Abstract
Cardiomyopathies are a major cause of heart failure and one of the leading indication for cardiac transplantation. Hypertrophic cardiomyopathy (HCM) is the second most frequently encountered phenotype of cardiomyopathy in clinical practice after dilated cardiomyopathy (DCM). Early identification and prompt treatment of patients with HCM at increased risk of death is imperative in clinical management. Thus, this article aggregates available research-based evidence to provide a comprehensive review of HCM. The goal is to advance the knowledge and understanding of HCM and improve its diagnosis and management. The review is organized into eleven main sections. The initial sections introduce and define HCM, review forms of HCM, its epidemiology (risk and prevalence rates in defined populations), clinical signs and symptoms, prognosis and risk markers. The latter sections review the pathophysiology of HCM, the etiologic agents, differential diagnosis and clinical management of HCM. The review also includes a meta-analysis of diagnostic and clinical management methods.
Key words
cardiomyopathy, myocardium
Introduction
Teare made the seminal description of hypertrophic cardiomyopathy (HCM) as a clinical entity in 1958 in an article titled “Asymmetrical Hypertrophy of the Heart in Young adults”. Teare described asymmetric HCM of the interventricular septum in eight patients aged between 14 and 44 years [1]. Although a citation classic, McKenna and Sen-Chowdhry [2] report that Teare’s study was not the earliest historic description of HCM. The two trace the earliest clinical suggestion of HCM to two French pathologists’ description of asymmetric hypertrophy in interventricular septum in 1869 and later defined as a myocardial disease by Krehl in 1891 [2]. Over the past decades, extensive studies have examined HCM as a myocardial disorder and have provided cumulative findings and knowledge into phenotypes, pathogenesis and diagnosis of HCM [3]. The trend of recent studies have shifted from focusing on genetic etiology to novel genes and genetic mutations and recently to mutation distribution in diseased population [4].
Clinical definition of HCM
The American College of Cardiology Foundation (ACCF) and American Heart Association (AHA) Task Force on Practice Guidelines provide a widely cited clinicopathologic definition of HCM. The guidelines define HCM as a cardiac disorder marked by unexplained left ventricular (LV) hypertrophy (thickening) associated with non-dilated ventricular chambers in the absence of other cardiac or systemic disorders in sufficient amounts capable of producing LV hypertrophy. The definition also recognizes that genotype-positive patients could be phenotypically negative without overt hypertrophy [5]. The European Society of Cardiology (ESC) working group on myocardial and pericardial diseases also provides another widely cited definition of HCM. The ESC defines HCM as “the presence of increased left ventricular (LV) wall thickness that is not solely explained by flow-limiting coronary artery disease (CAD) or abnormal loading conditions” [6]. The definitions by ACCF/AHA and ESC establish the hallmark of HCM is LV hypertrophy (wall thickness) in the interventricular septum. Other characteristic pathological features are myocardial fiber hypertrophy, disorganized myocardial cells, interstitial fibrosis and thickened intra-myocardial coronary vessels [3].
The main classification of HCM are obstructive and non-obstructive HCM. Further, obstructive HCM can be described as sub-aortic and mid-ventricular based on the site of obstruction. On the other hand, non-obstructive HCM can be described as normal systolic function or impaired systolic function [3]. Figure 1 provides an illustration of the main and sub-classifications of HCM.
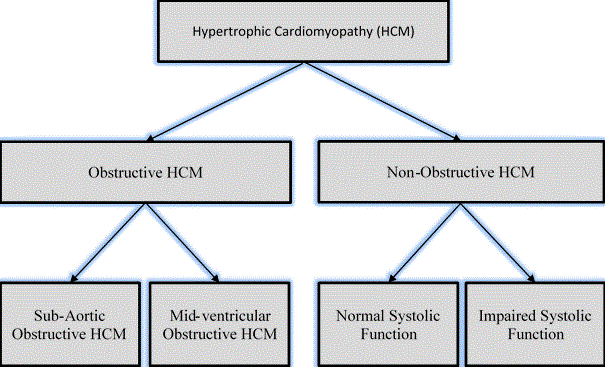
Figure 1: Forms of Hypertrophic Cardiomyopathy [3]
HCM is clinically categorized as obstructive and non-obstructive. The obstructive type is further described as sub-aortic or mid-ventricular based on the site of obstruction while the non-obstructive type is further described based on systolic function as either normal or impaired systolic function. Adapted from Kofflard, 1998, p. 3 [3].
Figure 2 provides a schematic representation of the two types of obstructive HCM: (a) sub-aortic and (b) mid-ventricular obstructive HCM based on the location of obstruction. Systolic opposition of the mitral leaflet against the septum produces sub-aortic obstruction while and muscular opposition in mid-ventricular area produces mid-ventricular obstruction [7].
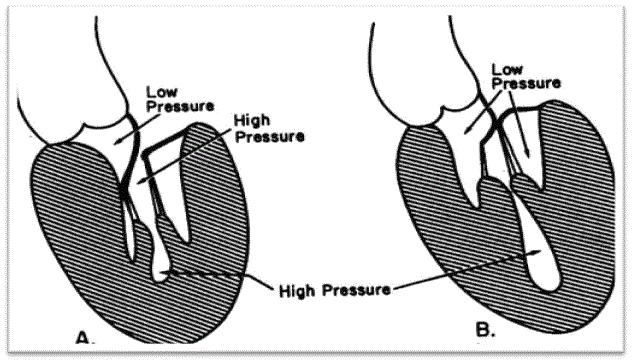
Figure 2: Sub-Aortic (A) and Mid-Ventricular (B) Obstructive HCM
Schematic representation of two possible sites of obstructive HCM. (A) Sub-aortic obstructive HCM produced by apposition of the mitral leaflet against the septum. (B) Mid-ventricular obstructive HCM produced by muscular apposition in the mid-ventricular region. Adapted from Falicov & Resnekov, 1977, p. 703 [7].
Obstructive HCM
Obstructive HCM is a variant of HCM characterized by enlarged cardiac myocytes causing hypertrophy of LV walls. The hypertrophy results into the obstruction of intraventricular blood flow. LV outflow tract (LVOT) narrowing is a characteristic manifestation of obstructive HCM and a frequent source of breathlessness, chest pain and syncope [8]. LVOT narrowing can result from anatomical (reduced cross-sectional LV outflow tract) and hemodynamic factors as summarized in Table 1.
Table 1. Factors Contributing to LV Outflow Tract Obstruction [8]
Causes |
Main Characteristic |
Anatomical Causes |
Basal septal thickness |
Mitral valve displaced anteriorly |
Mitral valve systolic anterior motion |
Abnormal insertion of the papillary muscles |
Elongated mitral valve |
Abnormal cooptation of the mitral valve leaflets |
Hemodynamic Causes |
Rapid early LV ejection |
Venturi forces acting on anterior mitral valve leaflet |
Flow drag |
Adapted from Li, Williams & Rakowski 2015
Sub-aortic obstructive HCM results from a combination of basal septal thickening, anterior displacement of the mitral valve and systolic anterior motion of the mitral valve contributing to LV outflow tract obstruction. Several mechanisms could induce systolic anterior motion of the mitral valve. The first is venturi forces, which results from the narrowing of LV outflow tract and subsequent increase in velocity and a decrease in pressure above the mitral valve causing the valve to displace towards the septum [9]. The second is flow drag. Altered anatomy in the mitral valve apparatus and anterior displaced papillary muscles pushes the mitral valve to protrude in the LV outflow tract exposing it to flow drag [9]. Flow drag leads to gradual development systolic anterior motion of the mitral valve [8].
Falicov and Resnekov [7] were the first to describe mid-ventricular obstructive HCM in 1977. It is rare form of HCM with an hourglass shaped LV cavity and distinct apical chamber [8]. Echocardiographic systolic pressure at the apical chamber is elevated but normal above the mid-ventricular obstruction compared to sub-aortic obstructive HCM where systolic pressure is elevated in the entire LV cavity [10]. Mid-ventricular obstructive HCM also does not present with mitral insufficiency but has greater symptom burden and an indicator of disease progression and end-stage HCM. It is a predictor of HCM-related sudden death and lethal arrhythmias [11,12]. Formation of apical aneurysm in mid-ventricular obstructive HCM is common, indicative of progression to end-stage disease and higher risk of stroke, sudden death or lethal arrhythmias [12].
Non-obstructive HCM is clinically defined as a variant of HCM characterized by the absence of LV outflow tract obstruction, with a gradient < 30 mmHg both under exercise conditions and at rest [8]. Its main classification are HCM with normal or impaired systolic function (end-stage phase). Depressed LV systolic function could result from myocardial ischemia and infarction due to many of non-obstructive HCM patients present with myocardial fibrosis [13]. Patients with non-obstructive HCM present with severe symptoms due to diastolic dysfunction and microvascular ischemia. These symptoms are difficult to treat and manage because of the lack of LV outflow tract obstruction that is usually the treatment target [8].
Apical HCM is another relatively rare variant of non-obstructive HCM characterized by thickening of the myocardium in the left ventricular apex. Very rarely the condition affects the right ventricular (RV) apex or both LV and RV apex [14,15]. Although initially believed to be confined in Japanese populations, apical HCM also affects non-Japanese populations but at very low prevalence rate (3%) compared to Japan (15%) [15]. Typical ventriculography findings associate apical HCM with spade-shaped LV cavity at end-diastole [14]. Many patients with apical HCM have favorable prognosis with annual mortality rate of 0.1%. However, fewer patients present with severe symptoms of diastolic dysfunction due to reduction of LV chamber. About 10% of the patients will present with apical infarction in the absence of coronary artery disease (CAD), which may result in the formation of apical aneurysm [16,17]. Whereas sudden cardiac death is less likely in isolated apical HCM [18], the formation of apical aneurysm is associated with adverse outcomes at 10.5% annual event rate including sudden cardiac death, progressive heart failure and thromboembolic events [17].
The first population-based epidemiologic study of idiopathic HCM [19] was in Olmsted County, Minnesota. It was based on a review of complete medical records of all diagnosed cases within a ten-year period (between 1975 and 1984). Diagnostic methods used were echocardiography, angiography and autopsy analysis. The incidence rates of HCM increased from 1.4 to 3.6 per 100,000 person-years in the first five years of the study. Age and sex-adjusted prevalence rates of HCM at January 1981 was 19.7 per 100,000 population [19]. The landmark Coronary Artery Risk Development in Young Adults (CARDIA) cohort study in 1995 [20] investigated the prevalence of echocardiographic-defined HCM in 4,111 men and women aged 23 to 35 years in urban areas. The overall prevalence rate of HCM was 0.17% (about 1 in 500 people), with a greater prevalence in males 0.26% than females (0.09%) and in blacks (0.24%) than in whites (0.10%) [20].
Two decades later, the prevalence of HCM in the general population is higher as explained by the discovery of its molecular basis and advancement in genetic testing [21]. Recent studies [22,23] have established 11 genes encoding more than 1,500 cardiac sarcomere mutations. Another four studies following the CARDIA study established increased prevalence rates of HCM in different defined populations as summarized in Table 2.
Table 2. Population-based Epidemiologic Studies of HCM Prevalence
1st Author, year [Ref #] |
Sample |
Age |
Male |
Prevalence |
Study Population |
Maron et al. 1995 [20] |
4,111 |
25-35 |
71% |
0.17% |
Urban population |
Maron et al. 1999 [24] |
15,137 |
57 |
48% |
0.19% |
Health examinations |
Maron et al. 2004 [25] |
3,501 |
60 |
50% |
0.23% |
American Indians |
Zou et al. 2004 [26] |
8,080 |
52 |
69% |
0.16% |
9 Chinese communities |
Maro et al. 2006 [27] |
6.680 |
55 |
68% |
0.19% |
East Africa hospitals |
Epidemiology by types of HCM
The epidemiology of obstructive and non-obstructive HCM depends on their clinical definitions [8]. The hallmark of non-obstruction HCM is the lack of LV outflow tract obstruction, indicated by LV outflow tract gradient < 30 mmHg. The prevalence of obstructive HCM (LV outflow tract ≥ 30 mmHg) is 25 to 30% in resting conditions and up to 70% of HCM patients under exercise conditions. Thus, non-obstructive HCM is prevalent in a third (30%) of HCM patients [28]. Sub-aortic obstructive HCM affects about 25% of patients diagnosed with HCM [3] whereas mid-ventricular obstructive HCM is a rare variant of HCM occurring in less than 10% of reported cases of HCM [8]. Apical HCM on the other hand is prevalent in Japan with reported rates of between 13% and 25% of the diagnosed cases of HCM and less than 3% in non-Asian populations [14].
Risk markers
Sudden cardiac death (SCD) is unpredictable in HCM patients. Detecting patients at high risk of developing SCD is important in informing appropriate clinical management. Detection involves identifying risk markers to determine HCM patients at risk of SCD [29].
Established risk markers
Family history of SCD
Family history of HCM-associated sudden cardiac death is a risk marker predicting an increased risk of death for the affected family members. The risk is more pronounced if the incidents of SCD in a family are high and occur at a relatively young age [30].
Syncope
Syncope is the transient and self-limiting loss of consciousness and muscular strength followed with spontaneous recovery [31]. In the absence of other causes, syncope is a significant risk factor for sudden cardiac death in HCM patients. Syncope has a higher risk if it occurs repeatedly, occurs in younger children or occurs with exertion [29].
Non-sustained ventricular tachycardia
Non-sustained ventricular tachycardia (NSVT) is tachycardia lasting < 30 seconds. Although specific evidence suggesting it is an independent risk factor for sudden cardiac death is inconclusive, it considered a marker for increased risk of lethal sustained arrhythmias and sudden cardiac death [29]. Frequent, faster and longer episodes of NSVT suggest increased risk for SCD [31]. For HCM patients aged ≤ 30 years, the predictive value of NSVT for sudden cardiac death increases [32].
Significant lv hypertrophy
Clinically significant LV hypertrophy indicated by wall thickness ≥ 30mm affects about 10% of HCM patients. An Italian study of 480 HCM patients found the risk of SCD has a correlation with LV wall thickness: 0% for ≤ 15mm and 1.8% for ≥ 30mm per year. The incidence of SCD double with every increase of 5 mm in LV wall thickness [33].
Abnormal blood pressure under provocative conditions
LV outflow tract obstruction causes 20-40% of HCM patient fail to augment their blood pressure under exercise provocation. About 15% of HCM patients under provocative or exercise conditions presented with abnormal blood pressure: < 20mm Hg increase in systolic blood pressure or > 20mm Hg decrease from peak value suffer from sudden cardiac death. In comparison, only 3% of those with normal blood pressures suffered sudden cardiac death [29].
Potential risk markers
LV apical aneurysm
The formation of apical aneurysm is a risk marker suggesting progression of HCM and associated risk of cardiac death, lethal arrhythmias or progressive heart failure [17]. Apical aneurysm is however very rare in HCM patients, estimated at 2% in patients with apical or mid-ventricular obstructive HCM. Still, there are reports of incidence rate of sudden cardiac death or heart transplantation therapy at 3.6% annually in apical and mid-ventricular patients [29].
LV outflow tract obstruction
LV outflow tract obstruction is an independent indicator of HCM progression into end-stage disease and a predictor of severe symptoms of heart failure, lethal arrhythmias or sudden cardiac death [35]. However, not all previous HCM studies have not clearly established that significant LV outflow obstruction always has an association with sudden cardiac death, with some studies strongly suggesting the lack of a significant association [36].
Late gadolinium enhancement on cardiac MRI
Late gadolinium enhancement (LGE) on cardiac magnetic resonance imaging (CMRI) quantifies myocardial fibrosis and is associated with elevated risk for sudden cardiac death. Positive predictive power of LGE on CMRI is low because of a great percentage (60%) of all HCM patients have LGE. However, it is the extent, rather than the present of LGE, that indicates the risk of sudden cardiac death but additional studies are needed to define the utility of LGE in predicting sudden cardiac death [37,38].
Clinical presentation
Signs and symptoms
The majority of HCM patients in unselected populations are asymptomatic for many years because of the slow progression of the disease [39]. In these patients, diagnosis is usually incidental during family screening. They live a normal functional life as the rest of the population but about 25% develop symptom or suffer from sudden cardiac death [40]. Patients with untreated obstructive HCM progressively develop multiple symptoms such as exertional angina, dyspnea, syncope, stroke and sudden cardiac death, which may be the first presentation of HCM for a small percentage of patients [3]. HCM-related deaths is often always the end-point of SCD, progressive hearth failure and ischemic stroke occasioned by aneurysm and lethal arrhythmias [29].
Clinical symptoms
The main clinical signs and symptoms of HCM are dependent upon the pathophysiologic processes leading to the development of HCM. Figure 3 presents an illustration of the clinical symptoms and their association with pathophysiologic processes of HCM.
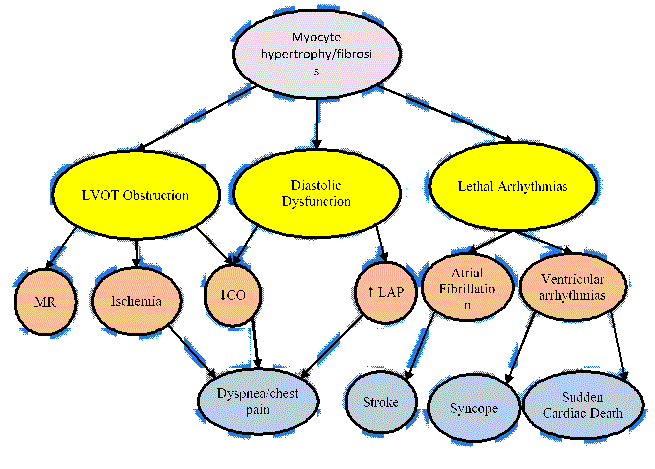
Figure 3: Clinical Symptoms and Associated Pathophysiologic Processes
The pathobiology mechanisms of HCM (LVOT obstruction, diastolic dysfunction and lethal arrhythmias in yellow) are associated with clinical symptoms of HCM: mitral regurgitation, ischemia, reduced cardiac output, increased left atrial pressure, atrial fibrillation and ventricular arrhythmias in green) and dyspnea, stroke, syncope and sudden cardiac death in blue). Adapted from Enriquez & Goldman, 2014, p.36). [29]
MR: Mitral regurgitation; CO: Cardiac Output; LAP: Left Atrial Pressure; LVOT: Left Ventricular Outflow Tract
In Figure 3, green and yellow color-coding represent pathophysiologic processes while blue represents clinical manifestations (signs and symptoms). The interplay of the three established pathophysiologic processes (LVOT obstruction, diastolic dysfunction and lethal arrhythmias), produce hemodynamic changes, which contribute to the symptoms and clinical picture of HCM. The pathophysiologic process of LV outflow tract obstruction and diastolic dysfunction increases LV systolic filling leading to mitral regurgitation and ischemia. Reduced cardiac output and increased LV end-diastolic pressure contribute to complaints of exertional angina and dyspnea. Lethal arrhythmias, such as atrial fibrillation and sustained ventricular arrhythmias contribute to symptoms of stroke, syncope and sudden cardiac death [29]. Since HCM pathophysiologic processes have different contribution to symptoms, clinical understanding of HCM requires clarification of the dominant process to inform the most effective therapeutic intervention [3].
Clinical progression
The clinical progression of HCM occurs in three main pathways: (a) progressive heart failure; (b) sudden cardiac death; and (c) lethal arrhythmias.
Progressive heart failure
Heart failure is a progressive conditioning in which cardiac muscles cannot pump blood in sufficient quantities to meet blood and oxygen demand of body tissues [41]. In the absence of other etiologies, heart failure is one pathway indicating the clinical progression of HCM. Heart failure may present with exertional angina, dyspnea or a combination of the two. The cause of heart failure has been associated with LV outflow tract obstruction and diastolic dysfunction [29].
Sudden cardiac death
Sudden cardiac death is death from cardiac causes that occurs with an hour of the onset of symptoms [42]. In the absence of other causative factors, sudden cardiac death is a significant clinical indicator of the clinical progression of HCM. Microvascular disorders may cause the development of myocardial fibrosis and ischemia and increase the predisposition of HCM patients to lethal arrhythmias especially atrial fibrillation and ventricular arrhythmias. Sudden cardiac death indicates increasing frequency, prolongation or sustenance of these lethal arrhythmias. Although the incidence of sudden cardiac death is 1% annually in HCM patients, it has a devastating effect since it occurs mainly among young asymptomatic HCM patients [29].
Atrial fibrillation
Atrial fibrillation refers to supraventricular arrhythmia with characteristic chaotic atrium contraction [43]. In HCM patients, atrial fibrillation affects 20%, and it is associated with an elevated risk of stroke, sudden cardiac death and heart failure. More than 20% of HCM patients with atrial fibrillation experience life-threatening thromboembolic events [44].
Prognosis
Although HCM is associated with a significant risk of sudden cardiac death, its prognosis has remained poorly or inconsistently defined. Reported annual mortality rates vary widely across studies between 1% and 4% mainly associated with different patient population used in the studies such as patients’ records in hospitals and retrospective studies of health and autopsy records [45]. Symptoms such as LV outflow tract obstruction, syncope and exertional angina, aneurysm and dyspnea indicate poor prognosis and the main predictors of sudden cardiac death [17,29,36]. On the other hand, specific subsets of HCM patients – the elderly, non-obstructive HCM and apical HCM – have shown favorable prognosis as well as have pharmacotherapy and heart transplantation [45]. Despite favorable prognosis for apical HCM, long-term observation reveals occasional incidents of sudden cardiac deaths, lethal arrhythmias and apical aneurysms [46].
Pathophysiology
The pathophysiology of HCM is a complex process marked with an interplay of multiple interrelated cardiac abnormalities. Frequently cited cardiac abnormalities are LV outflow tract obstruction, diastolic dysfunction, mitral regurgitation, myocardial ischemia and severe arrhythmias [47,48].
LV outflow tract obstruction
LV outflow tract obstruction (defined as LVOT gradient ≥ 30 mm Hg) is a well-established clinical feature of obstructive HCM characterized by LV hypertrophy – increased LV mass and thickened LV wall – especially at the intraventricular septum [49]. LV outflow-tract obstruction causes increased LV systolic pressure that is associated with multiple cardiac abnormalities. Frequent cardiac abnormalities include prolonged ventricular relaxation, elevated LV diastolic pressure, mitral regurgitation, decreased cardiac output and myocardial ischemia [6]. LV outflow tract obstruction is usually due to systolic anterior motion (SAM) of the mitral valve and mitral-septal contact. However, recent research-based evidence strongly suggest LV outflow tract obstruction occurs as a result of ventricular systole where blood flow against abnormal position of mitral valve apparatus causes a drag (venturi) forces that pushed mitral valve leaflets into the outflow tract [50,51,53]. LV outflow obstruction is also associated with muscular obstruction, where thickened papillary muscle abuts the septum or insert into the anterior mitral leaflet [53]. LV outflow tract obstruction varies depending on ventricle contraction and loading conditions. Increased LV contraction or reduced ventricular volume correlates with the degree of sub-aortic obstruction. However, at rest, patients have insignificant obstruction but is significant during provocative conditions such as exercise or pharmacologic provocation. LV outflow obstruction predicts HCM-related heart failure symptoms and is a major factor for predicting prognosis and efficacy of clinical management [47,54].
Diastolic dysfunction
Diastolic dysfunction is a major pathophysiologic abnormality in HCM patients. The dysfunction has a significant effect on ventricular relaxation and stiffness. Abnormal ventricular relaxation is due to systolic contraction loading associated with LV outflow tract obstruction, non-uniform ventricular contraction/relaxation, and delayed inactivation [55]. Myocardial ischemia causes and increase in chamber stiffness and affects ventricular relaxation. These alterations are associated with compensatory increase in late diastolic filling. Under exercise provocation, decrease in diastolic filling combined with myocardial ischemia exacerbates decreased diastolic filling causing exertional angina and subsequent dyspnea [6].
Myocardial ischemia
Some HCM patients may develop severe myocardial ischemia with or without infarction that is unrelated to coronary artery disease (CAD). Myocardial ischemia instead develops due to a mismatch between demand and supply. HCM patients irrespective of age may experience an increase in oxygen demand associated with LV hypertrophy and abnormal loading conditions. Some may present with abnormal coronary blood flow to LV myocardium attributed to mid-ventricular hypertrophy and luminal narrowing [56,57].
Mitral regurgitation
Mitral regurgitation is a common clinical feature due to LV outflow tract obstruction and a major contributor to symptoms of exertional dyspnea in HCM patients. It is a secondary pathophysiologic feature in most HCM patients [47]. The displacement of systolic anterior motion of the mitral valve leaflets secondary to LV outflow tract obstruction is the cause of mitral regurgitation whose severity correlates with LV outflow tract obstruction in some patients [47,48]. Lateral and posterior directed mitral regurgitation jet is dominant during mid to late systole. Anterior directed jet on the other hand indicates abnormality of the mitral valve leaflets [58]. Ventricular loading contraction and contraction affecting the degree of LV outflow tract obstruction also affect the extent of mitral regurgitation. Identification of other intrinsic abnormality of mitral valve leaflets is clinically important to inform therapeutic interventions [59].
Etiology
Mutations in the genes encoding cardiac sarcomeric protein is the dominant etiology for HCM in about 60% of teenagers and adults [60]. Other genetic disorders such as inherited metabolic, genetic syndromes, chromosomal disorders and neuromuscular disorders account for 5-10% of HCM in adults [61].
Mutation in the sarcomeric protein
Genetic mutation in beta-myosin heavy chain and myosin binding protein are the dominant genetic etiologies of HCM whereas cardiac troponin, tropomyosin alpha-1 chain and myosin light chain are less frequent genetic etiologies [5]. Mutation in genes encoding sarcomeric proteins present clinical manifestations earlier and associated with higher cases of family history of HCM-related sudden cardiac death compared to HCM without mutations [62,63]. Severe LV hypertrophy, microvascular abnormality and myocardial fibrosis have been associated with HCM patients with genetic etiology of sarcomeric protein mutation [62]. While there are reports of some mutations in sarcomeric proteins having a poor prognosis, these reports are based on small sample of patients with wide inter-study variations and limited by individual genetic mutations [64-67]. In addition, about 5% of HCM patients have HCM with multiple mutations in sarcomeric proteins [68,69]. However, the evidence of prognostic value of different sarcomeric protein mutation will improve as more data continue to be gathered in international databases [5].
Inherited metabolic disorders
Many metabolic disorders associated with LV hypertrophy are inherited in an autosomal recessive pattern with a few in an X-linked pattern [70]. Anderson-Fabry disease is the most frequent inherited metabolic disorders in 0.5 to 1% of HCM patients 35 years or older [71]. Metabolic disorders due to mutation of the adenosine monophosphate-activated protein kinase gene is also prevalent in 1% of HCM patients and Danon disease due to mutation in lysosome-associated membrane protein 2 is prevalent in 0.7-2.7% of HCM patients [72]. Inherited metabolic disorder is a rare genetic etiology of HCM but is associated with a greater prevalence in children and adolescent [5].
Neuromuscular disorders
Neuromuscular disorders is a rare etiology of HCM. However, HCM has been associated with some muscular dystrophies and inherited skeletal myopathies such as nemaline myopathy, and muscle weakness and contractures caused by mutations in in the four-and-half LIM domain-1 [73]. Mutation in the gene encoding desmin protein, which is a cause of dilated and restrictive cardiomyopathies, can also cause atrioventricular block and HCM [5].
Mitochondrial disorders
Inherited mitochondrial disorders causing HCM are in mitochondrial DNA. Their transmission is through heterogeneous patterns: autosomal dominant, autosomal recessive, X-linked or maternal. The dominant mutations for mitochondrial etiologies is the gene encoding the respiratory chain protein. Clinical manifestation of HCM due to mitochondrial disorder varies with age of onset and the extent and severity of the organ affected [5].
Other etiologies
Infiltrative inflammation such as cardiac amyloidosis, which causes wall thickening in LV and RV as well as in inter-atrial septum and atria-ventricular valves [74]. Endocrine disorders such as diabetes may cause ventricular hypertrophy in infants whose mothers are diabetic. In adults, untreated Pheochromocytoma [75] and acromegaly [76] may also cause LV hypertrophy. Chronic use of drugs, such as anabolic steroids and hydroxychloroquine can cause LV hypertrophy. However, drug-related LV hypertrophy is usually ≤ 15 mm [5].
Differential diagnosis
HCM has a diverse etiology, which in the absence of abnormal loading conditions, warrants a systematic diagnosis work-up to detect the specific etiologic agent. The 2014 ECS guidelines on diagnosis and management of HCM developed a standardized diagnostic work-up, which involves laboratory testing, family screening, electrocardiography (ECG), echocardiography (echo), cardiac magnetic resonance imaging (MRI) and genetic testing [6]. Figure 4 provides an illustration of a general approach to diagnostic work-up for HCM developed by the ESC.
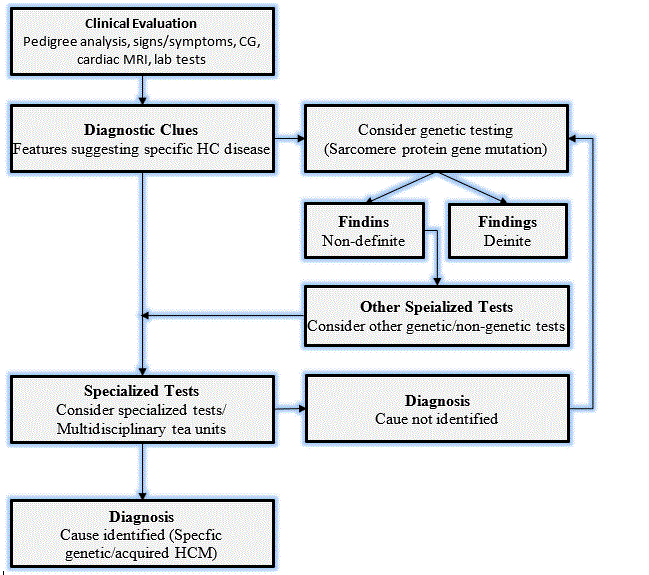
Figure 4: The ECS Recommended Standardized Diagnosis Work-up for HCM [6]
According to the ESC diagnostic guidelines for HCM, the primary diagnostic criteria is the presence of LV hypertrophy defined by LV wall thickness ≥ 15 mm measured by any imaging modality: echo, cardiac MRI, or cardiac CT in the absence of any abnormal loading condition. In some cases, genetic/non-genetic abnormalities present with smaller extent of LV hypertrophy (13 to 14 mm). In these patients, recommended diagnostic procedure should include other tests such as family history, ECG abnormalities, non-cardiac symptoms and multi-modal cardiac MRI [6]. The ECS diagnostic guidelines recommend the importance of differential diagnosis to confirm HCM by excluding dilated/hypokinetic LV or LV wall thinning presenting in the late phase HCM, physiological hypertrophy due to intense exercise, co-existent pathologies and isolated basal septal wall thickening in elderly patients as discussed hereinafter [6].
Family history
Family history provides important information for patients with a diagnostic possibility of HCM, particularly in a family with a history of heart disease at young age [77]. Analysis of family history involves constructing 3-4 generations of family pedigree. The clinical goal is to establish genetic origin of HCM as well as other family members at risk of developing HCM. Pedigree analysis should focus on incidents and age of presentation of sudden cardiac deaths, heart failure, heart transplantation and stroke [6]. Pedigree analysis also provides valuable information on the pattern of inheritance. Most genetic etiologies of HCM have an autosomal dominant inheritance pattern with incidents in every generation and a 50% risk of inheritance to offspring. In the absence of male-to-male transmission and that, males are the most severely affected, suggests X-linked inheritance pattern. When both parents are unaffected and consanguineous suggests autosomal recessive inheritance pattern. Finally, when only female parent transmit HCM, suggests mitochondrial DNA mutation [6].
Physical examination
In most instances, HCM patients are asymptomatic and diagnosis is incidental during screening. Some HCM patients present with symptoms such as exertional angina, dyspnea, syncope and palpitations. In addition to these symptoms, general physical extermination provides information about non-cardiac symptoms useful for specific diagnosis such as syndromic or metabolic etiologies. Table 3 provides a summary of signs and symptoms suggesting specific diagnosis.
Table 3. Signs and Symptoms Suggesting Specific Diagnostic Clues [78]
Signs and Symptoms |
Specific Diagnostic Clues |
Learning difficulties/mental retardation |
Mitochondrial disease, Danon disease, syndromic disease (Noonan/LEOPARD) |
Sensorineural deafness |
Mitochondrial disease with diabetes, Anderson-Fabry disease, LEOPARD syndrome |
Visual impairment |
Mitochondrial disease, Amyloidosis, Danon disease and Anderson-Fabry disease |
Gait disturbance |
Friedreich’s ataxia |
Sensory abnormalities |
Amyloidosis, Anderson-Fabry disease |
Muscle weakness |
Mitochondrial disease, Friedreich’s ataxia, glycogen storage disorders |
Palpebral ptosis |
Mitochondrial disease, Noonan/LEOARD syndrome |
Angiokeratomata, hypohidrosis |
Anderson-Fabry disease |
Electrocardiography
Electrocardiography (ECG) is the recommended initial test for known or suspected HCM at rest and during exercise. It is also recommended when there is a progressive change (worsening) of symptoms in already diagnosed HCM patients. It has been established that ECG abnormalities precede the occurrence of LV hypertrophy in carriers of genetic mutation of HCM [79]. Various combination of ECG abnormalities such as Q-waves, LVH, ST- and T- waves could suggest important diagnostic clues of HCM [80]. Although a sensitive marker of HCM, ECG is non-specific and interpretation of findings should be considered together with findings from echo and cardiac MRI. Table 4 provides a summary of ECG findings suggesting specific diagnosis. Table 4 presents findings of ECG suggesting specific diagnosis of HCM.
Table 4. ECG Findings Suggesting Specific Diagnosis [78]
Signs and Symptoms |
Specific Diagnostic Clues |
Pre-excitation |
Storage disease and mitochondrial disorders |
Short PR without pre-excitation |
Storage diseases (Anderson-Fabry disease) |
AV block |
Mitochondrial disorders, some storage disease (Anderson-Fabry disease), amyloidosis and desminopathies. |
Severe LV hypertrophy
(Sokolow score ≥ 50) |
Storage disease (Pompe and Danon) |
Low QRS voltage |
AL/TR amyloidosis |
Extreme QRS axis deviation |
Syndromic disease (Noonan syndrome) with severe basal hypertrophy. |
Giant negative T-wave >10 mm |
Involvement of LC apex |
Abnormal Q-wave |
Asymmetric LV distribution |
ST segment elevation |
Apical or distal hypertrophy |
Echocardiography
Echocardiography (echo) is an imaging modality used primarily to measure LV wall thickness [81]. Echo is used to measure LV outflow tract obstruction defined by LV outflow tract pressure gradient ≥ 50 mm Hg [82]. 2-D and Doppler echo are used to determine the severity and mechanism of LV outflow tract obstruction. When non-invasive images lack clarity, transoesophageal echo (TEE) or transthoracic echo (TTE) maybe considered in selected patients [5]. Echo is recommended in the initial assessment of all patients suspected with HCM, as a component of family screening, periodic (12-18 months) screening of offspring of HCM patients, and repeat screening for changes in clinical status of HCM patients [5]. Contrast TTE is recommended for assessment of LVOT obstruction when apical HCM/infarction or the severity of LV wall thickness is not known, or when other cardiac imaging modalities such as MRI is unavailable [5]. Table 5 provides a summary of echo findings alongside their diagnostic clues.
Table 5. ECHO Findings Suggesting Specific Diagnosis [78]
Signs and Symptoms |
Specific Diagnostic Clues |
Increased inter-atrial septum thickness |
Amyloidosis |
Increased AV valve thickness |
Amyloidosis, Anderson-Fabry disease |
Increased RV wall thickness |
Amyloidosis, Anderson-Fabry disease, myocarditis, Noonan syndrome |
Mild-moderate pericardial effusion |
Amyloidosis, myocarditis |
Concentric LVH |
Glycogen storage disease, Anderson-Fabry disease |
Severe Concentric LVH (≥ 30 mm) |
Danon disease, Pompe disease |
Global LV hypokinesia |
Mitochondrial disease, Amyloidosis, Danon disease, myocarditis, Anderson-Fabry disease |
RV Obstruction |
Noonan syndrome and associated abnormalities |
Echocardiography findings are able to differentiate different patterns of LV hypertrophy and LVOT gradient and velocity. Figure 5 presents an illustration of echocardiographic findings of key patterns of LV hypertrophy.
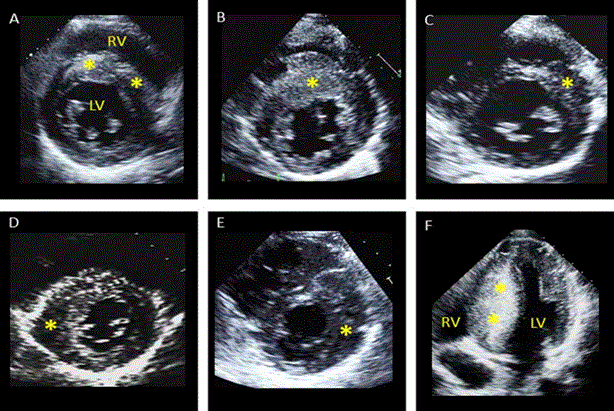
Figure 5: Echocardiographic Images Showing Patterns of LV Hypertrophy [83]
Patterns of LV hypertrophy (*). (A) LV hypertrophy involving the entire basal septum. (B) Marked LV hypertrophy of the anterior septum. (C) LV hypertrophy of the septum-anterior wall junction. (D)LV hypertrophy limited to sepal-posterior wall. (E) Anterior LV free wall thickening. (F) Massive LV hypertrophy of the entire septum from basal to apical regions. Adapted from Wigle et al., 1985, p. E2 [83].
Figure 5 shows six different images of echocardiography from HCM patients with different patterns of LV hypertrophy marked using asterisk symbol (*). The main patterns of LV hypertrophy in HCM include (A) LV hypertrophy affecting the entire basal septum; (B) affecting the anterior septum; (C) affecting septal-anterior wall junction; (D) affecting septal-posterior wall junction; (E) affecting anterior free wall thickening: and (F) affecting the entire septum right from the base to the apex [83]. Figure 6 further illustrates two examples of Doppler recordings of LVOT gradients of two HCM patients at rest and during Valsalva maneuver.
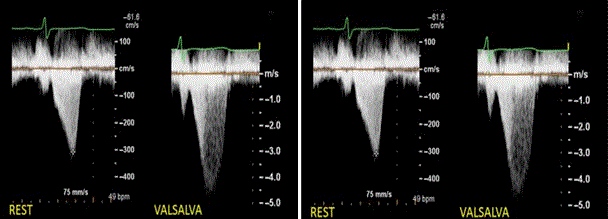
Figure 6: Doppler Recordings of LVOT Gradients of Two Patients.
Doppler recordings of LVOT gradients from two HCM patients. Left panel show only mild gradient at rest in HCM patient and increasing significantly with Valsava maneuver. Right panel shows LVOT velocity in a HCM patient increasing markedly with exercise. Adapted from Wigle et al., 1985, p. E5 [83].
Cardiac magnetic resonance
Several imaging modalities of cardiac magnetic resonance imaging (MRI) provide detailed diagnostic information about cardiac morphology and ventricular function as well as characterize myocardial tissue in HCM patients [84]. According to ACCF/AHA guidelines on diagnosis of HCM, cardiac MRI is indicated for patients with suspected HCM when echo findings are insufficient for a definite diagnosis. It is also recommended for HCM patients when additional information such as the degree of hypertrophy and anatomy is required for clinical management [85]. Cardiac MRI also provides additional information to define apical or aneurysm and suggests the need for alternative diagnosis for underlying disorders such as Anderson-Fabry disease and genetic phenocopies [86,87].
Cardiac MRI is indicated for diagnosis and assessment of concomitant cardiac disorders while cardiac CT is used in the diagnosis of concomitant CAD in HCM patients presenting with CAD, useful for informing changes in therapy. Cardiac-CT angiography is indicated in the assessment of CAD in HCM patients with chest discomfort and a low-likelihood of CAD. Single Positron emission tomography (SPECT) or positron emission tomography (PET) MRI is indicated to rule out CAD in HCM patients with low likelihood of CAD due to its high negative predictive value [5]. Late gadolinium enhancement (LGE) MRI has been shown to correlate with cardiac death, heart failure and all-cause mortality but evidence is inconclusive of its value in risk prediction of sudden cardiac death in HCM patients [37,88,89].
Cardiac MRI is useful in the differential diagnosis of HCM to suggest specific diagnoses based on magnetic properties, and distribution and degree of interstitial expansion. Reduced non-contrast T1 signal and posterolateral LGE suggests Anderson-Fabry disease [87,91]. Global or segmental LGE and specific pattern of myocardial LGE suggests cardiac amyloidosis and absence of fibrosis distinguished HCM from LV hypertrophy due to physiological adaptation of athletes [5,91]. Cardiac MRI images improve the assessment of LV hypertrophy especially in the anterolateral LV free wall [91]. Figure 7 illustrates cardiac MRI images of different severity of LV hypertrophy in HCM patients acquired using 1.5T Sonata, Siemens Medical Systems CMR scanner.
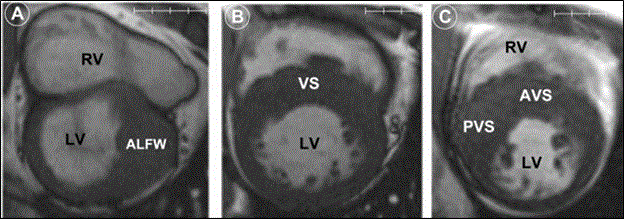
Figure 7: Cardiac MRI Images Showing Patterns of LV Hypertrophy in HCM patients [91].
Figure 7 shows HCM has diverse patters of LV hypertrophy as indicated by cardiac MRI short axis at the end of diastole. Figure 7 (A) shows LV hypertrophy at anterolateral free wall (ALFW) transitioning sharply to normal wall thickness at anterior septum and posterior free wall. (B) Moderate LV hypertrophy at anterior and posterior portions of ventricular septum (VS). (C) Predominant posterior ventricular septal hypertrophy (PVS) that extends to anterior ventricular septum (AVS). Adapted from Rickers et al., 2005, p. 859 [91].
Nuclear imaging
The use of nuclear imaging is not very common but it has been used to assess myocardial blood flow and defects in myocardial perfusion in HCM patients [92,93]. The major contribution of nuclear imaging to diagnosis of HCM is the detection of TTR-induced cardiac amyloidosis. TTR is a plasma transport protein produced in the liver and a precursor in the development of systemic and TTR-related amyloidosis [94,95]. There is growing evidence on the value of TTR-derived fibrils have avidity for bone tracers supporting its consideration for bone scintigraphy for HCM patients suspected with TTR-related amyloidosis. However, there are no evidence of cardiac uptake of tracers in HCM related to sarcomere proteins gene mutation [96]. Nuclear imaging have different modalities: myocardial perfusion, radionuclide angiography and gated SPECT (LV volumes and LVEF) and Coronary ?ow reserve by PET [97].
Genetic testing
HCM has been associated with eleven or more genes having more than 1000 mutations [61]. Of these, mutations in genes encoding β-myosin heavy chain and cardiac myosin binding protein C account for ≥ 50% of mutations in HCM [98]. Genetic testing has proved clinically valuable during family screening. Current ESC guidelines recommend genetic counselling during assessment of HCM patients. In this case, genetic testing on the index patient provides valuable information for family screening. If the index patient is diagnosed with a certain genetic mutation, family members will be screened for that mutation even in the absence of LV hypertrophy. However, genetic testing and family lacks clear guidelines on the intervals they should be done [29,99].
Meta-analysis of HCM diagnostic methods
Search strategy
Prospective and randomized controlled trials investigating the diagnosis of patients with HCM using various diagnosis methods were identified using a two-level search strategy. In the first strategy, public domain databases including MEDLINE, EMBASE and Cochrane Central Registry for Control Trials were searched using web-based search engines: PubMed, OVID and Google Scholar. The electronic search used a combination of broad based key words: hypertrophic cardiomyopathy, thickened left ventricular (LV) wall, diagnosis, echocardiography, cardiac MRI, electrocardiography and genetic testing. The second strategy involved identifying studies through a manual search of secondary sources including bibliographies of initially identified articles and a search of reviews, commentaries and proceedings from national cardiology meetings. All references were downloaded for consolidation and elimination of duplicates and further analysis.
Inclusion criteria
Studies were included in the meta-analysis if they satisfied the following inclusion criteria: (a) were published peer reviewed studies; (b) investigated at least one of the diagnostic methods for detecting HCM; (c) included at least one of the following diagnostic outcomes: (a) number of patients detected with HCM and the location of the LV hypertrophy. Case series examining one or two HCM patients and studies with only abstract and seminar papers with insufficient data to enable extraction were excluded. Each potential study was screened in a hierarchical pattern – title, abstract and full text – against the inclusion criteria and data extracted and recorded on Microsoft Excel Worksheet. The extracted data was first author, year of publication, diagnosis method used, number of patient detected and the location of LV hypertrophy. Table 6 provides a summary fo the extracted data from each of the included studies.
Table 6. Summary of Studies on HCM Diagnosis
1st Author [Ref. No.] |
Year |
Sample |
Method |
% Patients Detected |
Symptom/Location of Hypertrophy |
Rickers et al. [95] |
2005 |
48 |
CMRI |
100 |
Anterolateral LV free wall |
Amano et al. [100] |
2005 |
23 |
LGE-MRI |
100 |
Interventricular septal wall |
Villa et al. [101] |
2016 |
30 |
LGE-MRI |
100 |
Interventricular septal wall |
Moon et al. [102] |
2004 |
10 |
CMRI |
100 |
Apical |
Suzuki et al. [103] |
1999 |
13 |
Nuclear MRI |
100 |
Lateral wall, anterior-lateral wall & septal-anterior wall |
Maron et al. [54] |
1981 |
21 |
2-D Echo |
76 |
Posterior ventricular septum, anterior/lateral left ventricular free wall and ventricular septum |
Shapiro et al. [104] |
1983 |
89 |
2-D Echo |
100 |
Septum and free and posterior wall in the proximal and distal LV |
Klues et al. [105] |
1993 |
37 |
2-D Echo |
84 |
Mitral valve dimensions |
Joshi et al. [106] |
2011 |
98 |
Doppler Echo (standing) |
76 |
Standing is not as provocative maneuver as Valsalva or treadmill exercise |
Adabag et al. [32] |
2005 |
178 |
24-h Holter ECG |
94 |
Negative predictive value (95%) of SCD |
Charron et al. [107] |
1997 |
155 |
Echo/ECG |
100 |
LVH/ECG abnormalities |
Savage et al. [108] |
1978 |
134 |
12-Lead ECG |
73 |
Unexplained ECG abnormalities suggests HCM |
Maron et al. [109] |
1983 |
153 |
ECG |
50 |
Non-specific but sensitive marker suggesting HCM |
Ingles et al. [110] |
2005 |
80 |
Genetic Testing |
29 |
Single, double and multiple gene mutation |
Rosenzweig et al. [111] |
1991 |
15 |
Genetic Testing |
PCR |
β cardiac myosin heavy-chain mRNA |
Total/Average |
|
1,084 |
|
84 |
|
2-D: Two Dimension; LV: Left Ventricular; LVH: Left Ventricular Hypertrophy; CMRI: Cardiac Magnetic Resonance Imaging; LGE-MRI: Late Gadolinium Enhancement Magnetic Resonance Imaging; PCR: Polymerase Chain Reaction; ECG: Electrocardiography; Echo: Echocardiography
In total, 15 randomized controlled studies (RCTs) investigating methods used in the differential diagnosis of HCM published between 1978 and 2016 were included in this meta-analysis. The most common researched diagnostic methods for HCM are cardiac magnetic resonance imaging (MRI), echocardiography, and genetic testing. The total patient sample in the 15 studies was 1,084 and the average definitive diagnosis rate was 84%. Five studies [95,100-103] investigated cardiac MRI as a diagnostic method for HCM. The combined patient sample of 124 in the five studies had 100% detection of LV hypertrophy. Two examined cardiac MRI, two LGE- MRI and the remaining one nuclear MRI. The five studies demonstrated that Cardiac MRI has important diagnostic value in detecting LV hypertrophy in different LV wall locations: anterolateral LV free wall [95], interventricular septal wall [100,101], apical [102], lateral wall, and septal anterior wall [103].
Another eight studies [54,104-106,32,107-109] examined the diagnostic value of echo and ECG. Of the combined 865 patient population in the eight studies, the overall detection rate was 84%. Four studies on echo [54,104-106] demonstrated diagnostic value in detecting LV wall hypertrophy in the posterior ventricular septum, lateral left ventricular free wall and ventricular septum. One study [105] suggested mitral value dimensions shows promising results in the diagnosis and prognosis of HCM. No exercise (standing) was also shown to have no significant provocative value compared to Valsalva maneuver and exercise [106]. Four ECG studies [32,107-109] suggest ECG abnormalities require additional diagnostic tests because of a high negative predictive value of 95% for SCD. Finally, genetic testing [110,111] is an uncommon diagnostic method, but useful during family screening to identify at-risk family member based on the identified genetic mutation.
In the present meta-analysis, the characteristic morphological feature and the cornerstone of diagnosis of HCM is hypertrophied and non-dilated LV. The pothognomic morphological features makes cardiac imaging the mainstay of diagnosing HCM. The high success rate in the diagnosis of HCM based principally on imaging methods. In two-thirds of the studies (66.7%), two imaging modalities (echocardiography and cardiac MRI) were the primary diagnostic methods while electrocardiography and genetic testing were used in the remaining studies (33.3%). The importance of imaging is the ability to characterize abnormal morphological features of the LV associated with HCM as well as identify the location of the abnormality. In addition, the present findings reveal that imaging methods are able to reveal the location of LV hypertrophy – types of obstructive and non-obstructive HCM – to inform the choice of appropriate therapy. These findings are consistent with the 2014 ESC recommendation on standardized diagnostic work-up, which proposes cardiac imaging of distinctive morphological characteristics of the HCM. The recommendations further identify two preferable imaging modalities are two-dimensional echocardiography and cardiac MRI. Imaging methods are effective since the HCM phenotype has two characteristic morphological characteristics: hypertrophied and non-dilated LV occurring in the absence of underlying cardiac or systemic disease capable of producing the observable magnitude of hypertrophy (≥ 15mm in adults or equivalent relative to body surface areas in pediatric) [5].
Whereas the present findings support the value of echocardiography and cardiac MRI in the diagnosis of HCM, other factors such as genetic testing and ECG abnormalities are also important in raising clinical suspicion of HCM or supporting the confirmatory diagnosis of HCM [28,99]. Genetic testing is now commercially available and the ESC diagnostic guidelines have supported its inclusion as a part of clinical assessment of patients with HCM. Although the usefulness in diagnosis is limited, genetic analysis is valuable in detecting first-degree family members (where the index patient having a definitive pathogenic mutation) who are at the highest risk of developing HCM and instituting appropriate prophylactic therapies [98]. In addition, to genetic testing, 12-lead ECG has also been demonstrated to be clinically useful in raising suspicion of HCM in patients without LV hypertrophy as well as identifying patterns such as Wolff-Parkinson-White syndrome suggesting certain phenocopies of HCM [5,79,80]. 12-lead ECG may provide patterns mimicking myocardial infarction, which indicate evidence of the diagnosis and may be present in young adults prior to the clinical manifestation of evidence of wall thickening on echocardiography [5].
In summary, the diagnosis of HCM relies principally on pathognomonic morphological features of LV hypertrophy and non-dilation characterized using either 2D-echocardiography or cardiac MRI. Imaging methods also are effective in identify the location of hypertrophy (type of HCM). Although imaging methods have a high rate of successful diagnosis, they are limited in their ability to identify individual at a higher risk of developing HCM. In this regard, genetic testing of first-degree patients where the index had been diagnosed with definitive pathogenic mutation (at the highest risk) and ECG abnormalities long before LV hypertrophy develops are valuable in identifying patients at a higher risk as well as identifying the potential underlying etiologies.
Clinical management of HCM targets to alleviate symptoms of HCM such as heart failure, sudden cardiac death, syncope and atrial fibrillation. Although general clinical management maybe broadly classified into pharmacologic and non-pharmacologic therapies [29], the ESC clinical management guidelines classified treatment based on the presence of obstructive physiology or heart failure symptoms. The primary approach is pharmacologic support, and in the event that the patient is refractory to medication, non-pharmacologic support maybe considered [5]. Figure 8 provides a schematic illustration to the ESC clinical management guidelines for HCM.
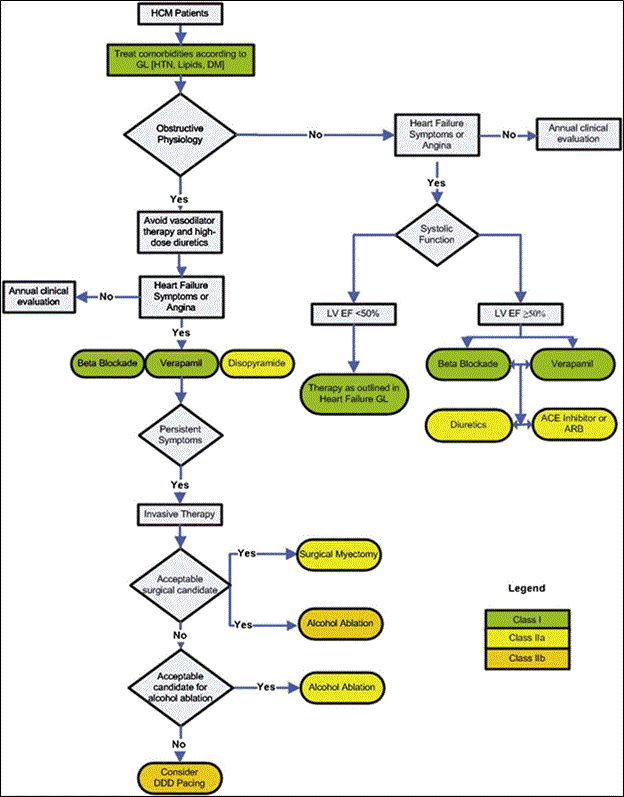
Figure 8: The 2011 ACCF/AHA Clinical Management Algorithm for HCM
The main diagnosis target is the presence of LVOT obstruction, the presence of heart failure or dyspnea or systolic function. These factors depend on the type of medication: Class I, Class IIa or Class IIb medication.
Pharmacotherapy is the most frequently used therapy for HCM, mostly obstructive HCM. The clinical intention is to manage symptoms related to LV outflow tract obstruction. The mainstay of the therapy is a combination of three types of drugs: β-adrenergic antagonist, calcium channel blockers and antiarrhythmic drugs. These drugs act by slowing the heart rate to improve LV outflow tract obstruction and symptoms. A slow heart rate improves LV filling and decreases myocardial oxygen demand [29].
β-adrenergic antagonist was discovered in the 1960s as and effective drug in the treatment and management of obstructive HCM [112]. They are the first line treatment for symptomatic patients with demonstrated efficacy in the reducing LVOT gradient especially during exercise, and alleviating HCM-associates symptoms mainly dyspnea, angina, palpitation and syncope [113].
Calcium channel blockers have also been shown to provide promising results in treating obstructive HCM. Verapamil is a frequently prescribed calcium channel blocker, which improves LVOT gradient and diastolic function. However, verapamil vasodilatory properties are associated with negative side effects such as hypotension, increases outflow gradient and pulmonary edema limiting its prescription and actual use [29].
Antiarrhythmic drug are a third class of drugs used to alleviate HCM-related symptoms. A frequently prescribed antiarrhythmic drug is disopyramide, which acts by producing negative inotropic effect causing an improvement in resting LVOT gradient and the alleviation of heart failure symptoms even more than β-adrenergic antagonist. However, in most cases, it is prescribed with β-adrenergic antagonist or calcium channel blockers to improve treatment and prognostic value [114].
The focus of non-pharmacologic therapy is to treat obstructive HCM. The therapy is uncommon, indicated in about 5% of obstructive HCM patients to control symptoms associated with LVOT obstruction. Non-pharmacologic therapy – surgical myectomy, alcohol septal ablation or dual chamber pacing – is usually indicated for HCM patients with refractory symptoms after pharmacotherapy [29].
Surgical myectomy has been the mainstay surgical therapy for the relief of symptoms of LVOT obstruction. The procedure also corrects valvular and subvalvular abnormalities [115]. Although early studies suggested high operative mortality (4-6%), recent studies suggest improvement in the safety of the surgical procedure with reduced (1%) perioperative mortality [116]. Reported serious complication for surgical myectomy is ventricular septal defects (VSDs) common associated with HCM patients with mild septum hypertrophy [117]. However, surgical myectomy has favorable long-term prognosis marked with abolished LVOT gradient (90-95%), a significant reduction in heart failure symptoms, absent mitral regurgitation and thus no need for additional mitral valve surgery [118].
Transcatheter ablation of the septum using ethanol is another non-pharmacologic therapy for obstructive HCM. Since the first procedure performed in 1994, the use of alcohol ablation has grown considerably [118]. In the procedure of alcohol ablation, coronary angiography is carried out and ethanol injected into proximal septal perforators causing directed myocardial infarction and a subsequent reduction in LVOT gradient and symptoms [119,120]. Complications for alcohol ablation include peri-operative mortality (1-4%), ventricular arrhythmias (5%), AV block (10-20%) [29]. In addition, it takes 3-months for alcohol ablation to register clinically significant results. Despite the complications, alcohol ablation reduces LOVT gradient, symptoms, and improve survival rates [121].
Dual chamber pacing with short AV delays has been shown to induce asynchrony, which alleviates subaortic gradient and symptoms of heart failure [122]. Recent randomized controlled trials have however indicated that dual chamber pacing is only beneficial to a small proportion of HCM patients because of significant placebo effect with pacemaker implantation. In light of these findings, the ACCF/AHA guidelines only recommend dual chamber pacing for HCM patients presenting with refractory symptoms, LVOT obstruction and indicated for septal reduction therapy [29].
A systematic review and meta-analysis was conducted to evaluate the current clinical management methods of HCM. Studies were searched from online databases PubMed, EMBASE and Google Scholar. A combination of search terms used included hypertrophic cardiomyopathy, clinical management, treatment, medical therapy, myectomy and ablation. Studies were included if they were peer-reviewed, investigated at least one clinical management method of HCM, and provided data on treatment method, clinical endpoint and survival or improvement. Studies with only abstract providing insufficient information or case series with fewer cases (statistical insufficiency to support findings) for management were excluded. Table 7 provides a summary of data for the included studies.
Table 7. Summary of Studies on Clinical Management of HCM.
1st Author [Ref. No.] |
Year |
Sample |
Method |
Clinical Endpoint |
Survival/ Improvement |
Findings |
Kunkala et al. [123] |
2013 |
56 |
Septal myectomy |
LVOT gradient |
-- |
Septal myectomy relieves intraventricular gradients and related symptoms of midventricular obstruction |
|
Minakata et al. [124] |
2005 |
56 |
Septal myectomy |
Heart transplant survival free |
90 |
Safe and efficacious in symptomatic pediatric patients |
|
Orme et al. [125] |
2013 |
239 |
Septal myectomy |
Recurrent syncope survival |
82 |
Septal Myectomy reduces recurrent syncope and increases survival |
|
Quintana et al. [126] |
2015 |
31 |
Septal myectomy |
SCD |
100 |
Septal myectomy avoids scarring and diastolic dysfunction found in alcohol septal ablation |
|
Theodoro et al. [127] |
1996 |
25 |
Septal myectomy |
SCD |
96 |
Safe and effective in reducing cardiac symptoms and LVOT on pediatric patients |
|
Bonow et al. [128] |
1983 |
42 |
Verapamil |
LV diastolic filling |
83 |
Improves hemodynamic and improves atrial systole to LV filling |
|
Nistri et al. [129] |
2012 |
27 |
β blockers (Nadolol/ Bisoprolol) |
LVOT gradient |
85 |
β blockers prevent hemodynamic burden associated exercise LVOT obstruction |
|
Setaro et al. [130] |
1990 |
20 |
Verapamil |
Heart Failure symptoms |
NA |
Preserves systolic function and diastolic filling |
|
Sherrid et al. [131] |
2013 |
737 |
β blockers/ Verapamil |
LVOT obstruction relief |
46 |
Some patients how refractory HCM symptoms |
|
Maron et al. [132] |
2000 |
128 |
ICD |
SCD |
98 |
ICD are effective in preventing arrhythmias and subsequently SCD. |
|
Vriesendorp et al. [133] |
2013 |
134 |
ICD |
SCD |
97 |
ICD prevents arrhythmias and SCD |
|
Faber et al. [134] |
2005 |
242 |
Alcohol ablation |
LVOT gradient |
88 |
Reduces left atrial size and septal thickness |
|
Faber et al. [135] |
2007 |
312 |
Alcohol ablation |
LVOT gradient |
91 |
Effective in reducing symptoms and LVOT gradient |
|
Hess et al. [136] |
2009 |
72 |
Alcohol ablation |
LVOT gradient |
80 |
Alcohol ablation is a promising first line non-pharmacologic therapy |
|
Osterne et al. [137] |
2003 |
22 |
Alcohol ablation |
LVOT gradient |
82 |
Effective and safe in treating obstructive HCM |
|
Total |
|
2,143 |
|
|
86 |
|
|
After a review of studies against the inclusion criteria, 15 randomized clinical trials that specifically investigated clinical interventions used in the treatment and management of HCM were included in this meta-analysis. The studies were published between 1983 and 2015. The 15 studies recruited a combined patient population of 2,143. The studies revealed the efficacy of the current clinical interventions in the management of HCM with an overall survival rate or improvement in symptoms in 86% of the total HCM patients. The main clinical management interventions studied were septal myectomy [123-127], pharmacotherapy [128-131], ICD [132-133] and alcohol ablation [134-137]. The studies reveal the main treatment target is LVOT pressure gradient [123,129,134-137]. The others are symptoms related to LVOT obstruction [124,125], sudden cardiac death (SCD) [127,128,132,133], and hemodynamics such as LV diastolic filling [129].
The studies on septal myectomy demonstrate its efficacy in relieving intraventricular gradients [123], relieving cardiac symptoms [127] and the reduction of recurrent syncope and increased heart-transplant free survival rate [124,125]. Studies on pharmacotherapy indicate effectiveness of a combination of β-blockers (Nadolol, bisoprolol) and antiarrhythmic (Verapamil) drugs. The target is relieving LVOT obstruction [129,131], LV diastolic filling [128] and heart failure symptoms [130]. Heart transplantation, especially ICD, prevents lethal arrhythmias and sudden cardiac death. They are usually indicated in case of refractory symptoms after pharmacotherapy [132,133]. Finally, alcohol ablation primarily targets relieving LVOT gradient and associated symptoms [134-137]. It is the first line non-pharmacotherapy effective in the treatment of obstructive HCM. If alcohol ablation fails, septal myectomy [126] or ICD should be considered [128].
Hypertrophic Cardiomyopathy (HCM) is a well-recognized phenotype of cardiomyopathy with well-established and standardized clinical management guidelines. The standardized guidelines have contributed to improvement in its therapeutic success rates [5]. In the present meta-analysis, clinical management of HCM demonstrate they are efficacious, with event-free survival or improvement in symptoms in up to 86% of the total 2,143 patients included in the analysis. The findings further reveal that the current clinical management approaches primarily target treating LVOT obstruction (high LVOT pressure gradient > 30 mm Hg), symptoms associated with LVOT obstruction, sudden cardiac death and abnormalities in LV hemodynamics.
Pharmacotherapy is considered the first line treatment for HCM patients with LVOT obstruction. The present findings indicate the main medications are a combination of beta-blockers and antiarrhythmic drugs. They are effective in relieving LVOT obstruction and alleviating associated symptoms. Medications also improved hemodynamics (LV diastolic filling) and heart failure symptoms [128,130]. According to Enriquez and Goldman [29] medical therapy improved LVOT obstruction and LV filling by slowing heart rate. For HCM refractory to medical therapy, the first-line non-pharmacological treatment is alcohol ablation [126]. Although effective in reducing LVOT gradient, symptoms and survival rates, alcohol ablation takes longer (about three months) to register clinically significant outcomes [121].
Septal myectomy (targeting LVOT obstruction) is usually considered when medical therapy and alcohol ablation have failed to alleviate symptoms or LVOT gradient. In the present analysis, septal myectomy was the most common non-pharmacologic therapeutic method [123,127]. The method was efficacious in relieving LVOT gradient, cardiac symptoms associated with LV obstruction (high LVOT gradient) and reduces recurrent syncope [123,127]. Septal myectomy also conveyed protective effect against the need for heart transplant, improving heart-transplant free survival [124,125]. Prior studies suggest since its initial clinical use in the 1960, experience has improved safety of surgical procedure with significant reduction in peri- and post-procedural complications [115,117]. Furthermore, Sigwart [118] reports that, since septal myectomy eliminates LVOT obstruction it conveys a favorable long-term prognosis with reduction in symptoms of heart failure and mitral regurgitation [118]. When medical, therapy, alcohol ablation and septal myectomy do not produce the required clinical outcomes, device therapy (ICD) or heart transplantation may be indicated to prevent lethal arrhythmias and sudden cardiac death [133,132].
In summary, the current clinical management approaches for HCM depend on the presence or absence of LVOT obstruction and impaired systolic function. However, the mainstay of treatment is relieving LVOT gradient and associated symptoms. The first-line treatment is pharmacotherapy. If the LVOT gradient or symptoms are refractory to medication, non-pharmacological therapy are alcohol ablation and septal myectomy. Device therapy (ICD) and heart transplantation are the last-line of HCM treatment targeting relieving lethal arrhythmias and prevention of sudden cardiac death.
First described as a clinical entity in 1958, hypertrophic cardiomyopathy is now well recognized by the European Society of Cardiology (ESC) and the American Heart Association (AHA) as distinct clinical phenotype of cardiomyopathy. It is distinguished from other phenotypes by its characteristic hypertrophied and non-dilated LV not explained by other cardiac or systemic disorders. HCM is categorized as obstructive characterized by sub-aortic or mid-ventricular obstruction, and non-obstructive, characterized by normal or impaired LV systolic function. The obstructive type is the most prevalent accounting for up to 70% of HCM patients. The main risk factors for developing HCM are a family history of sudden cardiac death, recurrent syncope in young children or under exertion, frequent, faster and longer episodes of non-sustained ventricular tachycardia, significant LV hypertrophy and abnormal blood pressure under exertion. The main reason for clinical visit are dyspnea, stroke, syncope or sudden cardiac death. Markers for clinical progression or for ominous prognosis are atrial fibrillation, progressive heart failure and sudden cardiac death. The pathophysiologic mechanisms of HCM are LVOT, diastolic dysfunction, myocardial ischemia and mitral regurgitation. The main etiologic factors for HCM are genetic mutations in sarcomere protein, inherited metabolic disorders, neuromuscular disorders, and mitochondrial disorders. The mainstay of HCM diagnosis is echocardiography or cardiac magnetic resonance imaging of the LV structure, while genetic testing and physical examination provide additional clues for identifying patients at the greatest risk of HCM. Clinical management methods focus on management of LVOT obstruction (gradient) and LVOT symptoms. The first-line therapy is medication. Alcohol ablation and septal myectomy are indicated when HCM symptoms are refractory to medication. Device therapy and heart transplantation are the last-line for prevention against lethal arrhythmias or sudden cardiac death.
References
- Teare D (1958) Asymmetrical hypertrophy of the heart in young adults. Br Heart J 20: 1-8. [crossref]
- McKenna WJ, Sen Chowdhry S (2008) From Teare to the present day: a fifty-year odyssey in hypertrophic cardiomyopathy, a paradigm for the logic of the discovery process. Revista Espanola De Cardiologia 61: 1239-1244.
- Kofflard M (1998) New perspectives in Hypertrophic Cardiomyopathy.
- Arad M Seidman JG, Seidman CE (2002) Phenotypic diversity in hypertrophic cardiomyopathy. Human Molecular Genetics 11: 2499-2506.
- Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, et al. (2011) ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy. Circulation
- Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, et al. (2014) 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy. Kardiologia Polska 72: 1054-1126.
- Falicov RE, Resnekov LE (1977) Mid ventricular obstruction in hypertrophic obstructive cardiomyopathy. New diagnostic and therapeutic challenge. British Heart Journal 39: 701-705
- Li Q, Williams L, Rakowski H (2015) Natural history of untreated hypertrophic cardiomyopathy. In Hypertrophic Cardiomyopathy pp: 9-22.
- Wigle ED (1987) Hypertrophic cardiomyopathy: a 1987 viewpoint. Circulation 75: 311-322. [crossref]
- Sherrid MV, Delia E, Mogtader A, Dwyer EM (1993) An echocardiographic study of the fluid mechanics of obstruction in hypertrophic cardiomyopathy. JACC 22: 816-825.
- Efthimiadis GK, Pagourelias ED, Parcharidou D, Gossios T, Kamperidis V, et al. (2013). Clinical characteristics and natural history of hypertrophic cardiomyopathy with midventricular obstruction. Circulation Journal 77: 2366-2374.
- Minami Y, Kajimoto K, Terajima Y, Yashiro B, Okayama D, et al. (2011) Clinical implications of midventricular obstruction in patients with hypertrophic cardiomyopathy. JACC 57: 2346-2355.
- Factor SM, Butany J, Sole MJ, Wigle ED, Williams WC, et al. (1991) Pathologic fibrosis and matrix connective tissue in the subaortic myocardium of patients with hypertrophic cardiomyopathy. JACC 17: 1343-1351.
- Ates M, Kwong RY, Lipton MJ, Tatli S (2006) Apical hypertrophic cardiomyopathy: Diagnosed by cardiac magnetic resonance imaging. Texas Heart Institute Journal 33: 408.
- Yusuf SW, Bathina JD, Banchs J, Mouhayar EN, Daher IN (2011) Apical hypertrophic cardiomyopathy. World J Cardiol 3: 256-259. [crossref]
- Eriksson MJ, Sonnenberg B, Woo A, Rakowski P, Parker TG, Wigle ED, et al. (2002) Long-term outcome in patients with apical hypertrophic cardiomyopathy. Journal of the American College of Cardiology 39: 638-645.
- Maron MS, Finley JJ, Bos JM, Hauser TH, Manning WJ, et al. (2008) Prevalence, clinical significance, and natural history of left ventricular apical aneurysms in hypertrophic cardiomyopathy. Circulation 118: 1541-1549.
- Stainback RF (2012) Apical hypertrophic cardiomyopathy. Texas Heart Institute Journal 39: 747-749.
- Codd MB, Sugrue DD, Gersh BJ, Melton LJ (1989) Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. A population-based study in Olmsted County, Minnesota, 1975-1984. Circulation 80: 564-572.
- Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, et al. (1995) Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Circulation 92: 785-789.
- Semsarian C, Ingles J, Maron MS, Maron BJ (2015) New perspectives on the prevalence of hypertrophic cardiomyopathy. JACC 65: 1249-1254.
- Maron BJ, Maron MS, Semsarian C (2012) Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. JACC 60: 705-715.
- Seidman JG, Seidman C (2001) The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell 104: 557-567. [crossref]
- Maron BJ, Mathenge R, Casey SA, Poliac LC, Longe TF (1999) Clinical profile of hypertrophic cardiomyopathy identified de novo in rural communities. JACC 33: 1590-1595.
- Maron BJ, Spirito P, Roman MJ, Paranicas M, Okin PM, et al. (2004) Prevalence of hypertrophic cardiomyopathy in a population-based sample of American Indians aged 51 to 77 years (the Strong Heart Study). The American Journal of Cardiology 93: 1510-1514.
- Zou Y, Song L, Wang Z, Ma A, Liu T, et al. (2004) Prevalence of idiopathic hypertrophic cardiomyopathy in China: a population-based echocardiographic analysis of 8080 adults. The American Journal of Medicine, 116: 14-18.
- Maro EE, Janabi M, Kaushik R (2006) Clinical and echocardiographic study of hypertrophic cardiomyopathy in Tanzania. Trop Doct 36: 225-227.
- Maron MS, Olivotto I, Zenovich AG, Link MS, Pandian NG, et al. (2006) Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation, 114: 2232-2239.
- Enriquez AD, Goldman ME2 (2014) Management of hypertrophic cardiomyopathy. Ann Glob Health 80: 35-45. [crossref]
- McKenna W, Deanfield J, Faruqui A, England D, Oakley C, et al. (1981) Prognosis in hypertrophic cardiomyopathy: role of age and clinical, electrocardiographic and hemodynamic features. JACC 47: 532-538.
- Walsh K, Hoffmayer K, Hamdan MH (2015) Syncope: diagnosis and management. Curr Probl Cardiol 40: 51-86. [crossref]
- Adabag AS, Casey SA, Kuskowski MA, Zenovich AG (2005) Spectrum and prognostic significance of arrhythmias on ambulatory Holter electrocardiogram in hypertrophic cardiomyopathy. JACC 45: 697-704.
- Monserrat L, Elliott PM, Gimeno JR, Sharma S, Penas-Lado M, McKenna WJ (2003) Non-sustained ventricular tachycardia in hypertrophic cardiomyopathy: an independent marker of sudden death risk in young patients. JACC 42: 873-879.
- Spirito P, Bellone P, Harris KM, Bernabò P, Bruzzi P, Maron BJ (2000) Magnitude of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. New England Journal of Medicine 342: 1778-1785.
- Maron MS, Olivotto I, Betocchi S, Casey SA, Lesser JR, et al. (2003) Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. New England Journal of Medicine 348: 295-303.
- Elliott PM, Gimeno JR, Tomé MT, Shah J, Ward D, et al. (2006) Left ventricular outflow tract obstruction and sudden death risk in patients with hypertrophic cardiomyopathy. European Heart Journal 27: 1933-1941.
- Rubinshtein R, Glockner JF, Ommen SR, Araoz PA, Ackerman MJ, et al. (2009) Characteristics and clinical significance of late gadolinium enhancement by contrast-enhanced magnetic resonance imaging in patients with hypertrophic cardiomyopathy. Circulation: Heart Failure 109.
- Chiribiri A, Conte MR, Bonamini R, Gaita F, Nagel E (2011) Late gadolinium enhancement and sudden cardiac death in hypertrophic cardiomyopathy. Journal of the American College of Cardiology 57: 1402.
- McKenna WJ, Deanfield JE (1984) Hypertrophic cardiomyopathy: an important cause of sudden death. Arch Dis Child 59: 971-975. [crossref]
- Elliott P1, McKenna WJ (2004) Hypertrophic cardiomyopathy. Lancet 363: 1881-1891. [crossref]
- National Lung, Heart and Blood Institute. (2017). What Is Heart Failure? Retrieved from https://www.nhlbi.nih.gov/health/health-topics/topics/hf
- Zipes DP1, Wellens HJ (1998) Sudden cardiac death. Circulation 98: 2334-2351. [crossref]
- Calkins H, Kuck KH, Cappato R, Brugada J, Camm AJ, et al. (2012) HRS/EHRA/ECAS expert consensus statement on catheter and surgical ablation of atrial fibrillation: recommendations for patient selection, procedural techniques, patient management and follow-up, definitions, endpoints, and research trial design: a report of the Heart Rhythm Society (HRS) Task Force on Catheter and Surgical Ablation of Atrial Fibrillation. Developed in partnership with the European Heart Rhythm Association (EHRA), a registered branch of the European Society of Cardiology (ESC) Europace 14: 528-606.
- Olivotto I, Cecchi F, Casey SA, Dolara A, Traverse JH, et al. (2001) Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation 104: 2517-2524.
- Kofflard MJ, Waldstein DJ, Vos J, Folkert J (1993) Prognosis in hypertrophic cardiomyopathy observed in a large clinic population. The American Journal of Cardiology 72: 939-943.
- Maron BJ (2002) Hypertrophic cardiomyopathy: a systematic review. JAMA 287: 1308-1320. [crossref]
- Wigle ED, Rakowski H, Kimball BP, Williams WG (1995) Hypertrophic cardiomyopathy. Clinical spectrum and treatment. Circulation 92: 1680-1692. [crossref]
- Wigle ED, Sasson Z, Henderson MA, Ruddy TD, Fulop J, et al. (1985) Hypertrophic cardiomyopathy. The importance of the site and the extent of hypertrophy. A review. Progress in Cardiovascular Diseases, 28: 1-83.
- Noureldin RA, Liu S, Nacif MS, Judge DP, Halushka MK, et al. (2012) The diagnosis of hypertrophic cardiomyopathy by cardiovascular magnetic resonance. Journal of Cardiovascular Magnetic Resonance 14: 17-30
- Sherrid MV, Gunsburg DZ, Moldenhauer S, Pearle G (2000) Systolic anterior motion begins at low left ventricular outflow tract velocity in obstructive hypertrophic cardiomyopathy. JACC 36: 1344-1354.
- Sherrid MV (1998) Dynamic Left Ventricular Outflow Obstruction in Hypertrophic Cardiomyopathy Revisited: Significance, Pathogenesis, and Treatment. Cardiology in Review 6: 135-145.
- Shah PM, Taylor RD, Wong M (1981) Abnormal mitral valve coaptation in hypertrophic obstructive cardiomyopathy: proposed role in systolic anterior motion of mitral valve. JACC 48: 258-262.
- Maron BJ, Nishimura RA, Danielson GK (1998) Pitfalls in clinical recognition and a novel operative approach for hypertrophic cardiomyopathy with severe outflow obstruction due to anomalous papillary muscle. Circulation 98: 2505-2508.
- Maron BJ, Gottdiener JS, Bonow RO, Epstein SE (1981) Hypertrophic cardiomyopathy with unusual locations of left ventricular hypertrophy undetectable by M-mode echocardiography. Identification by wide-angle two-dimensional echocardiography. Circulation 63: 409-418.
- Bonow RO, Dilsizian V, Rosing DR, Maron BJ, Bacharach SL, et al. (1985) Verapamil-induced improvement in left ventricular diastolic filling and increased exercise tolerance in patients with hypertrophic cardiomyopathy: short-and long-term effects. Circulation 72: 853-864.
- Maron MS, Olivotto I, Maron BJ, Prasad SK, Cecchi F, et al. (2009) The case for myocardial ischemia in hypertrophic cardiomyopathy. Journal of the American College of Cardiology 54: 866-875.
- Maron BJ, Wolfson JK, Epstein SE, Roberts WC (1986) Intramural (“small vessel”) coronary artery disease in hypertrophic cardiomyopathy. Journal of the American College of Cardiology 8: 545-557.
- Wigle ED, Adelman AG, Auger P, Marquis Y (1969) Mitral regurgitation in muscular subaortic stenosis. JACC 24: 698-706.
- Zhu WX, Oh JK, Kopecky SL, Schaff HV, Tajik AJ (1992) Mitral regurgitation due to ruptured chordae tendineae in patients with hypertrophic obstructive cardiomyopathy. JACC 20: 242-247.
- Lopes LR, Zekavati A, Syrris P, Hubank M, Giambartolomei C, et al. (2013) Consortium Genetic complexity in hypertrophic cardiomyopathy revealed by high-throughput sequencing. Journal of medical genetics 50: 228-239.
- Watkins H, Ashrafian H, Redwood C (2011) Inherited cardiomyopathies. N Engl J Med 364: 1643-1656. [crossref]
- Lopes LR, Rahman MS, Elliott PM (2013) A systematic review and meta-analysis of genotype–phenotype associations in patients with hypertrophic cardiomyopathy caused by sarcomeric protein mutations. Heart 99: 1800-1811.
- Olivotto I, Girolami F, Ackerman MJ, Nistri S, Bos JM, (2008) Myofilament protein gene mutation screening and outcome of patients with hypertrophic cardiomyopathy. In Mayo Clinic Proceedings 83: 630-638.
- Pasquale F, Syrris P, Kaski JP, Mogensen J, McKenna WJ, et al. (2011) Long-term outcomes in hypertrophic cardiomyopathy caused by mutations in the cardiac troponin T gene. Circgenetics
- Watkins H, McKenna WJ, Thierfelder L, Suk HJ, Anan R, et al. (1995) Mutations in the genes for cardiac troponin T and a-tropomyosin in hypertrophic cardiomyopathy. New England Journal of Medicine 332: 1058-1065.
- Moolman JC, Corfield VA, Posen B, Ngumbela K, Seidman C, et al. (1997) Sudden death due to troponin T mutations. J Am Coll Cardiol 29: 549-555. [crossref]
- Torricelli F, Girolami F, Olivotto I, Passerini I, Frusconi S, et al. (2003) Prevalence and clinical profile of troponin T mutations among patients with hypertrophic cardiomyopathy in tuscany. The American Journal of Cardiology 92: 1358-1362.
- Girolami F, Ho CY, Semsarian C, Baldi M, Will ML, et al. (2010) Clinical features and outcome of hypertrophic cardiomyopathy associated with triple sarcomere protein gene mutations. JACC 55: 1444-1453.
- Ingles J, Doolan A, Chiu C, Seidman J, Seidman C, et al. (2005) Compound and double mutations in patients with hypertrophic cardiomyopathy: implications for genetic testing and counselling. Journal of Medical Genetics 42: e59-e59.
- Coats CJ, Elliott PM (2013) Genetic biomarkers in hypertrophic cardiomyopathy. Biomark Med 7: 505-516. [crossref]
- Elliott P, Baker R, Pasquale F, Quarta G, Ebrahim H, et al. (2011) Prevalence of Anderson–Fabry disease in patients with hypertrophic cardiomyopathy: the European Anderson–Fabry Disease Survey. Heart 97: 1957-1960.
- Murphy RT, Mogensen J, McGarry K, Bahl A, Evans A, et al. (2005) Adenosine monophosphate-activated protein kinase disease mimicks hypertrophic cardiomyopathy and Wolff-Parkinson-White syndrome. JACC, 45: 922-930.
- Friedrich FW, Wilding BR, Reischmann S, Crocini C, Lang P, et al. (2012) Evidence for FHL1 as a novel disease gene for isolated hypertrophic cardiomyopathy. Human Molecular Genetics 21: 3237-3254.
- Falk RH (2005) Diagnosis and management of the cardiac amyloidoses. Circulation 112: 2047-2060. [crossref]
- Huddle KR, Kalliatakis B, Skoularigis J (1996) Pheochromocytoma associated with clinical and echocardiographic features simulating hypertrophic obstructive cardiomyopathy. Chest 109: 1394-1397.
- Hradec J, Marek J, Petrásek J (1988) The nature of cardiac hypertrophy in acromegaly: an echocardiographic study. Cor Vasa 30: 186-199. [crossref]
- Wigle ED (2001) Cardiomyopathy: The diagnosis of hypertrophic cardiomyopathy. Heart 86: 709-714. [crossref]
- Rapezzi C, Arbustini E, Caforio AL, Charron P, Gimeno BJ, et al. (2012) Diagnostic work-up in cardiomyopathies: bridging the gap between clinical phenotypes and final diagnosis. A position statement from the ESC Working Group on Myocardial and Pericardial Diseases. European Heart Journal 34: 1448-1458.
- Ryan MP, Cleland JG, French JA, Joshi J, Choudhury L, et al. (1995) The standard electrocardiogram as a screening test for hypertrophic cardiomyopathy. The American Journal of Cardiology 76: 689-694.
- Konno T, Shimizu M, Ino H, Yamaguchi M, Terai H, et al. (2004) Diagnostic value of abnormal Q waves for identification of preclinical carriers of hypertrophic cardiomyopathy based on a molecular genetic diagnosis. European Heart Journal 25: 246-251.
- Pantazis A, Vischer AS, Perez-Tome MC, Castelletti S (2015) Diagnosis and management of hypertrophic cardiomyopathy. Echo Res Pract 2: R45-53. [crossref]
- Wigle ED, Sasson Z, Henderson MA, Ruddy TD, Fulop J, et al. (1985) Hypertrophic cardiomyopathy. The importance of the site and the extent of hypertrophy. A review. Progress in Cardiovascular Diseases 28: 1-83.
- Pandian NG, Rowin EJ, Gonzalez AMG, Maron MS (2015) Echocardiographic profiles in hypertrophic cardiomyopathy: imaging beyond the septum and systolic anterior motion. Echo Research and Practice 1: 1-7.
- O’Hanlon R, Assomull RG, Prasad SK (2007) Use of cardiovascular magnetic resonance for diagnosis and management in hypertrophic cardiomyopathy. Current Cardiology Reports 9: 51-56.
- Gange CA, Link MS, Maron MS (2009) Utility of cardiovascular magnetic resonance in the diagnosis of Anderson-Fabry disease. Circulation 120: e96-e97.
- Maceira AM, Joshi J, Prasad SK, Moon JC, Perugini E, et al. (2005) Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation, 111(2), 186-193.
- Moon JC, Sachdev B, Elkington AG, McKenna WJ, Mehta A, et al. (2003) Gadolinium enhanced cardiovascular magnetic resonance in Anderson-Fabry disease 1: evidence for a disease specific abnormality of the myocardial interstitium. European Heart Journal 24: 2151-2155.
- Prinz C, Schwarz M, Ilic I, Laser KT, Lehmann R, et al. (2013) Myocardial fibrosis severity on cardiac magnetic resonance imaging predicts sustained arrhythmic events in hypertrophic cardiomyopathy. JCC 29: 358-363.
- Bruder O, Wagner A, Jensen CJ, Schneider S, Ong P, et al. (2010) Myocardial scar visualized by cardiovascular magnetic resonance imaging predicts major adverse events in patients with hypertrophic cardiomyopathy. JACC 56: 875-887.
- Sado DM, White SK, Piechnik SK, Banypersad SM, Treibel T, et al. (2013) The identification and assessment of Anderson Fabry disease by cardiovascular magnetic resonance non-contrast myocardial T1 mapping Circimaging 112: 1-10
- Rickers C, Wilke NM, Jerosch HM, Casey SA, Panse P, et al. (2005) Utility of cardiac magnetic resonance imaging in the diagnosis of hypertrophic cardiomyopathy. Circulation 112: 855-861.
- Bravo PE, Zimmerman SL, Luo HC, Pozios I, Rajaram M, et al. (2013) Relationship of delayed enhancement by magnetic resonance to myocardial perfusion by positron emission tomography in hypertrophic cardiomyopathy. Circulation: Cardiovascular Imaging 6: 210-217.
- Fowler SJ, Narula J, Gurudevan SV (2006) Review of noninvasive imaging for hypertrophic cardiac syndromes and restrictive physiology. Heart Failure Clinics 2: 215-230.
- Rapezzi C, Quarta CC, Guidalotti, P. L., Pettinato, C., Fanti, S., Leone, O., ... & Gagliardi, C. (2011). Role of 99m Tc-DPD scintigraphy in diagnosis and prognosis of hereditary transthyretin-related cardiac amyloidosis. JACC: Cardiovascular Imaging, 4(6), 659-670.
- Rapezzi C, Quarta CC, Guidalotti PL, Longhi S, Pettinato C, et al. (2011) Usefulness and limitations of 99mTc-3, 3-diphosphono-1, 2-propanodicarboxylic acid scintigraphy in the aetiological diagnosis of amyloidotic cardiomyopathy. European journal of nuclear medicine and molecular imaging 38: 470-478.
- Quarta CC, Guidalotti PL, Longhi S, Pettinato C, Leone O, et al. (2012) Defining the diagnosis in echocardiographically suspected senile systemic amyloidosis. JACC: 5: 755-758.
- Nagueh SF, Bierig SM, Budoff MJ, Desai M, Dilsizian V, et al. (2011) American Society of Echocardiography clinical recommendations for multimodality cardiovascular imaging of patients with hypertrophic cardiomyopathy: endorsed by the American Society of Nuclear Cardiology, Society for Cardiovascular Magnetic Resonance, and Society of Cardiovascular Computed Tomography. Journal of the American Society of Echocardiography 24: 473-498.
- Ho CY (2010) Genetics and clinical destiny: improving care in hypertrophic cardiomyopathy. Circulation 122: 2430.
- Maron BJ, Seidman JG, Seidman CE (2004) Proposal for contemporary screening strategies in families with hypertrophic cardiomyopathy. Journal of the American College of Cardiology 44: 2125-2132.
- Amano Y, Kumita S, Takayama M, Kumazaki T (2005) Comparison of contrast-enhanced MRI with iodine-123 BMIPP for detection of myocardial damage in hypertrophic cardiomyopathy. American Journal of Roentgenology 185: 312-318.
- Villa AD, Sammut E, Zarinabad N, Carr WG, Lee J, et al. (2016) Microvascular ischemia in hypertrophic cardiomyopathy: new insights from high-resolution combined quantification of perfusion and late gadolinium enhancement. Journal of Cardiovascular Magnetic Resonance 18: 4-11.
- Moon JCC, Fisher NG, McKenna WJ, Pennell DJ (2004) Detection of apical hypertrophic cardiomyopathy by cardiovascular magnetic resonance in patients with non-diagnostic echocardiography. Heart 90: 645-649.
- Suzuki JI, Shimamoto R, Nishikawa JI, Yamazaki T, Tsuji T, et al. (1999) Morphological onset and early diagnosis in apical hypertrophic cardiomyopathy: A long-term analysis with nuclear magnetic resonance imaging. Journal of the American College of Cardiology 33: 146-151.
- Shapiro LM, McKenna WJ (1983) Distribution of left ventricular hypertrophy in hypertrophic cardiomyopathy: a two-dimensional echocardiographic study. Journal of the American College of Cardiology 2: 437-444.
- Klues HG, Proschan MA, Dollar AL, Spirito P, Roberts WC, et al. (1993) Echocardiographic assessment of mitral valve size in obstructive hypertrophic cardiomyopathy. Anatomic validation from mitral valve specimen. Circulation 88: 548-555.
- Joshi S, Patel UK, Yao SS, Castenada V, Isambert A, et al. (2011) Standing and exercise Doppler echocardiography in obstructive hypertrophic cardiomyopathy: the range of gradients with upright activity. Journal of the American Society of Echocardiography 24: 75-82.
- Charron P, Dubourg O, Desnos M, Isnard R, Hagege A, et al. (1997) Diagnostic value of electrocardiography and echocardiography for familial hypertrophic cardiomyopathy in a genotyped adult population. Circulation 96: 214-219.
- Savage DD, Seides SF, Clark CE, Henry WL, Maron BJ, et al. (1978) Electrocardiographic findings in patients with obstructive and nonobstructive hypertrophic cardiomyopathy. Circulation 58: 402-408.
- Maron BJ, Wolfson JK, Ciró E, Spirito P (1983) Relation of electrocardiographic abnormalities and patterns of left ventricular hypertrophy identified by 2-dimensional echocardiography in patients with hypertrophic cardiomyopathy. The American Journal of Cardiology, 51: 189-194.
- Ingles J, Doolan A, Chiu C, Seidman J, Seidman C, et al. (2005) Compound and double mutations in patients with hypertrophic cardiomyopathy: implications for genetic testing and counselling. Journal of Medical Genetics 42: e59-e59.
- Rosenzweig A, Watkins H, Hwang DS, Miri M, McKenna W, et al. (1991) Preclinical diagnosis of familial hypertrophic cardiomyopathy by genetic analysis of blood lymphocytes. New England Journal of Medicine 325: 1753-1760.
- Harrison DC, Braunwald E, Glick G, Mason DT, Chidsey CA, et al. (1964) Effects of beta adrenergic blockade on the circulation, with particular reference to observations in patients with hypertrophic subaortic stenosis. Circulation 29: 84-98.
- Fifer MA, Vlahakes GJ (2008) Management of symptoms in hypertrophic cardiomyopathy. Circulation 117: 429-439. [crossref]
- Spirito P, Seidman CE, McKenna WJ, Maron BJ (1997) The management of hypertrophic cardiomyopathy. N Engl J Med 336: 775-785. [crossref]
- Brown ML, Schaff HV (2008) Surgical management of obstructive hypertrophic cardiomyopathy: the gold standard. Expert Review of Cardiovascular Therapy 6: 715-722.
- Ommen SR, Maron BJ, Olivotto I, Maron MS, Cecchi F, et al. (2005) Long-term effects of surgical septal myectomy on survival in patients with obstructive hypertrophic cardiomyopathy. Journal of the American College of Cardiology 46: 470-476.
- Xin J, Shiota T, Lever HM, Kapadia SR, Sitges M, et al. (2001) Outcome of patients with hypertrophic obstructive cardiomyopathy after percutaneous transluminal septal myocardial ablation and septal myectomy surgery. Journal of the American College of Cardiology 38: 1994-2000.
- Sigwart U (1995) Non-surgical myocardial reduction for hypertrophic obstructive cardiomyopathy. Lancet 346: 211-214. [crossref]
- Park TH, Lakkis NM, Middleton KJ, Franklin J, Zoghbi WA, et al. (2002) Acute effect of nonsurgical septal reduction therapy on regional left ventricular asynchrony in patients with hypertrophic obstructive cardiomyopathy. Circulation 106: 412-415.
- Van Dockum WG, Beek AM, Folkert J, Jurrien M, Bondarenko O, et al. (2005) Early onset and progression of left ventricular remodeling after alcohol septal ablation in hypertrophic obstructive cardiomyopathy. Circulation 111: 2503-2508.
- Jensen MK, Almaas VM, Jacobsson L, Hansen PR, Havndrup O, et al. (2011) Long-term outcome of percutaneous transluminal septal myocardial ablation in hypertrophic obstructive cardiomyopathy: a Scandinavian multicenter study. Circulation: Circinterventions 1: 45-110.
- Maron BJ, Maron MS, Wigle ED, Braunwald E (2009) The 50-year history, controversy, and clinical implications of left ventricular outflow tract obstruction in hypertrophic cardiomyopathy: from idiopathic hypertrophic subaortic stenosis to hypertrophic cardiomyopathy. Journal of the American College of Cardiology 54: 191-200.
- Kunkala MR, Schaff HV, Nishimura RA, Abel MD, Sorajja P, et al. (2013) Transapical approach to myectomy for midventricular obstruction in hypertrophic cardiomyopathy. The Annals of Thoracic Surgery 96: 564-570.
- Minakata K, Dearani JA, O’Leary PW, Danielson GK (2005) Septal myectomy for obstructive hypertrophic cardiomyopathy in pediatric patients: early and late results. The Annals of Thoracic Surgery 80: 1424-1430.
- Orme NM, Sorajja P, Dearani JA, Schaff HV, Gersh BJ, et al. (2013) Comparison of surgical septal myectomy to medical therapy alone in patients with hypertrophic cardiomyopathy and syncope. The American Journal of Cardiology 111: 388-392.
- Quintana E, Sabate RA, Maleszewski JJ, Ommen SR, Nishimura A, et al. (2015) Septal myectomy after failed alcohol ablation: does previous percutaneous intervention compromise outcomes of myectomy? The Journal of Thoracic and Cardiovascular Surgery 150: 159-167.
- Theodoro DA, Danielson GK, Feldt RH, Anderson BJ (1996) Hypertrophic obstructive cardiomyopathy in pediatric patients: results of surgical treatment. The Journal of Thoracic and Cardiovascular Surgery 112: 1589-1599.
- Bonow RO, Frederick TM, Bacharach SL, Green MV, Goose PW, et al. (1983) Atrial systole and left ventricular filling in hypertrophic cardiomyopathy: effect of verapamil. The American Journal of Cardiology 51: 1386-1391.
- Nistri S, Olivotto I, Maron MS, Ferrantini C, Coppini R, et al. (2012) ß blockers for prevention of exercise-induced left ventricular outflow tract obstruction in patients with hypertrophic cardiomyopathy. The American journal of cardiology 110: 715-719.
- Setaro JF, Zaret BL, Schulman DS, Black HR, Soufer R (1990) Usefulness of verapamil for congestive heart failure associated with abnormal left ventricular diastolic filling and normal left ventricular systolic performance. The American journal of cardiology 66: 981-986.
- Sherrid MV, Shetty A, Winson G, Kim B, Musat D, et al. (2013) Treatment of obstructive hypertrophic cardiomyopathy symptoms and gradient resistant to first-line therapy with beta-blockade or verapamil. Circheartfailure
- Maron BJ, Shen WK, Link MS, Epstein AE, Almquist AK, et al. (2000) Efficacy of implantable cardioverter–defibrillators for the prevention of sudden death in patients with hypertrophic cardiomyopathy. New England Journal of Medicine 342: 365-373.
- Vriesendorp PA, Schinkel AF, Van CJ, Willems R, Jordaens LJ, et al. (2013). Implantable cardioverter-defibrillators in hypertrophic cardiomyopathy: patient outcomes, rate of appropriate and inappropriate interventions, and complications. American Heart Journal 166: 496-502.
- Faber L, Seggewiss H, Gietzen FH, Kuhn H, Boekstegers P, et al. (2005) Catheter-based septalablation for symptomatic hypertrophic obstructive cardiomyopathy. Zeitschrift für Kardiologie 94: 516-523.
- Faber L, Welge D, Fassbender D, Schmidt HK, Horstkotte D, et al (2007) One-year follow-up of percutaneous septal ablation for symptomatic hypertrophic obstructive cardiomyopathy in 312 patients: predictors of hemodynamic and clinical response. Clinical Research in Cardiology 96: 864-873.
- Hess OM, Streit S (2009) Alcohol Ablation of Hypertrophic Cardiomyopathy. Current Best Practice in Interventional Cardiology 5: 117-125.
- Osterne EC, Seixas TN, Paulo FW, Osterne EC, Gomes OM (2003) Percutaneous transluminal septal alcoholization for the treatment of refractory hypertrophic obstructive cardiomyopathy: initial experience in the Federal District. Arquivos Brasileiros De Cardiologia 80: 369-378.