapy consisted of discontinuation of alendronate and tenofovir. A substitution was not necessary. Three months later the Fanconi syndrome had completely regressed.
Immediately after discontinuation of tenofovir, the hepatitis B viral load rose briefly from 78 to 1857 U/ml and then fell back to 174 U/ml three months later. An increase in the transaminases was not observed.
Osteomalacia improved with a reduction of the alkaline phosphatase from 265 U/L to 157 U/l. Within a single year, the bone density increased by 20% in the lumbar spine and by 17% in the femoral neck. The walking speed increased from walking not possible to 1.32 m/s. The pain improved from 10/10 to 4/10 in the VAS score.
However, the bony deformities remained.
Maintaining antiviral therapy for chronic hepatitis B, which is no longer absolutely necessary, led to kidney damage. However, excretory function was well maintained, and the tubular dysfunction was not recognizable through the serum creatinine, but only through the low uric acid and glucosuria. The accompanying unrecognized loss of phosphate caused by the kidney damage then led to the development of a mineralization disorder of the bone via hypophosphatemia. The osteomalacia softening of the skeleton then finally led to the bending of the bones, fractures, knee joint arthrosis and pain.
- Even low lab results may indicate disease.
- A normal serum creatinine does not rule out significant kidney disease.
- A normal urine phosphate concentration does not rule out phosphate loss via the kidneys.
- Not every low bone density is due to osteoporosis.
- Not every osteomalacia is caused by a vitamin D deficiency.
- Antiviral therapy should not be carried out without monitoring of the renal function parameters mentioned above.
- Phosphaturia is not an irrelevant detail. Overlooking phosphaturia can have serious skeletal consequences.
The patient gave informed consent.
Ethical approval statement was not required for this manuscript.
This work was not funded.
None.
- Ahmed A, Arora D (2019) Ultrasound guided neurolysis of six genicular nerves for intractable pain of knee osteoarthritis: a case series. Pain Pract 19: 16-26.
- Thacher TD, Pludowski P, Shaw NJ, Zulf Mughal M, Munns CF, et al. (2016) Nutritional rickets in immigrants and refugee children. Public Health Rev 37: 1-10.
- Priemel M, Domarus C, von Klatte DO, Kessler S, Schlie J, et al. (2010) Bone mineralization defects and vitamin D deficiency: histomorphometric analysis of iliac crest bone biopsies and circulating 25-hydroxyvitamin D in 675 patients. J Bone Mineral Res 25: 305-312.
- Fukumoto S, Ozono K, Michigama T, Minagawa M (2015) Pathogenesis and diagnostic criteria for rickets and osteomalacia – proposal by an expert panel supported by the Ministery of Health, Labour and Welfare, Japan, the Japanese Society for Bone and Mineral Research, and the Japan Endocrine Society. J Bone Mineral Res 33: 467-473.
- Macdonald H, Aspray TJ (2014) Vitamin D supplements and bone mineral density. Lancet 383: 1292.
- Dawson-Hughes B, Harris SS, Dallal GE (1997) Effect of calcium and vitamin D supplementation on bone density in men and women 65 years of age or older. New Engl J Med 337: 670-676.
- DVO S3 Guideline 2003 (2003) Osteologie 2: 54-137.
- Bolland M, Grey A, Avenell A (2018) Effects of vitamin D supplementation on muskuloskeletal health. Lancet Diabetes Endocrinol 6: 847-858.
- Bhan A, Qiu S, Rao S (2018) Bone histometry in the evaluation of osteomalacia. Bone reports 8: 125-134.
- Peach H, Compston J, Vedi S, Horton L (1982) Value of plasma calcium, phosphate and alkaline phosphatase measurements in the diagnosis of histological osteomalacia. J Clin Pathol 35: 625-630.
- Fanconi G (1936) The early infantile nephrotic-glycosuric dwarfism with hypophosphatemic rickets. Pediatrics yearbook 147: 299-338.
- Murphy R, Stafford R, Petrasovits B, Boone M, Valentovic M (2017) Establishment of HK-2 cells as a relevant model to study tenofovir –induced cytotoxicity. Int J Mol Sci 18: 531-546.
- McLaughlin M, Guerrero BS, Merker A (2018) Renal effects of non-tenofovir antiretroviral therapy in patients living with HIV. Drugs in Context 7: 212519.
- Verhelst D, Monge M, Meynard J-L, Fouqueray B, Mougenot B, et al. (2002) Fanconi syndrome and renal failure induced by tenofovir: a first case report. Am J Kidney Dis 40: 1331-1333.
- Gupta S, Anderson A, Ebrahimi R, Fralich T, Graham H, et al. (2014) Fanconi Syndrome accompanied by renal function decline with tenofovir disoproxil fumarate: A prospective, case control study of predictors and resolution in HIV-infected patients. Plos 0ne 9: e92717.
- Medland N, Chow, E, Walker R, Chen M, Read T, et al. (2018) Incidence of renal fanconi Syndrome taking antireroviral therapy including tenofovir disoproxil fumarate. Int J STDAIDS 29: 227-236.
- AWMF Leitlinie Hepatitis-B-Virusinfektion. www.awmf.org/leitlinien/detail/II/021-011.html