The better understanding of the diverse mechanisms leading to the hemorrhagic transformation of an ischemic stroke is the crucial point in the prevention of this altogether common phenomenon. The different individual risk factors include the anatomical variability of collateral blood supply, age, genetics, body weight, etiology of the occlusion, side of the occluded vessel, renal status, stroke severity, history of high serum glucose or hypertension and hypertension or hyperglycemia at the onset of the stroke, ferritin level, INR, antiplatelet usage or the platelet count etc. All these might contribute to the process of hemorrhagic transformation. These risk factors have been identified by retrospective epidemiological studies and are useful to identify patients who are at high risk for hemorrhagic conversion and help the decision between conservative, thrombolytic or more advanced treatments such as thrombectomy. The modulations of potential molecular targets participating in the process seem to be promising in animal models, but human trials are lacking the breakthrough success so far, both in extending the time frame of the thrombolysis and in replacing the recombinant tissue plasminogen activator (rtPA) as thrombolytic agent.
So far, the careful preselection of patients eligible for thrombolytic therapy is the best way to prevent hemorrhagic transformation. The intensive research in the field revealing small but important molecular details are continuously contributing to the better understanding of hemorrhagic transformation with the intention of finding new agents that co-administrated to the rtPA or completely replacing that could decrease the risk of secondary bleeding.
The aim of this review is to give comprehensive insight into the process of hemorrhagic transformation of ischemic stroke, starting with basic overview of the penumbra concept and the collateral supply followed by discussion of the recanalisation-reperfusion-hemorrhagic transformation process after vessel occlusion also highlighting the importance of timing and ratio of recanalisation spontaneously and in case of thrombolytic agent administration. We overview the risk factors of hemorrhagic transformation, which may be helpful to physicians to identify high risk patients before rtPA administration. The last part of the work is dealing with future therapeutic possibilities based on the so far revealed molecular mechanisms of hemorrhagic transformation.
hemorrhagic transformation, stroke, blood-brain barrier, risk factors, molecular background
Abbreviations
rtPA: recombinant tissue plasminogen activator, BBB: blood–brain barrier, CT: computed tomography, HI-1 - hemorrhagic infarction 1, HI-2: hemorrhagic infarction 2, PH-1: parenchymal hematoma 1, PH-2: parenchymal hematoma 2, CS: collateral status, MCA: middle cerebral artery, ACA: anterior cerebral artery, ECASS: European Cooperative Acute Stroke Study, MCAO: middle cerebral artery occlusion, DWI: diffusion weighted imaging, PWI: perfusion weighted imaging, MMP: matrix metalloproteinas, PDGF: platelet-derived growth factor, PDGFR: platelet-derived growth factor receptor, HSP70: heat shock protein 70, CBV: cerebral blood volume, NCCT: non contrast CT, NOX: NADPH oxidase, NIHSS: National Institutes of Health Stroke Scale, AHT: asymptomatic hemorrhagic transformation, ROS: reactive oxygen species, DM: diabetes mellitus, NADPH oxidase: Nicotinamide adenine dinucleotide phosphate oxidase, NO: nitrogen monoxide, HMGB1: high mobility group box 1, VEGF: vascular endothelial growth factor, MT-MMP: membrane-type matrix metalloprotease, NSE: Neuron specific enolase, CSF: cerebrospinal fluid, OCLN: occludin, CLDN5: claudin 5, ZO-1: zona occludens 1, FDP: fibrin degradation products, APC: Activated protein C, EPCR, endothelial cell protein C receptor, PAI-1: plasminogen activator inhibitor 1, TAFI: thrombin-activatable fibrinolysis inhibitor, PAI-1: Plasminogen activator inhibitor-1, VAP-1: Vascular adhesion protein-1, SSAO: semicarbazide-sensitive amine oxidase, 15d-PGJ2: 15-Deoxy-Delta-12,14-prostaglandin J2, PPARγ: peroxisome proliferator-activated receptor gamma, A2M: alfa-2-macroglobulin, ASPECTS score: Alberta stroke program early CT score, HARM: hyperintense Acute Reperfusion Marker, ADC: apparent diffusion coefficient, FITC albumin: fluorescein isothiocyanate albumin, MAP2: anti microtubule associated protein, SOD1: superoxide dismutase 1, MARCH7: membrane associated ring CH 7, EGFR: endothelial growth factor receptor, MAP kinase: mitogen-activated protein kinase, TGF-ß: Transforming growth factor- ß, IRAK3: interleukin-1 receptor associated kinase 3, INP5P: inozitol phosphate-5-phoshpatase, LRP: lipoprotein receptor protein, GM-CSF: Granulocyte macrophage colony-stimulating factor, TIPM: tissue inhibitors of metalloproteinases, CCR2: C-C chemokine receptor type 2, CNS: central nervous system, MO/MP activation: monocyte/macrophage activation, COX: Cyclooxygenase, tMCAO: transient middle cerebral artery occlusion, TLR4: toll-like receptor 4, EPC: endothelial progenitor cell, SFK: SRC family kinase, GRE: Gradient-recalled echo, Bcl-2 gene: B-cell lymphoma 2 gene, AQP4: Aquaporin 4, TEER: transendothelial electrical resistance, TJ: tight junction, NPD-1: neuroprotectin D1, DHA: docosahexaenoic acid, PUFA: polyunsaturated fatty acid, ICAM-1: intercellular adhesion molecule-1, GSK-3β: Glycogen synthase kinase 3β, COMP: cartilage oligomeric matrix protein, TXNIP: thioredoxin-interacting protein, SMTP: Stachybotrys microspora triprenyl phenol, GLUT-1: Glucose transporter 1
The hemorrhagic transformation (HT) is a complication of ischemic stroke. In the clinical practice, it is indeed regarded as a complication, since it is the most feared consequence of thrombolytic therapy, although altogether the hemorrhagic transformation occurs quite often after ischemic stroke, between 10-40% depending on the individual influencing factors, suggesting that it is rather part of a normal process. We have to make difference between 2 main histological categories. The hemorrhagic infarct, which is actually petechial bleeding accounts for 89% of all HTs and has no effect on the prognosis or shows positive association with long term clinical outcome, since it is considered as sign of recanalisation. However, the parenchymal hematoma, 11% of all HTs, is definitely a complication, due to its detrimental effect on the prognosis.
Thrombolysis was the only available causal treatment for stoke patients until 2015, when the FDA approved the mechanical thrombectomy as effective treatment. Depending of various circumstances, unfortunately only less than 5-20% of all stroke patients are eligible for rtPA (recombinant tissue plasminogen activator) treatment, due to the narrow time window of the administration. Out of the time window, the risk of PH is getting so high, that risks overwhelm the benefits.
Most of the researches today are making huge efforts to achieve the extension of the time frame by preventing HT via BBB (blood–brain barrier) protection, recognizing the importance of complementary therapies beside rtPA. The better understanding of the underlying mechanisms and risk factors of this complex process will hopefully lead to more precise preselection of patients for thrombolytic therapy, newer therapeutic targets and to newer possibilities in the stroke management.
Reperfusion means restoration of the downstream capillary blood flow by recanalisation of the occluded vessel. The reperfusion can occur in anterograde way by spontaneous or therapeutic recanalisation or in retrograde way via collateral circulation. The aim of the recanalisation therapies (rtPA, intra-arterial fibrinolysis, neurothrombectomy) is the restoration of anterograde blood flow in the penumbra, including the microcirculation. Recanalisation and reperfusion have been regarded as one unit, but according the novel observations there are only a modest connection between them, indicating that recanalisation and reperfusion should not be used as synonyms. These observations can be explained with the no reflow phenomenon, which describes the situation where after a successful recanalisation of the occluded big vessel, in consequence of ischemia induced microvascular endothelium activation and microvascular clot formation, reperfusion at least partly will not occur. On the other hand reperfusion can occur without recanalisation as a result of retrograde filling due to better collateral circulation, although this process is hugely depending on the site of the occlusion, whereas a proximal occlusion is accompanied by more impaired collateral circulation [1]. Reperfusion and recanalisation is regarded to be a dynamic process that occurs gradually over hours with periods of partial reperfusion and the patency of the occluded vessel fluctuates during the recanalisation process [2,3].
With the aim to assess the rate and influencing factors on the recanalisation and reperfusion rate 381 patients with large-vessel occlusion was investigated. The patient group involved ones treated with rtPA or treated conservatively. Partial or complete recanalisation was achieved in 121 out of 210 (58%) patients after intravenous rtPA administration. The recanalisation success was more likely if the patient had atrial fibrillation [4] - maybe because cardioembolic clots consists more fibrin and are more likely to reopen by plasmin proteolysis than thrombosis of an atherosclerotic plaque – or in absence of early ischemic changes on CT as sign of better collaterals or smaller core. The significant extracranial (EC) stenosis or occlusion correlated negatively with the recanalisation success, since the decreased blood flow towards the clot is accompanied by decreased amount of rtPA reaching the clot and in addition the clot washout is weaker as well. In non-rtPA treated cases the spontaneous recanalisation rate was only 37% and was positively associated with history of hypercholesterinaemia, proximal occlusion, and inversely connected with decreased consciousness, extracranial or basilar artery pathology. It is important to mention that presence of recanalisation was assessed 24 hours after onset, and it is well known that recanalisation can occur even a week after onset [5]. Based on earlier studies it seems to be obvious that success and the benefit of reperfusion depends on the timing, the core size, the penumbra/core ratio and the collateral supply [6-11]. Further studies reported that milder baseline stroke deficits, elevated systolic blood pressures, normal glucose values, smoking history, absence of atrial fibrillation, distal vessel occlusion, and thrombus length positively predicts post-thrombolytic recanalisation. Considering these risk factors in individual cases, perhaps patients with smaller chance for reperfusion/recanalisation could benefit from more aggressive interventions, such as intra-arterial fibrinolysis [12-18].
Reperfusion and recanalisation can occur spontaneously up to several weeks after stroke onset, though according to the time is brain concept, it is irrelevant regarding the clinical outcome unless it happens within several hours after onset [19]. To demonstrate the dynamic of the spontaneous recanalisation a study investigated the timing of recanalisation after cardioembolic stroke. Out of 53 patients 10 showed spontaneous recanalisation in the first 6 hours, without HT. 19 patients showed recanalisation between 6-12 hours, with a HT rate of 60%. 15 patients showed no recanalisation in the first 48 hours, and accordingly the HT was lower, just 20%. All of the 3 patients with recanalisation between 24-48 hours showed HT. These results imply that reperfusion after 6 hours from occlusion is an independent predictor of HT. Another sub-analysis of this very study showed, that HI-1, HI-2, PH-1 were not related neither to early nor to 3 month outcome. Only PH-2 was associated with worse clinical outcome. In this study, all PH-2 occurred in the 6-12 h interval [20,21].
In order to, get more detailed picture of the underlying mechanism of the HT, spontaneously hypertensive rats were used to assess the dynamic of the reperfusion induced HT and lesion evolution. Due to the spontaneous hypertension of the rat models, 100% of the animals suffered HT. The results show that after 30 minutes MCA occlusion by day one 3/13, by day two 6/13, by day three 11/13 by day four 12/13, by day seven 13/13 animals presented HT. It means that the 92.3% of HT occurred within the first 4 day. The lesion volume increased from day 1 to day 2 as a result of vasogenic edema and decreased to its original size between day 4 and 7. The hemorrhage volume increased progressively, which meant an ECASS (European Cooperative Acute Stroke Study) class (HI-1, HI-2, PH-1, PH-2) evolution in 61,5% of the animals from a milder into a more severe category. This result suggests that HT is a quite dynamic process, which can last up to a week. HI-1 occurred most frequently by the periphery of the lesion and HI-2, PH-1, PH-2 were located rather central in the infracted area. Just PH-2 was associated with clinical symptoms. Only PH-2 was associated with worse clinical outcome.[1] As for the dynamic of the BBB breakdown, initial BBB disruption occurred in 3-4 h following the 2h MCAO in rat models with a peak at the 5th hour. Surprisingly, at 48h after MCAO, a delayed, more profound and expanded BBB permeability increase was observed. The supposed molecular background of this observation is detailed in the X/5 paragraph [22]. The vast majority of HT after rtPA treatment occurs within the first 24 hours [23].
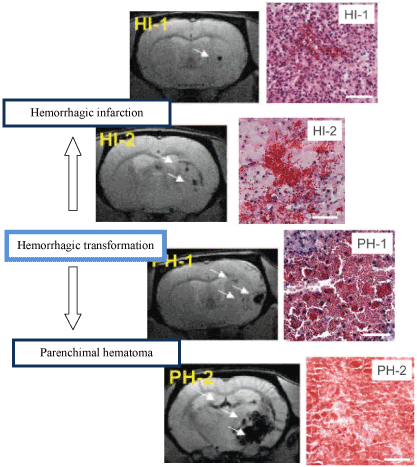
Figure 1. ECASS classification of HT (HI-1, HI-2, PH-1, PH-2). The left coloumn shows gradient-recalled echo images. Bleeding is hypointense (white arrows). The right coloumn shows images of hematoxylin-eosin–stained tissue. Bleeds are red; surrounding tissue is blue (nuclei) or pink (cytoplasm). HI-1 is a petechial bleeding occurring most frequently along the lesion periphery. HI-2: confluent petechial hemorrhage, located mainly int he infarct core. PH-1: real hematoma (<30% lesion volume) with or without mild space occupying effect . PH-2: hematoma (>30% lesion volume) with significant space occupying effect and perihematomal edema [21].
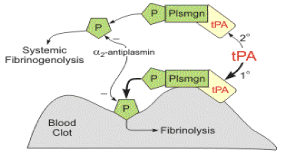
Figure 2. 1. tPA binds to fibrin on the surface of the clot. 2. Activates fibrin-bound plasminogen. 3. Plasmin is cleaved from the plasminogen associated with the fibrin. 4. Fibrin molecules are broken apart by the plasmin and the clot dissolves. Fibrin degradation products are released (modified from http://www.cvpharmacology.com/thrombolytic/thrombolytic).
A study from 2013 reported, that the hyperdense artery sign as a reliable indicator of occlusion disappears only in 55% of the patients after rtPA administration, suggesting a 55% recanalisation rate of thrombolysis [24]. The analysis of 5324 cases, where recanalisation was defined by CT or MR angiography as the resolution of dense artery sign, reports that out of 5324 cases 2592 (48,7%) showed recanalisation and 2412 (45,3%) not.
The best 3 month outcome could be expected in the patient group with 2 hours post treatment neurological improvement (NI) accompanied by vessel recanalisation. Both recanalisation and early NI are independent predictors of good 3 month functional outcome. After that, the patient group with neurological improvement in spite of persistent occlusion presented the second best prognostic prospects. Patients without neurological improvement despite recanalisation and the group without improvement and without recanalisation delivered the worst prognosis. Based on these findings a further, more intensive intervention, such as intra-arterial thrombolysis or neurothrombectomy warrants to be considered even in case of neurological improvement after rtPA, when reperfusion is not occurred, or the result is not satisfactory, despite the fact that the recent stoke treatment protocol declares clearly, that early NI is an exclusion criteria of further intravenous (IV) or intra-arterial (IA) thrombolysis, since early NI is an accepted sign of recanalisation after IVT, and has association with good 3 month prospects.
This very study supports the previous results of earlier studies, stating that even delayed recanalisation with mechanical thrombectomy or with other methods is related to better 3 month functional outcome [25]. For instance this earlier study ascertained that delayed (>6h) recanalisation has a favorable outcome regarding the DWI lesion expansion and can save peripheral cortical areas, in comparison to patients without recanalisation, implying that the collateral circulation may remain sufficient during a longer time interval [26]. Another systematic review and meta-analysis from 2012 support the known fact about rtPA treatment, the earlier the administration the better the outcome, although a subgroup of patients with individual characteristics and risk factors could definitely benefit from an extended rtPA administration interval [27].
As for the recanalisation rate and time course after intravenous thrombolysis, the following implies that the recanalisation itself is a dynamic process and the patency of the occluded vessel might fluctuate during the first 24 hours after treatment. Of 160 patients treated with rtPA, 82 patients (51.3%) showed recanalisation in the first 2 hours after rtPA administration monitored by transcranial Doppler. From these 82 patients 67 (81.7%) presented persistent recanalisation, which was investigated at 24 hours after rtPA treatment by computed tomographic angiography (CTA). From the original 160 cases altogether 84 (52.5%) showed recanalisation by the CTA at 24 hours regardless of the early recanalisation. As for the 3-month outcome the results imply that all kind of recanalisation in the first 24 hours is strongly associated with better outcome at 3 months [3].
Administered rtPA in the same way like endogen rtPA increases the MMP-2 [28], MMP-3 [29], MMP-9 levels in the brain [30]. It binds to protease activated receptor 1 on endothelial cells activating NF-κB pathway altering the vascular permeability and raising the MMP-9 level. Furthermore, rtPA due to its proteolytic activity removes an amino-terminal CUB domain from PDGF-CC homodimer causing its activation. The activated PDGF-CC than triggers PDGFR-α surface tyrosine kinase in the neurovascular unit, especially on astrocyte end feet, since it expresses a great number of PDGFR-α, increasing the BBB leakage [31-33].
Active rtPA injected in the cerebrospinal fluid of non-ischemic mice caused increased permeability of the BBB however administered intravenously was not followed by BBB leakage, suggesting that rtPA perform its negative effect on the abluminal side of the neurovascular unit.
The usage of Imatinib, a non specific tyrosine kinase inhibitor, co-administered to rtPA was associated with decreased BBB permeability, significantly smaller infarct size and lower HT rate, implying that imatinib interferes with PDGF signaling and PDFG signaling interferes with the BBB integrity.
rtPA is also capable of binding to the lipoprotein receptor protein on endothelial cells, increasing the emission of MMP-3 and MMP-9 [28,30,34]. Last but not least rtPA can support the degranulation of neutrophils into the blood, increasing the blood MMP-9 level [35].
This recent study investigated the time dependent effect of rtPA after 90min transient MCAO on spontaneously hypertensive rats. The first group of rats got no rtPA, the second got rtPA at the time of the recanalisation, and the third got rtPA 4 hours after recanalisation. MR imaging was performed 4 times, first during the occlusion and 3,6,24 hours after reperfusion. The BBB permeability on MR was examined as well, to assess the dynamic of the BBB opening. During the occlusion the permeability values were low in all groups. On the 3h MR image the second group presented the highest permeability values, but on the 6 h image the third group showed the highest BBB breakdown. On the 24h image the values were relatively decreased in comparison to earlier time points, but were even higher than during the occlusion. According to the observations above the highest HT (36,4%) and mortality rate (63,6%) was found in case of delayed rtPA administration. HT occurred mostly 6 h after reperfusion, when BBB permeability values were the highest. In the second group both the HT rate and the mortality was 10%. In the first group there was no observed HT or death [36]. These results are in agreement with other studies [37,38].
Penumbra is the so-called tissue at risk. This area is thought to be functionally silent and metabolically metastable but potentially salvageable if recanalisation occurs in a short period of time, and a full recovery can be expected. The size of the penumbra is individually different as a consequence of the hugely individual functional performance of the collateral system [39].
Approximately 8-12 hours pass by from the onset of the ischemic stroke till the infarcted area reaches its final size. There are several individual factors, such as site of vessel occlusion, degree of ischemic preconditioning, richness and molecular characteristics of collaterals [251,252], systemic blood pressure, blood volume, serum glucose, that influence this process. In general, the bigger the penumbra the bigger the potential benefit of rtPA administration.
The usage of rtPA has a recommended 4,5-hour time frame after stroke onset. This limit is necessary because of the ischemia induced BBB defect, which gets more severe in the course of time, resulting in an increased ratio for HT, especially for PH2. (parenchymal hematoma). However the „tissue at risk” could be salvaged between 8-12 hours, individually after stroke onset. That is why the researches today are pointed at the rtPA time window extension through BBB protection or improving the newer recanalisation methods like neurothrombectomy or trying to find alternatives to rtPA [19].
Collateral flow
Collateral flow system contributes enormously to the clinical outcome and tissue fate after an ischemic stroke event. In case of MCA (middle cerebral artery) occlusion collateral blood flow is supplied from the ACA (anterior cerebral artery) through the circle of Willis and through leptomeningeal anastomoses between cortical branches of MCA and ACA, and via extra and intracranial communication. Study results on rats with artificial induced transient ischemia showed strong correlation between the extent of collaterals and the ischemic core size. The molecular penumbra can be visualized by the presence of HSP70 Heat Shock Protein in neurons, which protein is the end product of natural defending pathways protecting the cell from further ischemic damage. The degree of collateral supply was inversely associated with molecular penumbra size and with final core size, which means the better the collateral supply the bigger the uninjured area. Acute MRI-defined penumbra is expected to be larger than molecular penumbra after 24 h of reperfusion due to the progressive recruitment of penumbra into the infarct core. The topographical mapping of the molecular penumbra revealed multiple patchy areas with irregular distribution and was not limited to the perilesional zone. Though, the finally infarcted area invariably consisted in one single lesion [39].
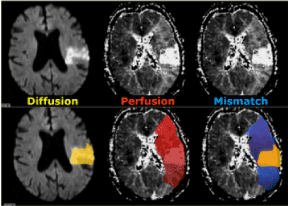
Figure 3. On the left side we have a diffusion MR image indicating the area with irreversible changes (dead issue). In the middle there is a large area of vital but hypoperfused tissue. On the right the diffusion-perfusion mismatch is indicated in blue. This is the tissue at risk that could be potentially saved with therapy.
(modified from http://www.radiologyassistant.nl/en/p483910a4b6f14/brain-ischemia-imaging-in-acute-stroke.html)
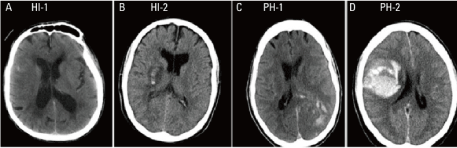
Figure 4. The four subtypes of hemorrhagic transformation (CT imaging) [253].
Better the collateral status, the bigger the penumbra and the smaller the core. That means more potentially salvageable tissue when the patient undergoes rtPA treatment. The better collateral status is associated with better recanalisation grade as well. Worse collateral status (CS) means bigger risk for worsening symptoms, increased severity and altogether worse clinical outcome. The penumbra is a mismatch between the diffusion CT image and perfusion CT image, so bigger the area with perfusion but without diffusion means bigger penumbra. Therefore, worse state of pial collateral system is associated with larger diffusion abnormality. The side of the clot and the CS are both independent factors of the clinical outcome among patients treated with rtPA. Favorable CBV (cerebral blood volume), NCCT (non contrast CT), and MTT ASPECTS (mean transit time Alberta Stroke Program Early CT Score) at arrival and favorable 3 month outcome were associated with good collateral status. Poor collateral status is associated with proximal occlusion and low NIHSS (National Institutes of Health Stroke Scale) on admission, but the distal occlusion is not predictor of good collaterals [40].
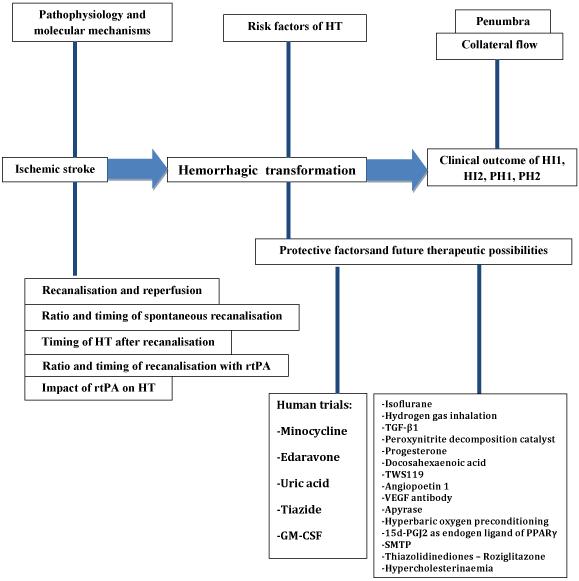
Figure 5. Overview figure about the process and the influencing factors of the hemorrhagic transformation after ischemic stroke. HT, hemorrhagic transformation; HI, hemorrhagic infarct; PH, parenchymal hematoma; rtPA, tissue plasminogen activator; TGF-β1, transforming growth factor β1; VEGF, vascular endothelial growth factor; 15d-PGJ2, 15-Deoxy-Delta-12,14-prostaglandin J2; PPARγ, peroxisome proliferator-activated receptor γ; SMTP, Stachybotrys microspora triprenyl phenol.
The performance of the collateral supply is worse when the occlusion occurs in the proximal part of the vessel and the location of the thrombus was found to be stronger predictor of the favorable outcome than the collateral status itself [41]. The admission cerebral blood volume is also lower in case of proximal occlusion of the vessel. The conclusion is that proximal occlusion results in worse collateral supply and hence less the salvable penumbra [42]. In case of proximal vessel occlusion, smaller infarct core and better CS rapid intravascular treatment had significantly favorable impact on the clinical outcome [43].
A prospective cohort study involving 134 patients with proximal MCA occlusion reports that the longer the ischemia, the better the CS is. This implies that during the ischemic attack the recruitment of collaterals is a dynamic process. In the first hour a fast and sufficient collateral recruitment occur in approximately 75% of the cases to refill the distal part of the occluded vessel, and in the course of the next 24 hours a slow secondary recruitment contribute maintaining the blood flow. The patients with sufficient collaterals had similar outcome than the control group without occlusion. The quarter of the patients with low collateral performance had significantly worse outcome [44].
As for the blood pressure, mild hypertension is associated with better collateral supply, although shows an inverse connection with 3-month outcome [40].
In a retrospective study of 395 patients baseline NIHSS score, hemorrhagic transformation, total ischemic volume, final infarct size, and a modified Rankin Scale score was investigated regarding the grade of collateral supply and recanalisation status. The greatest collateral benefit occurred in patients who were recanalisation negative (R-). Good collateral status independently and inversely predicted all outcomes except hemorrhagic transformation in patients with recanalisation negative status and mRS >2 in patients who were recanalisation positive (R+). That means that in non-recanalisation status the collateral system had no effect on HT. It also means that a patient with recanalisation but without collateral supply more likely to suffer a HT, and with good collaterals the risk of HT is significantly lower. HT occurred in 174/395 (44.1%) of patients. Hemorrhagic infarction (HI 1/2) occurred in 133/174 (76.4%) patients overall, but no difference in hemorrhage type (HI versus PH) was found between collateral-positive and collateral-negative groups for patients who were R+ or R-. In multivariate analysis, collateral presence was inversely related to HT risk in patients who were R+, while proximal clot location and hyperglycemia were positively related to HT in patients who were R+. Total ischemic volume was significantly associated with HT in patients who were R- but not in those who were R+. The frequency of poor collaterals in the R+ group was 30% whereas in the R- group it was only 11%, this indicates that most patients with acute stroke demonstrate sufficient collateral supply and in this aspect they are eligible for recanalisation therapy. Hyperglycemia was associated with a higher risk of hemorrhagic transformation in patients who were recanalisation-positive. HT was not related to collateral status in R- cases [45].
The poor pial collateral circulation was found to be associated with higher risk and larger size of HT. Identification of patients with worse collateral supply by angiography would contribute to better preselection of patient, who would not make profit from recanalisation therapy due to increased HT risk [46].
Hemorrhagic transformation occurs in 10-40% after ischemic stroke, depending on the circumstances and individual influencing factors. Because of this high rate of occurrence a lot of studies tried to assess the impact of HT on short and long term clinical outcome, which is a cardinal question, especially because rtPA therapy hugely increases the risk of HT. It is difficult to compare different studies dealing with this topic since hemorrhagic transformation can be defined radiologically or clinically.
- Clinically symptomatic or asymptomatic HT: Symptomatic hemorrhage based on the ECASS III is defined as any intracranial blood which is identified as direct cause of minimum 4 National Institutes of Health Stroke Scale (NIHSS) points deterioration. HT is related to severe hypoperfusion and bigger core size. HT mostly occur in these areas, therefore it is important to emphasize that HT can stay asymptomatic regardless its volume and that severe early clinical condition can make it hard to identify the HT-related deterioration [47]. Among patients with PH 54,5% were symptomatic [48]. The incidence of symptomatic HT after thrombolysis is 6-20% [49].
- Radiological/Histological criteria: The European Cooperative Acute Stroke Study (ECASS):
- HI-1: punctuate or small petechial hemorrhage
- HI-2: confluent petechial hemorrhage
- PH-1: real hematoma (<30% lesion volume) with or without mild space occupying effect
- PH-2: hematoma (>30% lesion volume) with significant space occupying effect and perihematomal edema [50]
Symptomatic intracranial hemorrhage is strongly associated to rtPA administration, which is the most feared adverse effect of the therapy, since symptomatic HT is independently associated with poor outcome [51] and was an independent predictor of in-hospital mortality [52]. Regarding the consequences of PH-2, the following report state that HT is not a reliable indicator of poor outcome, except PH-2, which is associated with higher early and even 90 days mortality in both rtPA and conservative treated patients.[2] Even when rtPA is used in a proposed manner 2,9% of the treated patients are going to die as a result of hemorrhage. ECASS-I and ECASS-II trials show, that only PH-2 is related to worse clinical outcome [243], and HI-1, HI-2, PH-1 are rather associated with better outcome as an indicator of reperfusion [21]. Other studies support the observation above and report that patients with PH had a highly increased risk of 24-hour deterioration and 3-month mortality [50.54,55].
In case of conservative treatment, neither spontaneous HI nor PH correlated independently with poor 3 month prognosis. The explanation of this observation is perhaps the lack of rtPA treated patients, which therapy is well known associated with higher rate of PH. It is assumed that patients with thrombolysis-related PH may have poorer prognosis than patients with spontaneous PH. Just to see the distribution of different outcomes, out of the total number of 407 eligible patients 12,3%, 50 patients suffered HT. HI occurred in 33 patients (66%), PH in 17 patients (34%), 32 patients (64%) with asymptomatic HT, and 18 (36%) with symptomatic HT. Out of the 33 patients with HI, 6 (18.2%) were symptomatic and out of the 17 patients with PH, 12 (70.6%) were symptomatic. The timing course of the HT was the following. HT was observed within the first 3 days in 11 patients (22%), in day 4–7 in 20 patients (40%), in day 8–14 in 11 patients (22.0%) and in day 15–30 in 8 patients (16%) [56].
As for the asymptomatic HT (AHT), vast majority of the studies report that there is no evidence, that asymptomatic HT would worsen the 3-month clinical outcome [57] and even thrombolytic-therapy related AHT does not deteriorate short-term and long-term clinical outcome [58]. Although several studies remark, that in spite of the assumption that asymptomatic HT is not connected with early clinical deterioration, the 3-month outcome is maybe by factor 2 worse, than in case of patients without HT [59]. On the other hand, another study reported that asymptomatic hemorrhage was an independent negative factor of the outcome, but asymptomatic hemorrhage after t-PA thrombolysis seemed to be associated with neurological recovery, as a supposed sign of recanalisation and reperfusion [60]. The 90-day outcome in the light of HT type is the following. The best outcome was observed without HT, than in descending order among patients with HI-1, HT-2, PH-1, PH-2. These results suggest that maybe prevention of all types of HT go along with better prognosis, probably because of the edema and toxic metabolites of extravascular blood [61]. Furthermore, this study gives evidence that if the long term outcome is assessed, the risk of poor outcome in case of asymptomatic HT was 1.51-fold higher at 3 months and 1.44-fold higher at 1 year than for patients without HT and as expected this ratio in case of symptomatic HT was 2.42-fold and 2.25-fold. Neither the symptomatic nor the asymptomatic HT affected the stoke reoccurrence incidence, suggesting that asymptomatic HT is not a risk factor for further HT [62].
The aim of finding reliable predicting factors of HT is to identify the potential risk population for HT with the intention of safe rtPA therapeutic window extension or to exclude patients who would not benefit from thrombolysis.
Time to reperfusion and rtPA usage
HT incidence after rtPA administration is 10 times higher compared to spontaneous reperfusion [2,63]. According to the theory of rtPA induced HT, the reperfusion itself is the main reason and also the requirement of HT, beside further pathways like free radicals [64] or increased MMP expression [65].
Several animal and human studies clearly report: The later the reperfusion, the higher the HT rate. For instance, in a study on mice where the connection between the duration of the artificial MCA occlusion and the HT rate was analyzed, 0,5 h occlusion caused no HT, ischemia lasting 5 hours resulted in 81,8% HT rate and 54,5% mortality rate and remarkably enough the permanent occlusion without reperfusion caused 18,2% HT incidence and 18,1% mortality rate. HT without recanalisation is a consequence of retrograde collateral flow. The observations above suggest the fact that reperfusion must have crucial role in the mechanism of HT but maybe not the reperfusion itself, but the time to reperfusion and thus the degree of BBB damage determines the HT rate [20,66].
However, a recent meta-analysis reported that reperfusion does not contribute significantly to HT neither with rtPA nor without rtPA, but in accordance with earlier studies, HT occurred twice as often with rtPA therapy. This observation implies that maybe further mechanisms beside reperfusion play more important role, than it was thought before [67]. Maybe the transportation of rtPA to the ischemic area and not the reperfusion itself results HT - since rtPA is supposed to act on the abluminal site of the neurovascular unit. This study also admits that other independent factors of HT such as ischemia severity and time to treatment play an important role as well. The duration of occlusion and the subsequent HT rate had a very strong correlation [24].
A further analysis of different aspects of HI and PH revealed, that HI is tend to be related to perfusion and PH is rather related to non-reperfusion, which observation match to the fact that HI has a better and PH has a worse prognosis, possibly related to reperfusion status. That is why HI is often regarded as a good sign, as a sign of reperfusion. HI and PH may differ in some other aspects too [67]. Furthermore, HI seems to be related to the severity and duration of the ischemia, while PH seems to be not [68].
Age
The exact way in which age contributes to HT is not clear, most likely the unspecific changes in the vasculature structure and the worse collateral supply is responsible. Elevated release of ROS and increased BBB permeability was also observed. Age is definitely a risk factor of HT, particularly in case of rtPA treatment, although altogether even elderly over 80 benefit from rtPA treatment [69]. Some other studies imply, that the dementia has effect on the stroke severity based on several molecular mechanisms [240,241,244,246,247].
Stroke severity/NIHSS (National Institutes of Health Stroke Scale)
According to the ECASS III classification of HT, symptomatic HT is defined by an increase of at least 4 points in the NIHSS [70]. Stroke severity, that correlates with the infarct size is independent and one of the most reliable predictive factor for HT [70]. Higher NIHSS on admission is associated with increased risk for symptomatic HT [71].
Systolic blood pressure
Acute high blood pressure is proved to contribute to HT by increasing the BBB permeability. Reducing blood pressure below 185/110mmHg before rtPA administration is part of the guidelines [72,73].
Hypertension history
Chronic hypertension alters vasculature structure also in the brain. This remodeling process involve increased vascular resistance, reduced vascular compliance and impaired collateral circulation and further contributes to BBB disruption with the tendency of increased ROS and MMP generation [2]. Apparently there is an interesting connection between age and hypertension. In chronic hypertensive rats there was no difference of HT rate between young and old animals, though among normotensive rats a higher incidence of HT occurred in the older group of rats, implying that in normotensive cases age might has higher influence on HT [21].
Hyperglycemic or spontaneously hypertensive rat models make out good candidates to investigate the background mechanisms of HT, new neuroprotective and BBB protective agents, because of their tendencies to higher HT rate.
Glucose
Acute high glucose level is strikingly associated with higher rate of BBB disruption, HT and worse outcome in all animal models and in human retrospective studies as well. 30-60% of all patients with stroke have elevated admission serum glucose level, as a result of increased sympathetic activity after stroke. A smaller part of the patients has manifest diabetes mellitus. Advantages and disadvantages of serum glucose level correction in acut stroke patients is under debate, since overcorrection related hyperglycemia can be fatal.
Effect of increased glucose level at the time of stroke has two sides. During the ischemia, higher level of serum glucose has the potential to maintain the ATP level in the neurons even in the lack of oxygen via anaerobic glycolysis which is inevitably followed by lactate acidosis, causing altogether longer cell survival interval, giving time for intervention. Lactic acidosis under a certain level has an assumed neuroprotective effect due to inhibiting NADPH oxidase, but after a while severe low pH causes protein denaturation itself, although there is no evidence that the negative effect of hyperglycemia is preventable by normalizing the pH. In contrast, during the reperfusion high glucose level has definitive destroying effect on the BBB, because in case of reperfusion the by hypoxia and acidosis blocked NADPH oxidase is released from inhibition, and its superoxide production is going to be enhanced by hyperglycemia.
The neurons are able to metabolize exclusively glucose, but not fatty acids. In details, glucose is the exclusive substrate in production of NADPH, which has central and biphasic role regarding the ROS formation and elimination. NADPH is part of the pathway producing glutathione, which is strongly antioxidant. In addition, NAHPH is used by nitric oxide synthase to generate NO and by NADPH oxidase to produce superoxide. Oxygen free radicals in ischemia-reperfusion are generated in mitochondrial electron transport chain as well. This process is absolutely glucose-dependent since all reducing equivalents passing through the mitochondrial electron transport chain originate from glucose.
Other mechanisms of glucose exacerbated stoke are supposed to involve enhanced glucose-sodium transporter activity, increased abnormal protein glycosylation and intensified post ischemic inflammatory response. As expected in the core region, where no blood flow can be detected, the serum glucose level had no influence. Supported by the results of several experimental studies, the high serum glucose indeed did not exacerbate the core injury and might rather have beneficial effect. In the area of the penumbra, between the core and the non-ischemic tissue, where decreased but present blood flow is detectable; the higher glucose can have its above discussed negative effect.
The excess of glucose over oxygen forces neurons to continue glucose metabolism in an anaerobic way causing lactate acidosis. This pathway produces just 1/16 part of the ATP/glucose molecules that the aerobic pathway would be able to generate. Penumbra is a dynamic and unstable area, where a relatively high blood-flow can wash the lactate out. Due to this heterogeneous effect it is hard to predict the effect of hyperglycemia in ischemic brain tissue, nevertheless in most cases the negative effect surely dominates. In the reperfusion phase via ROS production, hyperglycemia has undeniably negative impact [74].
Further studies give evidence to the theory above. Among patients with proved diffusion/perfusion mismatch, admission hyperglycemia was strongly associated with infarct size, progression of penumbra into definitive infarct, and lactate peaks in the area of penumbra. In comparison, among patients showing no diffusion-perfusion mismatch admission hyperglycemia was not associated to the parameters mentioned above [75]. As expected, in case of lacunar infarcts, where end arterial vascular areas are occluded and the collateral supply is weak, hyperglycemia had no notable negative but had rather positive effect [76].
Evidences show that high glucose level has direct effect on endothelial cells causing increased edema formation, hemorrhage transformation and reduced microvascular reflow. The underlying mechanisms include increased endothelial protein kinase C activation, amplified inflammatory responses and elevated superoxide generation [77-79].
Hyperglycemia also contributes to rtPA induced HT and poor outcome, even in the combination of mild hyperglycemia and low dose rtPA [80,81]. This effect could be suspended in rat models with the NADPH oxidase inhibitor apocynin in ischemia-reperfusion models suggesting that hyperglycemia enhanced HT is mediated by NADPH produced superoxide [82].
Based on the observation that hyperglycemia increased the HMGB-1 level in brain tissue early after stroke onset and that all of the negative effects of hyperglycemia mentioned above were suspended by the administration of a specific HMGB-1 inhibitor, glycyrrhizin it is suspected that extracellular HMGB-1 contributes to the hyperglycemia deteriorated clinical outcome and neurological deficit, larger infarct volume, edema and BBB disruption [83].
Several human studies proved as well that hyperglycemia have unfavorable effect on infarct growth and poor outcome with or without thrombolysis [84,85]. An ongoing trial at the time of this review called the Stroke Hyperglycemia Insulin Network Effort (SHINE), tries to provide novel information about the risks and benefits of hyperglycemia management in acute stroke patients [86].
Diabetes
History of high glucose level also contributes to higher HT incidence in several supposed ways [87]. Most likely the ROS induction [88], a modified leukocyte function [89], impaired BBB permeability [90], microvascular remodeling, altered vessel structure and angiogenesis via VEGF and peroxinitrit signaling [91], involving membrane-type matrix metalloprotease (MT-MMP) and c-src kinase activation [92] and further inflammatory cascades contribute to the HT [93]. A further study reports that after streptozotocin induced hyperglycemia and transient MCA occlusion the infarct volume and area was not altered by hyperglycemia, but the HT volume was significantly higher. The main cause was found to be the endothelial dysfunction, in particular, a mitochondrial dysfunction, which was provoked by MMP-9 in a ROS independent manner, causing caspase-3 activation. This key movement starts the apoptotic cascade. The process continues in fragmentation and vacuolation of the mitochondria with subsequently decreased ATP level and decreased mitochondrial membrane potential followed by decreased cell proliferation, worse regeneration capacity, severer BBB disruption, and aggravated HT volume [87].
HbA1c was found to be an important predictor of symptomatic HT after ischemic stroke [94].
Body weight
Increased body weight was found to be an independent predictor of symptomatic HT. Especially patients above 95 kg are at high risk [80].
Congestive heart failure
Congestive heart failure as a source of cardiac embolic stroke was observed to be associated with higher incidence of symptomatic HT and PH. The assumed background mechanism is the distal migration of embolic fragments that allow reperfusion of ischemic-weakened vessels [95].
Atrial fibrillation
Presence of atrial fibrillation in stroke patients most likely via poorer collateral blood supply is associated with more severe hypoperfusion and hence with increased infarct growth and volume, more frequent and more severe HT and worse stroke outcome [96].
Renal impairment, estimated GFR
Reduced eGFR did not increase the risk of symptomatic HT, but was associated with significantly increased HT rate, especially in strokes with large artery atherosclerosis but not in strokes with cardioembolic etiology [97].
Antiplatelet usage
An altered hemostasis contributes to HT when BBB disruption already occurred, and may make HI to evolve into PH. Antiplatelet or dual antiplatelet usage before rtPA treatment or antiplatelet use within the first 24 hours of rtPA treatment increases the risk of rtPA-related HT [95,98]. Dual therapy of aspirin and clopidogrel has strong association with the risk of symptomatic HT after rtPA treatment. Aspirin intake as monotherapy is an independent predictor of HT as well, however not as strong as the dual platelet inhibition [80].
Platelet count
Platelet count under 100.000/ µl is a contraindication for rtPA administration [99].
Anticoagulant/international normalized ratio/partial thromboplastin time
INR>1,7 is a contraindication for rtPA [99]. After cardioembolic stoke, secondary prophylaxis with administered heparin or enoxaparin hugely increases the risk of HT, though warfarin prophylaxis seems to be safe even if started shortly after stoke [100], though this question is quite complex and still under debate.
Blood markers
NSE: Neuron specific enolase has a 48 hour half life in serum and can be found in neurons and in the neuroendocrine system [101]. It is reported, based on serial NSE serum level measurements of 83 patients after stroke onset that the NSE level changing pattern significantly depends on the stroke mechanism. Specifically, a second peak of NSE level was associated with cardioembolism and hemorrhagic transformation. The first peak of NSE level is normally reached within the first 96 hours after onset, but the exact timing strongly depends on the size of the penumbra causing a continuous cell death and a continuous elevating or at least maintained NSA level [102]. NSE was found to be a significant predictor of lesion volume, early and 30-day functional outcome [103].
S-100 Protein: Measurement of serum S-100 protein levels in the first 10 days after stroke onset helps to predict long term clinical outcome and final infarct size [102]. The level of S-100 protein independently predicts HT before rtPA treatment, however with too low diagnostic accuracy to be useful in the preselection of patients for rtPA therapy [104,105].
MMP-9 level: Analysis of peripheral blood samples taken from stroke patients revealed an increased level of MMP-9 level. The elevation correlated with worse clinical outcome, bigger infarct size and higher rate of HT. MMP-9 level predicted the HT with the sensitivity of 87%, a specificity of 90%, a positive predictive value (PPV) of 61% and a negative predictive value (NPV) of 97%. The source of the MMP-9 elevation in early stroke is the over-expression of MMP-9 gene in neutrophil granulocytes. MMP-9 and neutrophil levels are elevated in the cerebrospinal fluid as well. Since rtPA itself promotes neutrophil degranulation, a peak of MMP-9 elevation can be detected 30 minutes after rtPA administration as well [105-107].
Fibronectin level: High plasma fibronectin level after ischemic stroke is significantly and independently associated with subsequent HT in case of rtPA administration, as a possible marker of the extension of BBB disruption. The elevated fibronectin in the serum arise at least from two sources. First the disruption of the BBB and second, as a natural response to injury, an increased fibronectin production leads to serum fibronectin elevation. Experimental animal studies have reported that endothelial cell injury induces fibronectin production to promote re-endothelialization. Furthermore, administration of synthetic fibronectin peptide V within 3 hours after reperfusion reduces the final infarct volume [107].
Tight junction proteins: Circulating principal TJ proteins like occludin (OCLN), claudin 5 (CLDN5), zona occludens 1 (ZO-1) are biomarkers of BBB damage and evaluation their serum levels in case of early stoke may help to screen patients who are at high risk for HT caused clinical deterioration, since they correlating with the extension of BBB breakdown and HT occurrence. The degradation and increased appearance of these proteins in the serum is mainly mediated by MMP-9 overexpression [105].
Fibrinogen level: The analysis of blood samples taken from stroke patients 2 hours after rtPA administration show a strong correlation between the fibrin degradation protein level and the PH occurrence rate. A supposed coagulopathy caused directly by the thrombolytic process involves excess fibrinogenolysis early after thrombolysis with the release of fibrin degradation products (FDP), acting as antithrombin and inhibiting fibrin polymerization. The risk of PH above 200mg/L of FDA level become significant, although already above 100mg/L the potential risk for PH is increasing. Hence patients with FDP > 100 mg/L at 2 hours after rtPA administration based on their prehemorrhagic antithrombotic state, new antithrombotic drugs should be avoided for 72 hours. FDP level is most likely dependent on the thrombolytic agent. FDP level was significantly higher in patients treated by streptokinase than in patients treated by rtPA, might contributing to the higher level of symptomatic HT rate in case of streptokinase administration [48].
Ferritin level: Elevated serum ferritin level in the first 24 hours after stroke onset is independently associated with HT and PH as well. In case of PH the risk is 4.9 times higher compared to patients with normal admission ferritin levels. Furthermore, ferritin level above 171.8 ng/ml was independently connected to symptomatic HT with a 5.7 times higher odds [108]. Also, earlier studies reported, that higher ferritin level is related to poor outcome and large lesion size in patients treated with rtPA. The supposed underlying mechanism of this neurotoxic effect caused by increased body iron storage involves production of hydroxyl radicals and endothelial injury [109]. The hypoxia induced superoxide radicals have the ability to release free, toxic iron from ferritin. Free iron generates further free radicals, initiates and propagates lipid peroxidation contributing to BBB breakdown. The deleterious effect on this pathways are assumed to be dependent on the concentration of tissue iron, hence serum ferritin as indicator of body iron saturation can predict HT rate [110].
Activated protein C: APC is a plasma serine protease with a well known anticoagulant effect and a recently recognized anti-inflammatory, anti-apoptotic, neuro- and vasculoprotective effect. These phenomenons are presumed to be based on endothelial cell protein C receptor (EPCR) and protease-activated receptor 1 activation leading to gene expression modulation in endothelial cells. These beneficial effects made APC eligible for further studies with the hope to extend rtPA window. Unfortunately, only in vitro and rodent models showed favorable results. In humans, an increased level of APC after 2 hours of rtPA treatment was strongly associated to HT. The elevation of the APC level after thrombolytic therapy was well known but now its negative influence on HT is clarified. Apparently, in humans the anticoagulant effect overwhelms the neuro- and vasculoprotective effect. The impact on the coagulation system involves irreversible inactivation of Va and VIIIa factors breaking the amplification of the coagulation process. In addition, it inactivates plasminogen activator inhibitor 1 (PAI-1) and weakens the activation of thrombin-activatable fibrinolysis inhibitor (TAFI), promoting fibrinolysis [111-113].
Thrombin activatable fibrinolysis inhibitor (TAFI): High baseline of TAFI level on admission were associated with higher rate of symptomatic HT [114] and was reported to be associated with better reaction on rtPA treatment with higher recanalisation rate, although TAFI inhibits fibrinolysis. The background of this apparent paradox is might be the TAFI consumption theory, which says that lower level of TAFI indicates higher affinity to fibrin and therefore high TAFI activity and elevated TAFI indicate low activity leading to fibrinolytic tendencies [115].
Plasminogen activator inhibitor-1 (PAI-1): Low baseline PAI-1 levels on admission are correlating with symptomatic HT after rtPA treatment. The underlying mechanism includes the altered fibrinolytic system and a tendency for bleeding disturbances [114]. High level of PAI-1 was found to be associated with worse answer to rtPA therapy with higher rate of recanalisation resistance [116].
Vascular adhesion protein-1 (VAP-1): The inflammatory response after stroke is mainly promoted and maintained by polymorphonuclear cell extravasation into the infarcted area. In this process participates the VAP-1 protein which is an important protein on the surface of endothelial cells – and circulating protein as well - involved in recruitment of lymphocytes and neutrophils via its semicarbazide-sensitive amine oxidase (SSAO) activity. Polymorphonuclear cells are indeed presented in big density around VAP-1 positive vessels in the infarcted area, therefore further studies are planned to examine a VAP-1/SSAO inhibitor with the purpose of extending the rtPA time window. There was no difference found regarding the serum VAP-1/SSAO activity between stroke patients without HT and control group, but elevated baseline VAP-1 level/SSAO activity measured before rtPA treatment strongly predicted PH and worse neurological outcome, making VAP-1 a possible candidate to efficiently preselect stroke patients eligible for rtPA therapy [117].
PDGF-CC isoforms: PDGF signaling pathway consists four ligands, PDGF A,B,C,D and two receptors, PDGFR alpha and beta. The ligands form functional homodimers, except for A and B that can form functional heterodimers as well. rtPA activates CC homodimers allowing CC dimers to trigger PDGFR alpha leading to increased BBB permeability, furthermore the blockage of PDGFR alpha with imatinib, which is tyrosine kinase inhibitor, can weaken the hemorrhagic complications caused by rtPA administration. All these reports point out that PDGF pathway has a central role maintaining BBB integrity. Serum PDGF-CC levels on admission and after 24 hours of rtPA treatment are associated with HT [33].
15d-PGJ2: Elevated plasma 15d-PGJ2 concentration is associated with good early and late neurologic outcome and with smaller infarct volume. 15d-PGJ2 is a non-enzymatic modified variant of PGD2. 15d-PGJ2 is an endogen ligand of PPARγ which is a nuclear hormone receptor playing an extended regulating role in several cellular signaling mechanisms, such as apoptosis, oxidative stress and neuroinflammation [118].
Uric acid: As an endogenous antioxidant: High uric acid level was significantly associated with lower rate of HT and with better clinical outcome among patients treated with rtPA. Uric acid, based on its antioxidant effect neutralizes ROS burden and all the complex mechanisms connected to reactive oxygen species in case of an ischemic stroke [119].
Calcium
Low serum calcium level at admission was significantly and independently associated with HT after rtPA treatment. Two underlying mechanism is assumed. First, calcium is an essential cofactor in the coagulation cascade; hence lower serum calcium level may promote coagulation disturbances causing bleeding complications. Second, decreased serum calcium level may induce vasoconstriction in the peri-ischemic zone leading to locally elevated blood pressure and HT [120].
Genetics
Leukocyte mRNA (Amphiregulin, MARCH7, SMAD4, IRAK3, INPP5D, MCFD2, VEGI) (see also X/4)
alfa-2-macroglobulin (A2M): Single nucleotide polymorphism rs669 (Val1000Ile) in A2M was associated with HT. It may induce decreased activated/inactivated A2M ratio and decreased A2M activity. A2M is a general protease inhibitor. After activation it sterically binds proteases such as rtPA, plasmin or MMP-9. Then, the complex binds to lipoprotein-related protein, followed by internalization and degradation. A2M was reported to play role in migration and degranulation of neutrophils as well. Elevated level of A2M in the CSF is a reliable sign of BBB disruption [121].
Factor XII: rs1801020 (4C>T) mutation of Factor XII predicted in-hospital death after rtPA administration due to increased factor XII activity. The observed phenomenon is assumed to be based on interference in the fibrinolytic and kinin-kallikrein pathways [121].
Factor XIII: Carriers of FXIII V34L polymorphism have 2.5-fold risk to die after rtPA treatment than patients with V/V genotype, since these patients show significantly higher HT rate. This polymorphism may affect the formation and structure of the fibrin clot [122].
Collagen IV: Mutations in the COL4A1 gene, which encodes procollagen type IV alpha 1 is promoting small vessel disease and intracerebral hemorrhage by altering the structure of the basal lamina. These mutations may influence the process of HT in ischemic stroke as well [123,124].
MMP-9: -1562C/T polymorphism in the matrix metalloproteinase-9 gene was found to be related to HT. T allele was a protecting factor of HT in the investigated population [125].
Survivin gene: 241C/T polymorphism in the promoter of the survivin gene is associated with lower risk of HT. Stroke patients presenting this mutation show less severe HT if the HT occur and have lower risk for PH. Survivin is an anti-apoptotic molecule inhibiting caspase-3 [126].
Neuroimaging
Infarct size/diffusion weighted imaging infarct volume: Increased lesion on DWI and increased delay on PWI is associated with higher rate of HT. Severe delay on PWI was proven to be more important factor for HT formation than tissue status on DWI [49,127].
Low cerebral blood flow or volume: Baseline very low cerebral blood volume on MRI imaging is a powerful predictor of PH after thrombolysis; furthermore it appears to be more powerful than DWI or PWI [128].
Early infarct sign: Alberta stroke program early CT score (ASPECTS) is a topographic CT scan score. The lower the ASPECTS score the bigger the infarcted area, thus bigger the risk of HT. ASPECT score is an independent predictor of HT with the sensitivity of 85,5% and with the specificity of 75% [69,129,130].
Dense cerebral artery sign: Hyperdense cerebral artery sign is independently predictive for HT after thrombolytic therapy [130,131].
Leukoaraiosis: White matter hyperintensities. Describes the nonspecific changes in the cerebral white matter frequently seen on CT and MRI in aged individuals and even young adults sometimes. Many patients can have leukoaraiosis without any associated clinical abnormality. However, underlying vascular mechanisms are suspected to be the cause of the imaging findings. Hypertension, smoking, diabetes, hyperhomocysteinemia, and heart disease are all risk factors for leukoaraiosis. Leukoaraiosis was proved to be a risk factor for symptomatic HT after rtPA administration [69,132].
MRI enhancement pattern: Early gadolinium enhancement on T1 weighted MR images of acute stroke patients are good predictors for subsequent symptomatic HT [133].
BBB permeability: Admission CT perfusion derived permeability-surface product maps show promising results in distinguishing patients who are likely to develop HT after ischemic stroke [134].
Hyperintense Acute Reperfusion Marker (HARM): HARM is the gadolinium enhancement of the cerebrospinal fluid and is regarded as a reliable sign of BBB disruption. Thrombolytic therapy is an independent factor of HARM, and HARM is an independent predictor of HT [135].
Apparent diffusion coefficient value: The percentage of low ADC values in ischemic areas destined to HT are greatly higher than in other lesion areas [125].
Collateral flow: After occlusion of a main artery, leptomeningeal backfilling can determine the size of the penumbra and the final lesion size as well. Poor collaterals are associated with larger infarct size and worse functional outcome at 3 months and this promotes higher HT rate, but just in case when recanalisation occurs, suggesting that reperfusion has a major role in HT. Assessment of baseline collateral supply can contribute to the decision leading to thrombolytic therapy [136].
Rating scores
Rating scores are suitable to identify patients who are at high risk for cerebral parenchymal hemorrhages, but alone none of them were sensitive enough to exclude otherwise eligible patient from rtPA treatment. According to a prospectively collected data of 3012 eligible patients, the SPAN-100 has the worse, and SEDAN has the best predictive power, though none of the scores reached better than moderate performance.
- HAT - Hemorrhage After Thrombolysis (NIHSS, glucose, early infarct sign, diabetes)
- MSS - Multicenter Stroke Survey (age, NIHSS, glucose, platelet count)
- SITS-SICH - Safe Implementation of Thrombolysis in Stroke (age, NIHSS, glucose, systolic BP, hypertension history, body weight, time to thrombolysis, antiplatelet)
- SEDAN score (blood sugar, early infract sign, dense artery sign, age, NIHSS)
- GRASPS score (glucose, race, age, systolic BP, NIHSS, sex)
- SPAN-100 index (age, NIHSS)
- iScore (age, NIHSS, glucose, sex, stroke subtype, atrial fibrillation, congestive heart failure, prior myocardial infarction, smoker, cancer, dialysis, prior disability) [137].
To provide a detailed picture about the structural changes during the BBB breakdown investigators used 3 different rat groups with different occlusion duration including rats with transient MCAO lasting 90 minutes, rats with permanent MCAO and embolic MCAO. Result analysis proceeded in multiple levels. Endothelium was visualized by fluorescence microscopy with the help of isoleticin B4, furthermore, FITC albumin was administered after 24 hours from ischemia onset. After that, in the histological process peroxidase conjugated anti FITC IgG was applicated with subsequent staining by diaminobenzidine to make the leaked FITC albumin visible for light and electron microscopy demonstrating the presence of the BBB leaking. Anti occludin, claudin-3, claudin-5 were used to make the TJs visible. Anti microtubule associated protein (MAP2) and anti heat shock protein 70 (HSP70) detection was used to define the molecular penumbra.
The results are the followings: Isolectin IB4 Antibody Staining showed endothelium alternations in areas of extravasation. This endothelium lost via apoptosis was found to be the main source of the BBB disruption. The endothelium loss had four good separable ultarstructural phases.
- First stage: Endothelial cell swelling, intracellular edema, no FITC-albumin extravasation.
- Second stage: Endothelial membrane fails to maintain the barrier function, the apical membrane becomes leaky, causing intracellular FITC-albumin-DAB grains, less cell edema and the basal membrane is intact.
- Third stage: Endothelial structure is completely lost, FITC albumin-DAB in the neuropil, beyond the basal membrane which is intact.
- Fourth stage: The basal membrane is degradated via MMP upregulation, causing erythrocyte extravasation, ending up in HT
In areas where FITC albumin extravasation was observed vessels showed stage 3 and stage 4 degeneration. Though, outer from the border zone of the tracer extravasation no stage 4 was observed but the vessels mostly presented stage 1 degeneration which corresponds to endothelium swelling. This swelling is partly the consequence of ischemia activated connexin-43 hemichannels activation. The results imply that stage 1 represents the initial phase of the BBB breakdown and that stage 3 or 4 is requirement of the extravasation. The tracer extravasation exceeded the by HSP70 and MAP2 defined molecular penumbra. The border zones of the tracer extravasation correspond to the outer layers of the penumbra. FITC extravasation border zone did not match with the molecular penumbra.
TJ proteins were unaltered in all groups. Occludin, ZO 1 and claudin ratio of the affected area did not differ from the contralateral hemisphere. The staining pattern was intact, however their special function is supposed to be altered [138]. Several studies proved that ischemic circumstances are causing TJ protein degradation followed by BBB breakdown. Surprisingly there were no difference in the grade of endothelium degeneration and FITC leaking in the transient and in the permanent group, which implies that loss of endothelial barrier is not a reperfusion injury [139,140]. This observation may serve as a base to the recent meta-analysis which found no connection between reperfusion and HT rate [67].
Due to resolution limits of imaging techniques, it is not surprising that autopsy reports show 71% HI rate among ischemic stroke cases. As for the mechanism of PH, it is likely that the primary bleeding of smaller vessels and the subsequent neuroinflammatory response is responsible for the secondary disruption of surrounding vessels [21,242]. Pathophysiology of ischaemic stroke and the subsequent HT involves various pathways that will be discussed in the further paragraphs.
The blood brain barrier comprises endothelial cells, pericytes, astrocytes and basal lamina. The basal lamina is basically built from collagen IV, laminin and fibronectin. The tight junctions between the cells are constituted of occludin, claudin-3, claudin-5, ZO-1, ZO-2 and ZO-3.
The time course of the BBB disruption can be assessed by contrast MRI. In the BBB disruption 2 peaks can be observed, the first take place within the first 2-6 hours after stroke onset, and is enhanced between 4-8 hours and between 12-16 hours after onset, which suggests that the infarct is rather a plastic and the reperfusion is a dynamic process, with hyperemic and hypoperfusion phases. The second peak is a delayed BBB disruption which occurs after 18-24 hours after onset and lasts for up to several weeks. The molecular mechanisms of the early and delayed disruption differ in several aspects. The early BBB breakdown is mediated by ROS, blood MMP-9 and MMP-3, and the delayed disruption after 18-24 hours is promoted by brain MMP-9, brain MMP-3 and several other brain proteins, vascular remodeling and neuroinflammation. Although the molecular pathways are different, they are not sharply separated; there are several overlaps and positive feedbacks. The point is that early HT does not predict the delayed HT and that the therapeutic targets supposed to be different [2].
ROS, like superoxide and peroxinitrit, arise mainly after reperfusion from different sources; from ischemic damaged cells or from inflammatory cells, as crucial part of the neuroinflammatory process. The ROS forming mechanisms include changes in intracellular mitochondria and in the NADPH oxidase pathway, MMP mediated proteolysis and the positive feedback of inflammatory mediators. Reperfusion causes a ROS burst with a subsequent greatly elevated level of ROS, compared to non-reperfusion mice, where a low initial level of ROS was followed by a slow increase in the first 3 hour. The early reperfusion mediated ROS burst can be effectively prevented by administration of ROS inhibitors in animal models, but unfortunately not in humans.
In addition, mice with SOD1 (superoxide dismutase 1) defect or NOX2 (nicotinamide adenine dinucleotide phosphate oxidase) show a dramatically reduced rate of BBB disruption as a consequence of milder ROS burden. Blood markers of ROS burden like F2 isoprostane or 3-nitrotyrosise are elevated after 6 respectively 3 hours after stroke onset. Both of them are end products of non enzymatic oxidation by ROS. The associated factors enhancing ROS injury are the followings: age, glucose level, diabetes mellitus, infarct size, congestive heart failure, renal impairment [2].
The natural reaction after an ischemic injury is the powerful activation of the immune system. After 30 minutes of ischemia occurs the first step, namely the leukocyte adhesion to the vascular endothelial cells of the affected area and by 6 hours the leukocyte infiltration. Both process increase capillary permeability. The plasma activity of MMP-9 in mice within 3-8 hours is increased. After stroke, the MMP descend from two sources, from the brain cells and from immune cells. In leukocyte depleted mice the early disruption did not occur, whereas in mice with brain MMP-9 deficit early disruption occurred, but not the delayed disruption. Conclusion is that the early sources of the MMP-9 are the leukocytes. MMP-9 inhibitors decrease the HT incidence. In humans, serum MMP-9 level rising early, and reaches its peak by 6-8 hours, and returns its basic level by 24 hours. Although a couple of studies have found increased level of MMP-9 after 24 hours, whose reason is unclear, but maybe the size of the lesion, co-morbid diseases or a biphasic elevation is responsible, in addition MMP-9 mRNA in leukocytes are also elevated by 3-5 hours and return to baseline by 24 hours. Depleting leukocytes with vinblastin or anti neutrophil antibody can decrease BBB breakdown and neutrophil activation with lipopolysaccharide can increase disruption. MMP-9 acts on the tight junctions and on the basal lamina. MMP-9 from the blood destroy directly the tight junctions or can be taken up by endothelial cells itself and then acting on the basal lamina. Another way is that infiltrating neutrophils emit MMP that reaches TJs and basal lamina. The level of TJ degradation products in blood is predictive for HT.
Brain MMP-2 level is increasing in 1-3 hours after onset and additionally remains elevated for days. This observation has a supposed role in early disruption, but its main effect contributes to the delayed HT. Possibly sources of MMP-2 are astrocytes, endothelial cells and leukocytes. Activators of MMP-2 expression are furin, thrombin, xanthine oxidase, MTI-MMP. All of them show an elevated serum level after stroke. MMP-2 knockout mice presented reduced HT rate, smaller hemorrhagic volume and better neurological function. MMP-2 seems to ha a role in both early and delayed form of HT.
Brain caveolin 1 membrane protein level is decreased during the ischemic attack, and according to a study on cav 1 knockout mice maybe caveolin in the brain tissue is associated with ROS and MMP-2 inhibition and contributes to the BBB integrity by influencing the claudin-5 TJ protein [2].
After analyzing the mRNA expression pattern in blood leukocytes 3 hours after ischemic stroke but before rtPA administration, 6 genes seem to be predictive to HT, though 29 different genes showed altered expression in patients who suffered HT.
Membrane associated ring CH 7
(MARCH7 or axotrophin) MARCH7 is an ubiquitin ligase which regulates membrane receptor expression is leukocytes and in other tissues as well. The increased serum level observed in stroke patients with HT may connected to downregulation of leukocyte surface proteins like MHC I and II, CD86 or ICAM1 and can shift T-cell response toward a Th17 pro-inflammatory response.
VEGF inhibitor
Expression was decreased in patients presenting HT. It may contribute to HT by altering the apoptosis of inflammatory cells via Caspase 8/ Fas ligand or decreasing the regeneration potential of damaged endothelium.
Amphiregulin
Expression was elevated. Amphiregulin is a ligand of EGFR and enhances neutrophil migration by altering E-Catherin in tight junctions, activating the MAP kinase pathway and promote MMP-9 and VEGF release inducing HT.
SMAD 4
Expression was also elevated, which is the target of TGF-ß and its function includes the regulation of the N-Catherin expression in endothelial cells, so stabilizing the BBB. No wonder that a hereditary lack of SMAD4 is related to hemorrhagic teleangiectasia and extra cellular matrix anomalies such as aneurysms and aortic dissection.
Two target genes of SMAD4 were also elevated, interleukin-1 receptor associated kinase 3 (IRAK3) which participate in TOLL-like receptor signaling and Inozitol phosphate-5-phoshpatase (INP5P), which regulates the proliferation of myeloid cells. TGF beta pathway of leukocytes including SMAD4 and IRAK3/INP5P most likely plays an important role in the process HT [141]. Although TGF beta/SMAD2 pathway in a subset of monocytes, that as part of the neuroinflammation migrates into the brain in the 1-7 days after stroke onset seems to enhance BBB integrity and is associated with a lower rate of HT [142].
Multiple coagulation factor deficiency 2
Gene expression was decreased. This gene manages the transport of factor V and VIII from endoplasmatic reticulum to Golgi apparatus, thus the lack of it results in bleeding disorder [141].
The molecular mechanism of BBB disruption after 18-24 hours from stroke onset differs from the early form; it is delayed, prolonged and more profound. Brain MMP-9,2,3,10,13,14 and other protease like plasmin, endogenous rtPA, urokinase, cathepsin, TNF alpha converting enzyme have a role in this process. In neutrophil depleted mice the MMP level in the brain remains unchanged after 24 hours from onset, suggesting that the source of the responsible MMPs is the brain itself. Brain MMP-9 is released in the first line from endothelial cells than from neurons, microglia, and astrocytes. The signaling pathways that promote MMP-9 release are the following ones: ROS, TLR4, NFkB, HMBG1 receptor, TNF alpha and IL-1. MMP-9 activation is mediated by MMP3, which turn proMMP-9 into activated MMP-9. MMP-3 is released from pericytes and endothelial cells within 24 hours after onset and especially in case of rtPA administration plays a central role. MMP-3 knockout mice show decreased rate of HT, and after administration of broad spectrum MMP inhibitor, the HT rate did not get better, suggesting how important the MMP-3 regulation pathway is. MMP-3 is promoted via inflammation or rtPA activated LRP (lipoprotein receptor protein) and NFkB pathway. MMP degrade the TJs, causing the appearance of its end product proteins in blood, such as claudin 5, occlusion, ZO-1, and destroy basal lamina as well [2].
Hyperglycemia induces MMP-3 activation and is associated with HT and worse outcome. MMP-3 inactivation in hyperglycemic rat models significantly reduced HT rate and improved the outcome [143].
TIPM-1 or TIMP-2 (tissue inhibitors of metalloproteinases) knockout studies on mice support the hypothesis that TIMP-1 and TIPM-2 inhibits MMP-9 activity and play a neuroprotective role in case of cerebral ischemia. In TIMP-1 knockout mice, BBB breakdown and leakage, neuronal apoptosis, and ischemic damage after ischemia was significantly higher. Mice without TIMP-2 gene showed increased BBB leakage and apoptosis without any change in the ischemic damage. TIMP-1 could be a potential target of neuroprotective therapy [144]. Also, the increase of the MMP9/TIMP1 ratio is significantly associated with symptomatic HT [145].
Inflammation after an ischemic event is a natural reaction of the body, simultaneously causing secondary brain damage and playing basic role in healing-repair mechanism, balancing between the early destructive inflammation and the regenerative environment. The delayed apoptotic cell demise in the border zone of the primary lesion core and the fact that the injury leads to the appearance of cells with characteristics of both microglia and astrocytes are intensively studied aspects of the ischemic process. [239,250].
Monocyte infiltration peaks in several days after onset, contributing to delayed HT. Peripheral monocytes are turning into macrophages and releasing MMP-9 and other inflammatory cytokines, maintaining the inflammation. During the monocyte infiltration they activate endothelial cells increasing their permeability. Microglia activation results in MMP-9 release. After augmentation of GM-CSF with rtPA in humans the incidence of HT did not increased, suggesting that maybe examination of different monocyte subtypes would be advantageous [2].
To assess the role of monocyte/macrophage infiltration, four different approaches were used and four groups of mice were formed: monocyte/macrophage depletion group, CCR2 inhibited, CCR2 knockout and bone marrow chimera group. Recently 2 different types of monocyte subpopulation have been identified. The first group with high level of the CCR2 chemokine receptor and the Ly6c antigen but low levels of CX3CR1, while the second group is characterized by the absence of CCR2, low Ly6c, and high CX3CR1. The first group of cells infiltrates the brain tissue in acute inflammation, the second group patrols in non-inflamed tissue. According to the functional role above, the 2 types of macrophages are the M1 type, activated in classical way with role in the inflammatory response and M2 type with repairing potential, activated in alternative ways. This study showed a worse outcome in CCR2 pathway deficient mice with higher rate of HT on day 3 and 6, with a decreased TGF beta, collagen 4, SMAD2 level (pathway of TGF signaling), causing bleeding from the border zone from thin walled and dilated neovessels, though when TGF beta was injected into the lesion border, the HT rate decreased. An inhibition of CCR2 with propagermanium or the monocyte/macrophage depletion model also ended in a higher rate of HT with the same TGF, SMAD, collagen 4 levels, showing the critical role of bone marrow-derived CCR2+ monocytes/macrophages in maintaining integrity of the neurovascular unit after ischemic stroke. These data suggest that monocyte/macrophage subtypes has crucial effect in preventing secondary HT and promoting repair. These observations highlight the fact that therapeutic approaches should concentrate on modulating the macrophage function instead of inhibiting the monocyte recruitment. This study revealed, that TGF signaling, as critical part of the tissue repair, is monocyte/macrophage dependent, and that early CCR2-dependent recruitment of peripheral inflammatory monocytes and their subsequent differentiation within the environment of the CNS lesion into non-inflammatory phagocytes is the connecting mechanism between post-ischemic inflammation and regeneration [146].
In contrast to that other studies on CCR2 pathway deficient mice state that CCR2 pathway has a harmful effect regarding the HT rate and outcome. In the deficient group of mice the infiltration of monocytes and neutrophils were lower, the BBB damage and the HT rate was decreased [147-149].
The following study was based on earlier promising results and aimed to extend the knowledge about influencing factors on MO/MP activation, differentiation and polarization. The examined factors were hyperglycemia as a frequent co-morbidity in stroke patients and the peroxisome proliferator-activated receptor gamma (PPARγ) activation. Artificial hyperglycemia was induced by streptozotocin. Hyperglycemic and PPAR knockout mice showed a shift toward inflammatory MO/PO response with a consequent increased rate of secondary HT caused by abnormal, leaky, thin walled, dilated neovessel formation and rupture. Though pharmacological PPAR activation via pioglitazone resulted in a reduced HT rate, which was independent from hyperglycemia but hugely dependent on MO/MP presence. These results altogether suggest that the inflammatory and anti-inflammatory-repair differentiation of macrophage subtypes basically determine the HT rate and therefore the clinical outcome. Interestingly PPAR modulation did influenced PH rate [150].
PPAR has influencing effect on neutrophil granulocytes as well. The supposed mechanism occurs after reperfusion as part of the early inflammatory answer. The inflammatory cells use glucose to be able to react quickly, but PPAR activation shift the energy usage toward fatty acid metabolism. Accordingly, it has effect on the cell metabolic activity, probably contributing to the inflammation weakening effect. PPAR has neuroprotective effect as well on several intracellular pathways, suggesting that PPAR take part in preventive mechanisms both in the first 24 hours and also after that by regulating the neuroinflammation [151].
Like in any other part of the body, also in the CNS an inflammation goes along with COX-2 mediated PGE2 elevation. Both the knocking-out of prosaglandin EP1 receptor and the usage of EP1 receptor antagonist, SC-51089 after tMCAO was associated with significant lower HT rate and BBB disruption as a consequence of significant reduced MMP3 and MMP9 levels, lower degree of neutrophil infiltration, and higher ZO-1 and occludin levels in the brain. This report proves an important link between neuroinflammation and BBB breakdown via MMPs. EP1 as a therapeutic target should be considered [152]. Further considerable mechanisms play role in the neuroinflammation in case of mixed and vascular dementia [248].
Vascular remodeling is natural part of the recovery process in the infarcted tissue, but unfortunately at the same time it promotes HT as well. Neovascularisation takes place with the leading of VEGF, MMP and other growth factors. New vessels incorporate into already existing vasculature, but these neovessels are weaker and leakier, predicting HT. In rodent, models suppression of hypoxia inducible factor 1 alpha resulted in the reduction of HT incidence. VEGF administration also in rodent studies within the 1 hour of stroke increased the severity of BBB disruption. However, when VEGF was given within 3-24 hours it reduced BBB permeability, increased pericyte coverage of brain capillary, enhanced angiogenesis and therefore increased cerebral blood flow and neurological recovery. Apparently, VEGF has biphasic role after stroke, and as expected, the recent observations show that MMP-9 has a biphasic role too. Early MMP inhibition cause reduced lesion size and increased TJ protein expression on endothelial cells and pericytes, implying a BBB protective effect, however delayed administration of MMP inhibitor in mice after 7 days from stroke onset, ended up in a suppressed remodeling and as a consequence in impaired functional recovery. Also angiopoetin seems to be involved in this process. Angiopoetin 1 is supposed to be associated with BBB disruption based on the fact that elevated serum level of angiopoetin 1 predicts HT after rtPA treatment, and angiopoetin 2 is likely to be related to neovascularisation. A damage associated molecular pattern molecule called high mobility group box 1 (HMGB1) is released by astrocytes and acts on HMGB1 receptor on endothelial progenitor cells, promoting angiogenesis and shows association with better long term outcome [2]. The effect of extracellular HMGB1 on HT is presumed to happen via TLR4 mediated MMP-9 activation [153]. HMGB1 contributes even to the hyperglycemia enhanced worse clinical outcome and BBB disruption [83].
ROS has a central role, not just in the early but in the delayed BBB breakdown as well. It has regulating effect on almost every known important pathways leading to BBB disruption, such as VEGF, hypoxia indictable factor 1 alpha, MMP activation, NFkB activity, protein kinase C activity, ERK1 and 2 activity, p38 MAP kinase activity. It participates in EPC (endothelial progenitor cell) mobilization, homing, differentiation, and as a result guides the whole angiogenesis and the vascular remodeling process. ROS stimulate endothelial cells to proliferate and migrate. Both in NOX1 and NOX2 (encode NADPH oxidase) knockout mice, the antigenic activity and neovascularisation were impaired [2].
Blood products getting out of vessels cause further damage in the ischemic area. To investigate the effect of thrombin in the brain tissue, experimentally thrombin was injected into the ventricle of rats, causing vascular leakage and brain edema at 24 hours [2]. Thrombin acts on protease activated receptors, like SRC kinase, causing phosphorylation cascade on NMDA receptors [154]. and causing neuronal death via calcium dysregulation, ending up in MMP and VEGF release and a consequent BBB opening. These processes were solved by the day 14, giving space to a long term repairing effect by phosphorylating cell cycle genes. This statement is supported by the observation, that SRC family kinase (SFK) inhibitor co-administered with thrombin prevented the BBB opening, though SFK treatment on day 2-6 after thrombin injection inhibited the above mentioned long term positive effects of thrombin [2].
The identification of patients, who has higher risk for HT is important before rtPA administration to possibly avoid PH-2, the most feared adverse effect of rtPA treatment. In the field of imaging, only the early ischemic sign on CT or MRI is used commonly for pre-selection of patients, in spite of its poor sensitivity and reproducibility. Treatment indications based on MRI showed altogether lower risk for HT than those CT based. It is known that increased lesion volume on DWI and increased delay on PWI are associated with increased rate of HT. Severe delay on PWI was proven to be stronger factor for HT formation than tissue status on DWI, tested with GRE. Gradient-recalled echo (GRE) has excellent specificity and sensitivity in visualizing intracerebral bleeding and was found at least as accurate as CT in detection of hemorrhage. GRE follow-up examination of stroke patients showed that HT type did not influence the NIHSS reduction in the first 7 days, moreover, deterioration in NIHSS was commonly observed during the early course of hospitalization regardless of the presence of HT. The relationship between radiological and clinical categories of HT was stronger in patients with milder on admission disabilities (initial NIHSS <14 points) than in patients with severe disabilities (>14 points) [49].
Some studies concentrate on early tissue status and subsequent HT, but others put emphasis on hypoperfusion MRI changes and subsequent HT with a result that implies, severe hypoperfusion on MRI (>8 sec) has bigger relevance in HT than DWI or early CT finding. rtPA administration should be considered in cases with severe hypoperfusion, regardless of the size of the core. Perfusion supporting interventions should be started not just for tissue salvation, but to decrease the HT risk, balancing with the fact, that aggressive recanalisation treatment is independently associated with HT [155].
Today unfortunately just 5% of all the patients suffering an ischemic stroke are eligible for rtPA administration, mostly because of the most feared adverse effect of rtPA, the HT. Unfortunately, the promising results in animal models in most cases are not translated into successful human trials due to the huge differences between animal models and the human body. At the time of this review rtPA remained the only approved treatment of stroke. None of the tried agents was be able to replace rtPA, but in case of co-administration some of them showed beneficial effect regarding the HT rate, also in human trials, such as minocycline, GM-CSF, uric acid or edaravone. As long as rtPA administration remains the only curative intervention, the aim of the future researches is the therapeutic window extension by preventing HT with the help of potential neuroprotective or BBB stabilizing agents [245,249]. The better understanding of molecular underlying mechanisms helps to find always newer potential molecular targets to prevent HT, since to date none of them reached breakthrough in human trials. Probably the different molecular pathways in early and in delayed hemorrhage and thus the different molecular targets are standing in way of success. Some of the main targets so far have proved biphasic role, which means that early blocking of MMP-9, and ROS prevent HT, but at the same time the recovery processes are inhibited as well. Another approach of HT prevention is the proper preselection of patients for rtPA treatment, considering the individual risk factors [156].
Minocyclin
The molecular acting mechanism of minocycline, which is a broad-spectrum tetracycline antibiotic was investigated in mice, that had genetically induced cerebral amyloid angiopathy ending in hemorrhagic stroke. The minocycline hugely modified the MMP and inflammatory pathways in a positive way and connecting to that, higher levels of microvascular tight junction and basal lamina proteins were found, proving its BBB protective effect, which could be utilized in ischemic stroke treatment, parallel to rtPA administration [157]. Furthermore, minocyclin was tested on spontaneous hypertensive rats as well. MMP-9 serum levels by 6 hours after stroke onset were lower, infarct size was smaller and HT rate was also lower, suggesting that minocycline is beneficial in extending the rtPA therapeutic window [158].
Minocycline was found to be potentially applicable in clinical usage. In this two, open-labeled evaluator-blinded trial 60, respectively 50 patients were enrolled, both with the conclusion that the patient group, which received minocycline daily for 5 days after stroke onset had significantly better NIHSS scores on day-90 control, however female patients did not show this positive result [159,160].
Absence of a definitive target that is responsible for the effect of minocycline is preventing the further results. Caspase-1, caspase-3, cyclo-oxygenase-2, inducible nitric oxide synthase, p38 mitogen-activated protein kinase, gelatinase, MMP-9 are supposed to participate in the molecular mechanism of minocycline [157,160].
Uric acid
Higher serum uric acid level was significantly associated with lower HT rate and better outcome among stroke patients with rtPA administration [121]. This recent meta analysis of 8131 acute ischemic stroke patients reports that high uric acid serum level on admission should be regarded as good prognostic marker, suggesting its positive impact on BBB damage. Uric acid as an endogen antioxidant is a promising therapeutic neuroprotective agent [161]. Based on promising animal studies human trials were begun. URICO-ICTUS, a double-bind phase III study was designed to investigate the safety and efficacy of uric acid in the management of acute stroke. Unfortunately, it failed to provide the expected therapeutic success, because administration of uric acid in addition to rtPA did not increased the rate of excellent outcome. Though, uric acid administration was found to be safe, it increased neither mortality nor hemorrhagic event or gout [162]. Secondary reanalysis were proceeded in different subpopulations, and it turned out, that in the woman subpopulation and among patients with hyperglycemia during acute stroke the uric acid administration with rtPA raised the number of excellent outcome and increased infarct growth in comparison to placebo. Hyperglycemia greatly increases ROS burden, and uric acid - as an antioxidant - is supposed to perform its positive effect in the way of ROS neutralizing [163,164].
Edaravone
The human usage of this free radical scavenger was based on promising animal studies that reported that edaravone significantly attenuates infarct size and HT rate in hyperglycemic rats [165]. Further studies found that this agent performs its beneficial effects via MMP-9 inhibition as well [166]. Since 2001 in Japan, edaravone is an approved treatment of ischemic stroke when administered within the first 24 hours of onset. Human studies show that edaravone can significantly suppress MMP-9 serum level after ischemic stroke [167]. On the other hand, edaravone was found to increase the HT rate in case of cardiogenic embolism treated with heparin and rtPA, though this did not impact the clinical outcome, on the contrary, the group treated with edaravone showed a higher rate of good outcome among patients suffered HI-1 and HI-2. The higher rate of HT is the consequence of the observation that beside the neuroprotective and endothelprotectiv effect of edaravone, the agent interferes with the fibrinolytic and coagulation system. It highlights the known fact, that ROS inhibit fibrinolysis and coagulation; ergo edaravone could reduce both the rtPA and heparin dose, preventing the negative effects of rtPA. Edaravone reaches the ischemic lesion when HT occurs [168].
A recent retrospective study compared 356 patients treated with edaravone and rtPA at the same time and 356 patients treated just with thrombolysis. After a logistic regression analysis, it could be ascertained that edaravone administration was significantly associated with lower mRS score at discharge, implying that it may improve early clinical outcome [169].
2021 Copyright OAT. All rights reserv
Isoflurane as a common used anesthetic
The role of isoflurane in the modulation of ischemia-reperfusion process and in HT is rather controversial. The effects of isoflurane were investigated both in case of preconditioning and postconditioning. In case of isoflurane preconditioning in rat models, short and even long term neurological outcome was improved, and the supposed neuroprotective effect of isoflurane was rested upon Bcl-2 induction [170]. To this neuroprotective effect may contributes the Notch signaling pathway as a central route of the inflammatory response and neuronal progenitor cell proliferation, differentiation [171]. The further molecular mechanisms of the pre and postconditioning may include lower level of inflammatory cytokines, decreased MMP-9 activation and ROS induced lipid peroxidation and due to that decreased tight junction protein degradation [172]. In case of postconditioning, the transforming growth-beta 1 pathway upregulation along with the c-Jun N-terminal kinase signaling pathway downregulation, which is part of the repair process, participates in the supposed neuroprotective effect [173]. Also mitochondrial adenosine 5′-triphosphate-sensitive potassium channels are assumed to be involved in the positive effect of isoflurane [174]. Isoflurane postconditioning was found to be effective in decreasing the infarct volume and HT rate in rtPA treated ischemic stroke model, proving its potential as candidate in future stroke therapy [175]. Though, other studies found no neuroprotective effect after preconditioning in case of intracerebral hemorrhage [176]. Moreover, in hyperglycemic rat models, postconditioned with isoflurane after stroke, one study found directly negative impact regarding the neurogical deficit when compared to the control group, with no influence on infarct size, brain edema and HT volume. It was accompanied by increased oxidative stress, possibly induced by decreased superoxide dismutase and catalase expression [177].
Hydrogen gas inhalation
For 2 hours after 90 minutes MCAO in hyperglycemic rat models was accompanied by decreased infarct size and HT rate and due to that improved neurological status. A remarkable aspect is, that 1 hour hydrogen inhalation did not decreased the infarct size but decreased HT rate, suggesting the direct effect of hydrogen on the HT process, independently from infarct size. The underlying mechanisms may include reduction of the expressions of 8OHG (nucleic acid oxidation marker), HNE and nitrotyrosine (markers of lipid peroxidation and protein nitrification), and the activity of MMP-9. However, the treatment had no significant impact on the decreased level of ZO-1, occludin, collagen IV in the brain, or on the increased expression of AQP4 which channel participates in the maintenance of BBB integrity and whose expression is increased in case of ischemia, suggesting that the prevention of HT in this study happened also in other ways beside MMP-9 inhibition or AQP4 downregulation [178].
Other agents administered with the purpose of ROS inhibition like spin-trap-agent NXY-059 [179] or N-t-Butyl-Phenylnitrone was ineffective. Further potential agents that are associated with lower HT incidence in animal models are the followings: activated protein C, PDGFR alpha antagonists, cilostazol (phosphodiesterase inhibitor), melatonin, fasudil (rho kinase inhibitor), fingolimod (sphingosine 1-phosphate receptor agonist), poly ADP ribose polymerase (PARP) inhibitors, sulfonylurea receptor 1 (Sur1) inhibitors (glyburide), 17-beta estradiol, FK506 (tacrolimus), VEGF inhibition and hyperbaric oxygen [180]. Erythropoietin exacerbates rtPA-related HT [2].
TGF-ß 1
Administered TGF-ß 1 in addition to rtPA after 3 hour of MCAO in rats reduced the basal lamina degradation, the BBB disruption and so the HT rate. The supposed mechanism of acting includes inhibited rtPA induced MMP2 and MMP9 elevation and upregulated PAI-1 expression. These pathways seem to be protective against BBB breakdown in the early stage of stroke [181].
Tiazide
Long term use of tiazide seems to be associated with lower initial NIHSS and better 3-month mRS score [182]. The possible cause of this observation is that hydrochlorotiazid therapy restores the dilatory capacity of cerebral arterioles, improving the collateral supply, hence leading to smaller infarct volume and lower HT rate. The increased blood pressure in the acute phase of stoke has positive effects on maintaining the collateral flow. The break of a long term tiazide usage after stroke onset may contribute to a better cerebral blood flow by fluid retention, better blood viscosity. Based on these results, tiazide as first line antihypertensive drug would be good choice [183].
FeTMPyP
Co-administration of peroxynitrite decomposition catalyst (Fe(III)tetrakis (1-methyl-4-pyridyl) porphyrin pentachlorideporphyrin pentachloride) to rtPA has beneficial effect on HT rate in rat models. The rtPA treatment after 5 hours of MCA occlusion caused severe HT, neurological deficit, MMP-9 and MMP-2 upregulation and elevated peroxynitrite level. Co-administered FeTMPyP prevented HT, reduced the peroxynitrite, MMP-9 and MMP-2 level likely due to the inhibition of the peroxynitrite induced MMP upregulation, furthermore improved neurological outcome [184].
Progesterone
A new era of the progesterone research was started when it got proved that progesterone is synthesized by neurons and glial cells as well, both in the central and peripheral nervous system, both in males and females [185], and has an assumed neuroprotective effect [186]. Since progesterone acts via a core binding protein and alters DNA transcription, its effect on neurons are accordingly diverse, like up-regulating the GABA neurotransmitter, decreasing lipid peroxidation and oxidative stress by its antioxidant effect, limiting the expression of inflammatory cytokines and decreasing cellular apoptosis. All of these, mechanisms result in significantly smaller infarct volumes, smaller extension of edema and altogether better neurologic outcomes on rat models [187-189]. Other studies proved that progesterone and its metabolite allopregnanolone inhibit MMP and VEGF upregulation, contributing to BBB maintenance [186].
A recent study based on the reports above, assessed the effect of progesterone on rtPA induced HT in rat models. The results are promising. Progesterone treatment attenuated rtPA-induced hemorrhage, brain edema, BBB breakdown and leakage and infarct volume. The reported mortality rates were 7.14% respectively 27.78% in the saline- and rtPA-treated groups, whereas no mortality was observed in case of rtPA and progesterone co-administration. Furthermore, the transendothelial electrical resistance (TEER) value, which is used to estimate the permeability of endothelial cells in vitro, was examined after hypoxia/reperfusion. Reduced TEER values indicate TJ disruption. rtPA administration decreased the TEER, whereas co-administered progesterone effectively protected the BBB integrity. The data supports the observation that increased level of MMP is accompanied by decreased level of occludin and claudin in the brain after hypoxia/perfusion and rtPA treatment [190].
VEGF is known to contribute to BBB breakdown in the acute phase of stroke by MMP upregulation and enhanced capillary permeability. The activation of VEGFR2 tyrosine kinase receptor through dimerization and phosphorylation leads to further activation of various intracellular pathways such as phospholipase C, protein kinase C, ERK1/2, and Src. Administration of anti-VEGF antibody inhibits the BBB breakdown [191]. Progesterone treatment significantly decreases the VEGF level in brain endothelial cell culture media as well. rtPA possibly contributes to the BBB disruption among others via VEGF upregulation, which seems to be preventable by progesterone administration [192].
DHA
Docosahexaenoic acid is an omega-3 essential fatty acid and one of the major easily obtainable long-chain polyunsaturated fatty acids (PUFAs) in the human diet. Neuroprotectin D1 (NPD-1) is a well-studied neuroprotective agent, that is actually a metabolite of DHA [193]. NPD-1 is part of a key survival signaling that upregulates anti-apoptotic and downregulates pro-apoptotic proteins, promoting neuronal survival in stress situation, and in addition it decreases the severity of the inflammatory response through downregulated inflammatory gene expression, decreased ROS burden, leukocyte adhesion and extravasation. Earlier studies report that in case of ischemic stroke long term intake of DHA or one time injection before stroke onset, eventually the administration after 3 hours of MCAO or direct infusion into the third ventricle after MCAO decreases the infarct size and improve the neurological outcome [194]. To assess the direct effect of DHA on HT, dextrose was injected 10 minutes before MCAO to induce hyperglycemia, reaching larger ischemic infarct size and more severe hemorrhage. DHA significantly decreased HT rate, most likely through BBB protection. Reduced erythrocyte extravasation, increased collagen IV amount and reduced expression of the intercellular adhesion molecule-1 (ICAM-1) was observed that was followed by maintained BBB integrity and decreased inflammatory response. 7 days neurological status was improved and infarct size was reduced up to 60% in comparison to the control group [195].
GM-CSF
Regarding the impact of GM-CSF on the HT process is still controversial. GM-CSF was found to be responsible for increased HT rate in rats when co-administered to rtPA. It provoked neutrophilia and MMP-9 induction [196]. In comparison, other in vitro and animal studies reported that GM-CSF has beneficial neuroprotective effect by reducing the infarct size and edema, blocking glutamate induced neurotoxicity and apoptosis, inhibiting neuroinflammation by decreasing IL-1 level. Furthermore GM-CSF activates endothelial cell migration and proliferation, mobilizes CD34+ progenitor cells from the bone marrow, contributing to angiogenesis and neovascularisation. This aspect suggests that endogen GM-CSF is part of neuroprotecion, BBB protection and delayed repair mechanisms. Comparing the effect of rtPA treatment with rtPA and GM-CSF co-administration on spontaneous hypertensive rats after temporary MCA occlusion show that GM-CSF administration contributes to decreased HT rate - proved by decreased hemoglobin content in the brain tissue - and to decreased infarct size. rtPA plus GM-CSF administration was accompanied by higher CD34+ cell number in the ischemic hemisphere, higher Ang-2 angiogenesis marker, vWF vasculogenesis marker and VEGF-2 compared to the group treated just with rtPA. Ang-2 participates in the ischemia induced neovessel formation by increasing the capillary diameter, helping the remodeling of the basal lamina and the growth of new blood vessels [197]. CD34+ endothelial progenitor cells are known to participate in BBB repair after ischemia and their presence is connected to better functional outcome. Circulating CD34+ cells after stroke are assumed to predict the long term clinical outcome [198]. Intravenous transplantation of endothelial progenitor cells was associated with better long term functional outcome too by promoting BBB repair [199].
Indeed, human studies report altogether beneficial connection between GM-CSF and stroke outcome. A meta-analysis of 711 patients showed significant positive impact of the GM-CSF regarding the NIHSS and mRS score, along with increased CD34+ cell mobilization from the bone marrow [200]. The administration of GM-CSF seems to be safe and well tolerated, suggesting that GM-CSF would be a good candidate co-administered to rtPA for daily stroke treatment [201].
Wnt/β-catenin pathway
Previous studies revealed that a disturbed Wnt/β-catenin pathway contributes to BBB breakdown. This is a pathway, that controls cell differentiation and fate hence it is important in maintaining BBB integrity in stroke. Glycogen synthase kinase 3β (GSK-3β), as part of the adenomatous polyposis coli/axin/GSK-3β complex, is involved in the ubiquitination and proteasomal degradation of β-catenin which is the key molecule of the Wnt/β-catenin pathway. Inhibiting GSK-3β with TWS119 is going to inhibit the degradation of β-catenin and therefore activate the Wnt/β-catenin signaling pathway. Indeed, using TWS119 parallel to rtPA had a significantly advantageous effect on the HT rate, infarct volume and brain edema. Modifying this pathway resulted in increased expression of claudin-3 and ZO-1, both are elemental part of the tight junction system however the detailed linking mechanism in this phenomenon remains unclear [202,203]. This pathway according to other reports also has role in the controlling of the neuroinflammation [204] and the angiogenesis [205].
Ang-1
Angiopoetin 1 is known to have a beneficial role in endothelial cell survival, vascular remodeling and in maintenance of the vascular stability, permeability. The mechanism of acting includes several ways such as VEGF pathway, stabilizing TJs, increasing glycocalyx in endothelial cells, shrinking intercellular spaces and altering the PDGF signaling pathway in pericytes. In addition, Ang-1 has neuroprotective effect as well. Its receptor, called Tie-2, can be found on endothelial cells, pericytes, and neuronal cells [206-211]. To assess the impact of Ang-1 on the HT process 4 different groups of rats were observed. The first group went through a permanent MCAO (HT rate: 17,4%), the second group got rtPA 1 hour after ischemia, so within the therapeutic window (HT rate: 6,7%), the third group got rtPA 4h after ischemia, so out of the time window (HT rate: 59%), and a control group. Ang1-positive vessel density was investigated. In the second group, the density was not decreased compared to the control group but a significant decline was observed in the third group and to a lesser degree but also in the permanent MCAO group. This result implies that decreased endogenous Ang-1 expression may play a central role in HT that occurs when rtPA is administered out of the therapeutic time frame. After that, COMP-Ang1 protein (COMP=cartilage oligomeric matrix protein) was administered parallel to rtPA treatment, that suppressed the hemorrhagic transformation process and the cerebral edema as well, however had no effect on the infarct size, so the neuroprotective effect of Ang-1 was not proved, in comparison to earlier studies. Ang-1 is a potential target for therapeutic BBB defense [212].
Apyrase
(APT102) Apyrase is a calcium-activated enzyme, which is naturally present in the human body, catalyzing the hydrolysis of ATP and ADP into AMP and inorganic phosphate. APT102 is a recombinant form of CD39 human apyrase. Apyrase was found to have an antithrombotic effect as a result of hydrolyzing extracellular ADP, inhibiting platelet aggregation with a minimally elevated risk for bleeding, because APT102 does not act direct on platelets, but in an indirect way via prothrombotic ways [156]. Apyrase has anti-inflammatory effect as well, reported in studies investigating the prevention of ischemia-reperfusion injury after lung, myocardial and brain infarction [213-215]. To assess the potential in APT102 in extending the time window of rtPA, aged rats were used to model the clinical conditions of the human population who suffer an ischemic stroke, since 72% of stroke patients are over the age of 65, and age is a strong risk factor of worse outcome, HT and stroke severity, as consequence of fragility of the BBB. 6 hours after MCAO rtPA and APT102 were co-administered. This combination was found to be significantly associated with decreased infarct volume, improved clinical outcome and prevention of HT, implying its beneficial effect in extending the time window till at least 6 hours from stroke onset. In case of endothelial injury or endothelial activation the local concentration of ATP and ADP increases, causing platelet activation, aggregation, fibrin deposition, worse regeneration circumstances and as a consequence microthrobus formation, hypoperfusion and just partial recanalisation. Inhibiting the endothelial activation prevents neutrophils from extravasation and that indirectly prevents MMP mediated BBB degradation. Thus APT102 requires reperfusion to perform its effect. These finding are in agreement with previous studies on CD39 knockout mice [216].
Hyperbaric oxygen preconditioning
It is known that hyperbaric oxygen preconditioning has neuroprotective effect via induction of the COX-2 enzyme during the preconditioning phase, that is followed by decreased level of this enzyme and its downstream targets in case of ischemia, extending the time window of neuronal survival. In addition, COX-2 inhibitor administration during the preconditioning phase eliminated the neuroprotective effect, proving the role of the COX-2 enzyme in hyperbaric oxygen preconditioning [217]. The presumed mediator of this phenomenon is the 15d-PGJ2 (15-deoxy-Δ12,14-prostaglandin J2), which is a modification of PGD2 by non-enzymatic dehydration. PGD2 is synthesized from arachidonic acid by COX-1 and COX-2, and PGD synthase. 15d-PGJ2 is an endogen ligand of PPARγ. It is a nuclear hormone receptor playing an extended regulating role in several cellular signaling mechanisms, such as apoptosis, oxidative stress and neuroinflammation. After the activation of PPARγ it forms a heterodimer with the PPAR-retinoid X receptor α (RXR) and binds to PPAR response elements in the promoter region of their target genes [218]. Earlier studies reported that activation of PPAR is associated with BBB prevention [219] and that hyperbaric oxygen preconditioning was accompanied by elevated 15d-PGJ2 and elevated PPARγ level [220]. 15d-PGJ2 proved its neuroprotective effect via anti-inflammatory and antioxidative ways in the following experiment. Hyperglycemic mice were investigated with 1 hour/day hyperbaric oxygen preconditioning for 5 consecutive days before MCA occlusion. 15d-PGJ2 and PPARγ levels were higher, along with lower serum levels of TNF-α and IL-1β, ZO-1, occludin and lower activity of MMP-2, MMP-9. Altogether a significant better functional outcome and a lower rate of HT were observed in hyperglycemic MCAO rats after hyperbaric oxygen preconditioning. Administration of PPARγ inhibitor GW9662 decreased the positive effect of preconditioning [221,222].
Further molecular pathways of HT prevention after hyperbaric oxygen preconditioning are assumed to involve Nod-like receptor protein 3 (NLRP3) inflammasome, which is a key component of the inflammatory response. It facilitates caspase-1 and interleukin-1β processing, in this way amplifying and maintaining the inflammatory response [223]. The activation cascade starts with excess level of ROS that oxidise thioredoxin into thioredoxin-interacting protein (TXNIP). TXNIP binds to NLRP3 causing its activation [224,225]. Based on the reports above hyperglycemic mice were undertaken a 1 hour hyperbaric oxygen therapy 5 days long before MCAO. The HT rate was significantly lower, though the infarct volume was unaltered. Different agents were administered to different groups of mice to investigate the steps of the signaling chain, such as thioredoxin-interacting protein small interfering RNA, Nod-like receptor protein 3 small interfering RNA and N-acetyl-l-cysteine as a general ROS scavenger. The NLRP3 level was decreased after preconditioning and was increased after siRNA administration. The results suggest that the neuroprotective effect of the hyperbaric preconditioning involves the ROS/TXNIP/NLRP3 pathway in hyperglycemic MCAO rats. The level of inflammatory molecules connected to the NLRP3 pathway such as interleukin-1β, IL-6, and TNF-α after preconditioning followed by MCAO were decreased and after siRNA administration were increased. The MMP-9 level altered in the same way suggesting that he BBB protective effect of preconditioning occurs through neuroinflammation inhibition as well [226].
SMTP
Approaching the problem of the HT from another point view, a new thrombolytic agent is now under investigation. A family of small molecules from the fungus Stachybotrys microspora was discovered which has a specific effect on plasminogen activation by modulating the conformation of the plasminogen molecule to be more susceptible to proteolytic activation. The most promising member of this family is the SMTP (Stachybotrys microspora triprenyl phenol). Since it is a plasminogen modulator, its effect depends on the endogenous plasminogen activator supply. Based on earlier rodent studies it has an assumed anti-inflammatory and neuroprotective effect too [227,228]. Studies on monkeys investigated the therapeutic potential of SMTP-7 with the help of photoirradiation-induced endothelial injury followed by thrombotic MCA occlusion. As a conclusion SMTP-7 was found to be efficient in reducing cerebral infarct size and improved neurologic deficit as a result of recanalisation due to its thrombolytic effect. The thrombolytic efficacy is comparable to rtPA, but in addition SMTP-7 decreased hemorrhagic transformation rate compared in the infarct area, compared to the vehicle control animal group. This observation is a consequence of the supposed anti-inflammatory effect. However further comparative studies between the rtPA and SMTP-7 are needed. As for the adverse haemostatic changes, no significant alternations following the SMTP-7 treatment (10 mg/kg) was observed in the heart rate, blood pressure, body temperature, blood gas and pH, haematocrit, blood glucose or in the activated partial thromboplastin time, fibrinogen, platelet aggregation, suggesting no involvement of hemodynamic changes or coagulation inhibition in the efficacy of SMTP-7 in the monkey stroke model. Only the fibrinolytic system was affected. SMTP-7 (10 mg/kg) also caused no significant changes in the fibrinolytic parameters such as plasminogen, a2-antiplasmin or D-dimer in the control monkey group, although, 2 times higher D-dimer level was observed referring to minimal mobilization of the fibrinolytic system. This result is in agreement with earlier studies on mice, where SMTP-7 did not increased the bleeding after tail amputation. The rtPA directly activates the plasminogen, though SMTP-7 acts in an on-demand way. The definitive advantage of SMTP-7 against rtPA is that a therapeutic dose of SMTP-7 did just minimally affect the fibrinolytic system; however a higher dose of SMTP-7 was significantly associated with bleeding complications [229].
Roziglitazone
Thiazolidinediones as insulin sensitizing agents, activate nuclear receptor peroxisome proliferator-activated receptor gamma (PPAR-γ). It was proven that PPAR-γ has a dramatically positive effect on infarct size, HT rate and neurological outcome. The underlying mechanism is assumed to be the decreased expression of inflammatory mediators, such as IL-1, cyclooxygenase-2, inducible nitric oxide synthase [230]. Further mechanisms of suppressing the inflammatory response are involved as well [231]. Rosiglitazone improved neurobehavioral outcome, reduced infarct volume and hemorrhagic transformation rate, inhibited hemoglobin leakage [232]. The efficacy was unchanged in case of administration before, at the time or after ischemia in rat MCAO model. The mechanism behind this phenomenon is assumed to be the inhibited collagen IV level decrease in the brain due to elevated GLUT-1 expression [233]. Decreased level of GLUT-1 receptor was observed on endothelium surface in case of ischemia contributing to BBB disruption. Rosiglitazone did not perform its effect by decreasing serum glucose level, because serum glucose levels remained unaltered. The glucose decreasing effect needs time to emerge [234].
Hypercholesterinaemia
Hypercholesterinaemia and statin treatment as proof of earlier hypercholesterinaemia, was found to be potential neuroprotective, most likely due to its antioxidant effect. Hypercholesterinaemia was associated altogether with better clinical recovery rate [235-237]. According to a meta-analysis all type of HT was associated with lower LDL cholesterol level, furthermore, symptomatic HT showed a connection with lower total cholesterol levels as well [238].
Supported by the Hungarian Brain Research Program (KTIA_13_NAP-A-II/7) and GINOP-2.3.2-15-2016-00043, Hungary.
The authors declare no conflict of interest.
- Angermaier A, Langner S (2016) Recanalization and reperfusion in acute stroke - More often different than alike. Clin Neuroradiol 26: 375-376. [Crossref]
- Yeo LL, Paliwal P, Teoh HL, Seet RC, Chan BP, et al. (2013) Timing of recanalization after intravenous thrombolysis and functional outcomes after acute ischemic stroke. JAMA Neurol 70: 353-358. [Crossref]
- Jickling GC, Liu D, Stamova B, Ander BP, Zhan X, et al. (2014) Hemorrhagic transformation after ischemic stroke in animals and humans. J Cereb Blood Flow Metab 34: 185-199. [Crossref]
- Vanacker P, Lambrou D, Eskandari A, Ntaios G, Cras P, Maeder P, et a. (2015) Improving the prediction of spontaneous and post-thrombolytic recanalization in ischemic stroke patients. J Stroke Cerebrovasc Dis 24: 1781-1786. [Crossref]
- Szabo K, Kern R, Gass A, Griebe M, Lanczik O, et al. (2008) Early spontaneous recanalization following acute carotid occlusion. J Neuroimaging 18: 148-153. [Crossref]
- Khatri P, Abruzzo T, Yeatts SD, Nichols C, Broderick JP, et al. (2009) Good clinical outcome after ischemic stroke with successful revascularization is time-dependent. Neurology 73: 1066-1072. [Crossref]
- Rha JH, Saver JL (2007) The impact of recanalization on ischemic stroke outcome: a meta-analysis. Stroke 38: 967-973. [Crossref]
- Olivot JM, Mlynash M, Thijs VN, Kemp S, Lansberg MG, et al. (2008) Relationships between infarct growth, clinical outcome, and early recanalization in diffusion and perfusion imaging for understanding stroke evolution (DEFUSE). Stroke 39: 2257-2263. [Crossref]
- Ogata T, Christensen S, Nagakane Y, Ma H, Campbell BC, et al. (2013) The effects of alteplase 3 to 6 hours after stroke in the EPITHET-DEFUSE combined dataset: post hoc case-control study. Stroke 44: 87-93. [Crossref]
- Davis SM, Donnan GA, Parsons MW, Levi C, Butcher KS, et al., (2008) Effects of alteplase beyond 3 h after stroke in the Echoplanar Imaging Thrombolytic Evaluation Trial (EPITHET): a placebo-controlled randomised trial. Lancet Neurol 7: 299-309. [Crossref]
- Alexandrov AV, Burgin WS, Demchuk AM, El-Mitwalli A, Grotta JC (2001) Speed of intracranial clot lysis with intravenous tissue plasminogen activator therapy: Sonographic classification and short-term improvement. Circulation 103: 2897-2902. [Crossref]
- Arnold M, Mattle S, Galimanis A, Kappeler L, Fischer U, et al. (2014) Impact of admission glucose and diabetes on recanalization and outcome after intra-arterial thrombolysis for ischaemic stroke: Research. Int J Stroke 9: 985-991. [Crossref]
- Kimura K, Iguchi Y, Yamashita S, Shibazaki K, Kobayashi K, et al. (2008) Atrial fibrillation as an independent predictor for no early recanalization after IV-t-PA in acute ischemic stroke. J Neurol Sci 267: 57-61. [Crossref]
- Moftakhar P, English JD, Cooke DL, Kim WT, Stout C, et al. (2013) Density of thrombus on admission ct predicts revascularization efficacy in large vessel occlusion acute ischemic stroke. Stroke 44: 243-245. [Crossref]
- Tsivgoulis G, Saqqur M, Sharma VK, Lao AY, Hoover SL, et al. (2008) Association of pretreatment ASPECTS Scores with rtPA-Induced arterial recanalization in acute middle cerebral artery occlusion. J Neuroimaging 18: 56-61. [Crossref]
- Arnold M, Liliane Kappeler, Krassen Nedeltchev, Caspar Brekenfeld, Urs Fischer, et al. (2007) Recanalization and outcome after intra-arterial thrombolysis in middle cerebral artery and internal carotid artery occlusion: Does sex matter? Stroke 38: 1281-1285.
- Kufner A, Nolte CH, Galinovic I, Brunecker P, Kufner GM, et al. (2013) Smoking-thrombolysis paradox: Recanalization and reperfusion rates after intravenous tissue plasminogen activator in smokers with ischemic stroke. Stroke 44: 407-413. [Crossref]
- Mendonça N, Rodriguez-Luna D, Rubiera M, Boned-Riera S, Ribo M, et al. (2012) Predictors of tissue-type plasminogen activator nonresponders according to location of vessel occlusion. Stroke 43: 417-421. [Crossref]
- Saver JL (2006) Time is brain--quantified. Stroke 37: 263-266. [Crossref]
- Molina CA, Montaner J, Abilleira S, Ibarra B, Romero F, et al. (2001) Timing of spontaneous recanalization and risk of hemorrhagic transformation in acute cardioembolic stroke. Stroke 32: 1079-1084. [Crossref]
- Henning EC, Latour LL, Hallenbeck JM, Warach S (2008) Reperfusion-associated hemorrhagic transformation in SHR rats: evidence of symptomatic parenchymal hematoma. Stroke 39: 3405-3410. [Crossref]
- Belayev L, Busto R, Zhao W, Ginsberg MD (1996) Quantitative evaluation of blood-brain barrier permeability following middle cerebral artery occlusion in rats. Brain Res 739: 88-96. [Crossref]
- (1997) Intracerebral hemorrhage after intravenous T-PA therapy for ischemic stroke. Stroke 28: 2109-2118. [Crossref]
- Kharitonova T, Thorén M, Ahmed N, Wardlaw JM, von Kummer R, et al. (2009) Disappearing hyperdense middle cerebral artery sign in ischaemic stroke patients treated with intravenous thrombolysis: clinical course and prognostic significance. J Neurol Neurosurg Psychiatry 80: 273-278. [Crossref]
- Kharitonova TV, Melo TP, Andersen G, Egido JA, Castillo J, et al. (2013) Importance of cerebral artery recanalization in patients with stroke with and without neurological improvement after intravenous thrombolysis. Stroke 44: 2513-2518. [Crossref]
- Humpich M, Singer OC, du Mesnil de Rochemont R, Foerch C, Lanfermann H, et al. (2006) Effect of early and delayed recanalization on infarct pattern in proximal middle cerebral artery occlusion. Cerebrovasc Dis 22: 51-56. [Crossref]
- IST-3 collaborative group, Sandercock P, Wardlaw JM, Lindley RI, Dennis M, et al. (2012) The benefits and harms of intravenous thrombolysis with recombinant tissue plasminogen activator within 6 H of acute ischaemic stroke (the Third International Stroke Trial [IST-3]): A randomised controlled trial. Lancet 379: 2352-2363. [Crossref]
- Kelly MA, Shuaib A, Todd KG (2006) Matrix metalloproteinase activation and blood-brain barrier breakdown following thrombolysis. Exp Neurol 200: 38-49. [Crossref]
- Suzuki Y, Nagai N, Umemura K (2011) Novel situations of endothelial injury in stroke--mechanisms of stroke and strategy of drug development: intracranial bleeding associated with the treatment of ischemic stroke: thrombolytic treatment of ischemia-affected endothelial cells with tissue-type plasminogen activator. J Pharmacol Sci 116: 25-29. [Crossref]
- Wang X, Lee SR, Arai K, Lee SR, Tsuji K, et al. (2003) Lipoprotein receptor-mediated induction of matrix metalloproteinase by tissue plasminogen activator. Nat Med 9: 1313-1317. [Crossref]
- Cheng T, Petraglia AL, Li Z, Thiyagarajan M, Zhong Z, et al. (2006) Activated protein C inhibits tissue plasminogen activator-induced brain hemorrhage. Nat Med 12: 1278-1285. [Crossref]
- Su EJ, Fredriksson L, Geyer M, Folestad E, Cale J, et al. (2008) Activation of PDGF-CC by tissue plasminogen activator impairs blood-brain barrier integrity during ischemic stroke. Nat Med 14: 731-737. [Crossref]
- Rodríguez-González R, Blanco M, Rodríguez-Yáñez M, Moldes O, Castillo J, et al. (2013) Platelet derived growth factor-CC isoform is associated with hemorrhagic transformation in ischemic stroke patients treated with tissue plasminogen activator. Atherosclerosis 226: 165-171. [Crossref]
- Yepes M, Sandkvist M, Moore EG, Bugge TH, Strickland DK, et al. (2003) Tissue-type plasminogen activator induces opening of the blood-brain barrier via the LDL receptor-related protein. J Clin Invest 112: 1533-1540. [Crossref]
- Cuadrado E, Ortega L, Hernández-Guillamon M, Penalba A, Fernández-Cadenas I, et al. (2008) Tissue plasminogen activator (t-PA) promotes neutrophil degranulation and MMP-9 release. J Leukoc Biol 84: 207-214. [Crossref]
- Zhang Y, Wang Y, Zuo Z, Wang Z, Roy J, et al. (2014) Effects of tissue plasminogen activator timing on blood-brain barrier permeability and hemorrhagic transformation in rats with transient ischemic stroke. J Neurol Sci 347: 148-154. [Crossref]
- García-Yébenes I, Sobrado M, Zarruk JG, Castellanos M, Pérez de la Ossa N, et al. (2011) A mouse model of hemorrhagic transformation by delayed tissue plasminogen activator administration after in situ thromboembolic stroke. Stroke 42: 196-203. [Crossref]
- Kano T, Katayama Y, Tejima E, Lo EH (2000) Hemorrhagic transformation after fibrinolytic therapy with tissue plasminogen activator in a rat thromboembolic model of stroke. Brain Research 854: 245-248. [Crossref]
- Beretta S, Cuccione E, Versace A, Carone D, Riva M, et al. (2015) Cerebral collateral flow defines topography and evolution of molecular penumbra in experimental ischemic stroke. Neurobiol Dis 74: 305-313. [Crossref]
- Rusanen H, Saarinen JT, Sillanpää N (2015) Collateral circulation predicts the size of the infarct core and the proportion of salvageable penumbra in hyperacute ischemic stroke patients treated with intravenous thrombolysis. Cerebrovasc Dis 40: 182-190. [Crossref]
- Saarinen JT, Rusanen H, Sillanpää N (2014) Collateral score complements clot location in predicting the outcome of intravenous thrombolysis. AJNR Am J Neuroradiol 35: 1892-1896. [Crossref]
- Sillanpää N, Saarinen JT, Rusanen H, Elovaara I, Dastidar P, et al. (2013) Location of the clot and outcome of perfusion defects in acute anterior circulation stroke treated with intravenous thrombolysis. AJNR Am J Neuroradiol 34: 100-106. [Crossref]
- Goyal M, Demchuk AM, Menon BK, Eesa M, Rempel JL, et al. (2015) Randomized assessment of rapid endovascular treatment of ischemic stroke. N Engl J Med 372: 1019-1030. [Crossref]
- Maas MB, Lev MH, Ay H, Singhal AB, Greer DM, et al. (2009) Collateral vessels on CT angiography predict outcome in acute ischemic stroke. Stroke 40: 3001-3005. [Crossref]
- Fanou EM, Knight J, Aviv RI, Hojjat SP, Symons SP, et al. (2015) Effect of collaterals on clinical presentation, baseline imaging, complications, and outcome in acute stroke. AJNR Am J Neuroradiol 36: 2285-2291. [Crossref]
- Christoforidis GA, Karakasis C, Mohammad Y, Caragine LP, Yang M, et al. (2009) Predictors of hemorrhage following intra-arterial thrombolysis for acute ischemic stroke: the role of pial collateral formation. AJNR Am J Neuroradiol 30: 165-170. [Crossref]
- Terruso V, D'Amelio M, Di Benedetto N, Lupo I, Saia V, et al. (2009) Frequency and determinants for hemorrhagic transformation of cerebral infarction. Neuroepidemiology 33: 261-265. [Crossref]
- Trouillas P, Derex L, Philippeau F, Nighoghossian N, Honnorat J, et al. (2004) Early fibrinogen degradation coagulopathy is predictive of parenchymal hematomas in cerebral rt-PA thrombolysis: a study of 157 cases. Stroke 35: 1323-1328. [Crossref]
- Kim JH, Bang OY, Liebeskind DS, Ovbiagele B, Kim GM, et al. (2010) Impact of baseline tissue status (diffusion-weighted imaging lesion) versus perfusion status (severity of hypoperfusion) on hemorrhagic transformation. Stroke 41: e135-142. [Crossref]
- Fiorelli M, Bastianello S, von Kummer R, del Zoppo GJ, Larrue V, et al. (1999) Hemorrhagic transformation within 36 hours of a cerebral infarct: relationships with early clinical deterioration and 3-month outcome in the European Cooperative Acute Stroke Study I (ECASS I) cohort. Stroke 30: 2280-2284. [Crossref]
- Hong KS, Kang DW, Koo JS, Yu KH, Han MK, et al. (2008) Impact of neurological and medical complications on 3-month outcomes in acute ischaemic stroke. Eur J Neurol 15: 1324-1331. [Crossref]
- Goldstein JN, Marrero M, Masrur S, Pervez M, Barrocas AM, et al. (2010) Management of thrombolysis-associated symptomatic intracerebral hemorrhage. Arch Neurol 67: 965-969. [Crossref]
- D'Amelio M, Terruso V, Famoso G, Di Benedetto N, Realmuto S, et al. (2014) Early and late mortality of spontaneous hemorrhagic transformation of ischemic stroke. J Stroke Cerebrovasc Dis 23: 649-654. [Crossref]
- Berger C, Fiorelli M, Steiner T, Schäbitz WR, Bozzao L, et al. (2001) Hemorrhagic transformation of ischemic brain tissue: Asymptomatic or symptomatic? Stroke 32: 1330-1335. [Crossref]
- Paciaroni M, Agnelli G, Corea F, Ageno W, Alberti A, et al. (2008) Early hemorrhagic transformation of brain infarction: rate, predictive factors, and influence on clinical outcome: results of a prospective multicenter study. Stroke 39: 2249-2256. [Crossref]
- Tan S, Wang D, Liu M, Zhang S, Wu B, et al. (2014) Frequency and predictors of spontaneous hemorrhagic transformation in ischemic stroke and its association with prognosis. J Neurol 261: 905-912. [Crossref]
- Libman R, Kwiakowski T, Lyden P, Grotta JC, Tilley BC, et al. (2005) Asymptomatic hemorrhagic transformation of cerebral infarction does not worsen long-term outcome. J Stroke Cerebrovasc Dis 14: 50-54. [Crossref]
- Weihua Jia, Xiaoling Liao, Yuesong Pan, Yilong Wang, Tao Cui, et al. (2015) Thrombolytic-related asymptomatic hemorrhagic transformation does not deteriorate clinical outcome: Data from TIMS in China. PLoS One 10: e0142381. [Crossref]
- Park JH, Ko Y, Kim WJ, Jang MS, Yang MH, et al. (2012) Is asymptomatic hemorrhagic transformation really innocuous? Neurology 78: 421-426. [Crossref]
- Kimura K, Iguchi Y, Shibazaki K, Aoki J, Terasawa Y (2008) Hemorrhagic transformation of ischemic brain tissue after t-PA thrombolysis as detected by MRI may be asymptomatic, but impair neurological recovery. J Neurol Sci 272: 136-142. [Crossref]
- Dzialowski I, Pexman JH, Barber PA, Demchuk AM, Buchan AM, et al. (2007) Asymptomatic hemorrhage after thrombolysis may not be benign: Prognosis by hemorrhage type in the canadian alteplase for stroke effectiveness study registry. Stroke 38: 75-79. [Crossref]
- Lei C, Wu B, Liu M, Chen Y (2014) Asymptomatic hemorrhagic transformation after acute ischemic stroke: is it clinically innocuous? J Stroke Cerebrovasc Dis 23: 2767-2772. [Crossref]
- National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group (1995) Tissue plasminogen activator for acute ischemic stroke. N Engl J Med 333: 1581-1587. [Crossref]
- Fraser PA (2011) The role of free radical generation in increasing cerebrovascular permeability. Free Radic Biol Med 51: 967-977. [Crossref]
- Gasche Y, Copin JC, Sugawara T, Fujimura M, Chan PH (2001) Matrix metalloproteinase inhibition prevents oxidative stress-associated blood-brain barrier disruption after transient focal cerebral ischemia. J Cereb Blood Flow Metab 21: 1393-1400. [Crossref]
- Lu A, Clark JF, Broderick JP, Pyne-Geithman GJ, Wagner KR, et al. (2009) Mechanical reperfusion is associated with post-ischemic hemorrhage in rat brain. Exp Neurol 216: 407-412. [Crossref]
- Horsch AD, Dankbaar JW, van der Graaf Y, Niesten JM, et al. (2015) Relation between reperfusion and hemorrhagic transformation in acute ischemic stroke. Neuroradiology 57: 1219-1225. [Crossref]
- Thomalla G, Sobesky J, Köhrmann M, Fiebach JB, Fiehler J, et al. (2007) Two tales: hemorrhagic transformation but not parenchymal hemorrhage after thrombolysis is related to severity and duration of ischemia: MRI study of acute stroke patients treated with intravenous tissue plasminogen activator within 6 hours. Stroke 38: 313-318. [Crossref]
- Whiteley WN, Slot KB, Fernandes P, Sandercock P, Wardlaw J (2012) Risk factors for intracranial hemorrhage in acute ischemic stroke patients treated with recombinant tissue plasminogen activator: a systematic review and meta-analysis of 55 studies. Stroke 43: 2904-2909. [Crossref]
- Terruso V, D'Amelio M, Di Benedetto N, Lupo I, Saia V, et al. (2009) Frequency and determinants for hemorrhagic transformation of cerebral infarction. Neuroepidemiology 33: 261-265. [Crossref]
- Selim M, Fink JN, Kumar S, Caplan LR, Horkan C, et al. (2002) Predictors of hemorrhagic transformation after intravenous recombinant tissue plasminogen activator: prognostic value of the initial apparent diffusion coefficient and diffusion-weighted lesion volume. Stroke 33: 2047-2052. [Crossref]
- Tejima E, Katayama Y, Suzuki Y, Kano T, Lo EH (2001) Hemorrhagic transformation after fibrinolysis with tissue plasminogen activator: evaluation of role of hypertension with rat thromboembolic stroke model. Stroke 32: 1336-1340. [Crossref]
- Jauch EC, Saver JL, Adams HP Jr, Bruno A, Connors JJ, et al. (2013) Guidelines for the early management of patients with acute ischemic stroke: A guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 44: 870-947. [Crossref]
- Robbins NM, Swanson RA (2014) Opposing effects of glucose on stroke and reperfusion injury: acidosis, oxidative stress, and energy metabolism. Stroke 45: 1881-1886. [Crossref]
- Parsons MW, Barber PA, Desmond PM, Baird TA, Darby DG, et al. (2002) Acute hyperglycemia adversely affects stroke outcome: a magnetic resonance imaging and spectroscopy study. Ann Neurol 52: 20-28. [Crossref]
- Uyttenboogaart M, Koch MW, Stewart RE, Vroomen PC, Luijckx GJ, et al. (2007) Moderate hyperglycaemia is associated with favourable outcome in acute lacunar stroke. Brain 130: 1626-1630. [Crossref]
- Cipolla MJ, Huang Q, Sweet JG (2011) Inhibition of protein kinase Cβ reverses increased blood-brain barrier permeability during hyperglycemic stroke and prevents edema formation in vivo. Stroke 42: 3252-3257. [Crossref]
- Martini SR, Kent TA (2007) Hyperglycemia in acute ischemic stroke: a vascular perspective. J Cereb Blood Flow Metab 27: 435-451. [Crossref]
- Ergul A, Li W, Elgebaly MM, Bruno A, Fagan SC (2009) Hyperglycemia, diabetes and stroke: focus on the cerebrovasculature. Vascul Pharmacol 51: 44-49. [Crossref]
- Mazya M, Egido JA, Ford GA, Lees KR, Mikulik R, et al. (2012) Predicting the risk of symptomatic intracerebral hemorrhage in ischemic stroke treated with intravenous alteplase: Safe implementation of treatments in stroke (SITS) symptomatic intracerebral hemorrhage risk score. Stroke 43: 1524-1531. [Crossref]
- Hafez S, Hoda MN, Guo X, Johnson MH, Fagan SC, et al. (2015) Comparative analysis of different methods of ischemia/reperfusion in hyperglycemic stroke outcomes: Interaction with tPA. Transl Stroke Res 6: 171-180. [Crossref]
- Won SJ, Tang XN, Suh SW, Yenari MA, Swanson RA (2011) Hyperglycemia promotes tissue plasminogen activator-induced hemorrhage by Increasing superoxide production. Ann Neurol 70: 583-590. [Crossref]
- Huang J, Liu B, Yang C, Chen H, Eunice D, et al. (2013) Acute hyperglycemia worsens ischemic stroke-induced brain damage via high mobility group box-1 in rats. Brain Res 1535: 148-155. [Crossref]
- Béjot Y, Aboa-Eboulé C, Hervieu M, Jacquin A, Osseby GV, et al. (2012) The deleterious effect of admission hyperglycemia on survival and functional outcome in patients with intracerebral hemorrhage. Stroke 43: 243-245. [Crossref]
- Ribo M, Molina CA, Delgado P, Rubiera M, Delgado-Mederos R, et al. (2007) Hyperglycemia during ischemia rapidly accelerates brain damage in stroke patients treated with rtPA. J Cereb Blood Flow Metab 27: 1616-1622. [Crossref]
- Bruno A, Durkalski VL, Hall CE, Juneja R, Barsan WG, et al. (2014) The stroke hyperglycemia insulin network effort (SHINE) trial protocol: a randomized, blinded, efficacy trial of standard vs. intensive hyperglycemia management in acute stroke. Int J Stroke 9: 246-251. [Crossref]
- Mishiro K, Imai T, Sugitani S, Kitashoji A, Suzuki Y, et al. (2014) Diabetes mellitus aggravates hemorrhagic transformation after ischemic stroke via mitochondrial defects leading to endothelial apoptosis. PLoS One 9: e103818. [Crossref]
- Won SJ, Tang XN, Suh SW, Yenari MA, Swanson RA (2011) Hyperglycemia promotes tissue plasminogen activator-induced hemorrhage by Increasing superoxide production. Ann Neurol 70: 583-590. [Crossref]
- M Morigi, S Angioletti, B Imberti, R Donadelli, G Micheletti, et al. (1998) Leukocyte-endothelial interaction is augmented by high glucose concentrations and hyperglycemia in a NF-kB-dependent fashion. J Clin Invest 101: 1905-1915. [Crossref]
- Hawkins BT, Lundeen TF, Norwood KM, Brooks HL, Egleton RD (2007) Increased blood-brain barrier permeability and altered tight junctions in experimental diabetes in the rat: contribution of hyperglycaemia and matrix metalloproteinases. Diabetologia 50: 202-211. [Crossref]
- Hou Q, Zuo Z, Michel P, Zhang Y, Eskandari A, et al. (2013) Influence of chronic hyperglycemia on cerebral microvascular remodeling: An in vivo study using perfusion computed tomography in acute ischemic stroke patients. Stroke 44: 3557-3560. [Crossref]
- Prakash R, Somanath PR, El-Remessy AB, Kelly-Cobbs A, Stern JE, et al. (2012) Enhanced cerebral but not peripheral angiogenesis in the goto-kakizaki model of type 2 diabetes involves VEGF and peroxynitrite signaling. Diabetes 61: 1533-1542. [Crossref]
- Tureyen K, Bowen K, Liang J, Dempsey RJ, Vemuganti R (2011) Exacerbated brain damage, edema and inflammation in type-2 diabetic mice subjected to focal ischemia. J Neurochem 116: 499-507. [Crossref]
- Rocco A, Heuschmann PU, Schellinger PD, Köhrmann M, Diedler J, et al. (2013) Glycosylated hemoglobin A1 predicts risk for symptomatic hemorrhage after thrombolysis for acute stroke. Stroke 44: 2134-2138. [Crossref]
- Larrue V, von Kummer R R, Müller A, Bluhmki E (2001) Risk factors for severe hemorrhagic transformation in ischemic stroke patients treated with recombinant tissue plasminogen activator: a secondary analysis of the European-Australasian Acute Stroke Study (ECASS II). Stroke 32: 438-441. [Crossref]
- Tu HT, Campbell BC, Christensen S, Desmond PM, De Silva DA, et al. (2015) Worse stroke outcome in atrial fibrillation is explained by more severe hypoperfusion, infarct growth, and hemorrhagic transformation. Int J Stroke 10: 534-540. [Crossref]
- Liu B, Wang D, Hao Z, Li D, Zhang J, et al. (2016) Reduction in estimated glomerular filtration rate (eGFR) results in an increased risk of spontaneous hemorrhagic transformation in patients with large-artery atherosclerosis stroke. Curr Neurovasc Res 13: 75-81. [Crossref]
- Chen S, Lu X, Zhang W, Han Z, Yang W, et al. (2016) Does Prior Antiplatelet Treatment Increase the Risk of Hemorrhagic Transformation and Unfavorable Outcome on Day 90 after Intravenous Thrombolysis in Acute Ischemic Stroke Patients? J Stroke Cerebrovasc Dis 25: 1366-1370. [Crossref]
- Jauch EC, Saver JL, Adams HP Jr, Bruno A, Connors JJ, et al. (2013) Guidelines for the early management of patients with acute ischemic stroke: A guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 44: 870-947. [Crossref]
- Hallevi H, Albright KC, Martin-Schild S, Barreto AD, Savitz SI, et al. (2008) Anticoagulation after cardioembolic stroke: to bridge or not to bridge? Arch Neurol 65: 1169-1173. [Crossref]
- Missler U, Wiesmann M, Friedrich C, Kaps M (1997) S-100 protein and neuron-specific enolase concentrations in blood as indicators of infarction volume and prognosis in acute ischemic stroke. Stroke 28: 1956-1960. [Crossref]
- Kim BJ, Kim YJ, Ahn SH, Kim NY, Kang DW, et al. (2014) The second elevation of neuron-specific enolase peak after ischemic stroke is associated with hemorrhagic transformation. J Stroke Cerebrovasc Dis 23: 2437-2443. [Crossref]
- Zaheer S, Beg M, Rizvi I, Islam N, Ullah E, et al. (2013) Correlation between serum neuron specific enolase and functional neurological outcome in patients of acute ischemic stroke. Ann Indian Acad Neurol 16: 504-508. [Crossref]
- Foerch C, Wunderlich MT, Dvorak F, Humpich M, Kahles T, et al. (2007) Elevated serum S100B levels indicate a higher risk of hemorrhagic transformation after thrombolytic therapy in acute stroke. Stroke 38: 2491-2495. [Crossref]
- Kazmierski R, Michalak S, Wencel-Warot A, Nowinski WL (2012) Serum tight-junction proteins predict hemorrhagic transformation in ischemic stroke patients. Neurology 79: 1677-1685. [Crossref]
- Turner RJ, Sharp FR (2016) Implications of MMP9 for blood brain barrier disruption and hemorrhagic transformation following ischemic stroke. Front Cell Neurosci 10: 56. [Crossref]
- Castellanos M, Leira R, Serena J, Blanco M, Pedraza S, et al. (2004) Plasma cellular-fibronectin concentration predicts hemorrhagic transformation after thrombolytic therapy in acute ischemic stroke. Stroke 35: 1671-1676. [Crossref]
- Choi KH, Park MS, Kim JT, Nam TS, Choi SM, et al. (2012) The serum ferritin level is an important predictor of hemorrhagic transformation in acute ischaemic stroke. Eur J Neurol 19: 570-577. [Crossref]
- Millan M, Sobrino T, Castellanos M, Nombela F, Arenillas JF, et al. (2007) Increased body iron stores are associated with poor outcome after thrombolytic treatment in acute stroke. Stroke 38: 90-95. [Crossref]
- Sorond FA, Ratan RR (2004) Ironing-out mechanisms of neuronal injury under hypoxic-ischemic conditions and potential role of iron chelators as neuroprotective agents. Antioxidants & Redox Signaling 2: 421-436.
- Mendioroz M, Fernández-Cadenas I, Alvarez-Sabín J, Rosell A, Quiroga D, et al. (2009) Endogenous activated protein C predicts hemorrhagic transformation and mortality after tissue plasminogen activator treatment in stroke patients. Cerebrovasc Dis 28: 143-150. [Crossref]
- Cheng T, Petraglia AL, Li Z, Thiyagarajan M, Zhong Z, et al. (2006) Activated protein C inhibits tissue plasminogen activator-induced brain hemorrhage. Nat Med 12: 1278-1285. [Crossref]
- Zlokovic BV, Griffin JH (2011) Cytoprotective protein C pathways and implications for stroke and neurological disorders. Trends Neurosci 34: 198-209. [Crossref]
- Ribo M, Montaner J, Molina CA, Arenillas JF, Santamarina E, et al. Admission fibrinolytic profile is associated with symptomatic hemorrhagic transformation in stroke patients treated with tissue plasminogen activator. Stroke 35: 2123-2127. [Crossref]
- Monasterio J, Bermúdez P, Quiroga D, Francisco E, Meneses B, et al. (2003) Plasma thrombin-activatable fibrinolytic inhibitor (TAFI) among healthy subjects and patients with vascular diseases: a validation study. Pathophysiol Haemost Thromb 33: 382-386. [Crossref]
- Ribo M, Montaner J, Molina CA, Arenillas JF, Santamarina E, et al. (2004) Admission fibrinolytic profile predicts clot lysis resistance in stroke patients treated with tissue plasminogen activator. Thromb Haemost 91: 1146-1151. [Crossref]
- Hernandez-Guillamon M, Garcia-Bonilla L, Solé M, Sosti V, Parés M, et al. (2010) Plasma VAP-1/SSAO activity predicts intracranial hemorrhages and adverse neurological outcome after tissue plasminogen activator treatment in stroke. Stroke 41: 1528-1535. [Crossref]
- Blanco M, Moro MA, Dávalos A, Leira R, Castellanos M, et al. (2005) Increased plasma levels of 15-deoxyDelta prostaglandin J2 are associated with good outcome in acute atherothrombotic ischemic stroke. Stroke 36: 1189-1194. [Crossref]
- Yu X, Shi J, Jiang C, Xu J, You S, et al. (2015) Association study of serum uric acid levels with clinical outcome and hemorrhagic transformation in stroke patients with rt-PA intravenous thrombolysis. Zhonghua Yi Xue Za Zhi 95: 2351-2354. [Crossref]
- Guo Y, Yan S, Zhang S, Zhang X, Chen Q, et al. (2015) Lower serum calcium level is associated with hemorrhagic transformation after thrombolysis. Stroke 46: 1359-1361. [Crossref]
- del Río-Espínola A, Fernández-Cadenas I, Giralt D, Quiroga A, Gutiérrez-Agulló M, et al. (2012) A predictive clinical-genetic model of tissue plasminogen activator response in acute ischemic stroke. Ann Neurol 72: 716-729. [Crossref]
- González-Conejero R, Fernández-Cadenas I, Iniesta JA, Marti-Fabregas J, Obach V, et al. (2006) Role of fibrinogen levels and factor XIII V34L polymorphism in thrombolytic therapy in stroke patients. Stroke 37: 2288-2293. [Crossref]
- Weng YC, Sonni A, Labelle-Dumais C, de Leau M, Kauffman WB, et al. (2012) COL4A1 mutations in patients with sporadic late-onset intracerebral hemorrhage. Ann Neurol 71: 470-477. [Crossref]
- Gould DB, Phalan FC, van Mil SE, Sundberg JP, Vahedi K, et al. (2006) Role of COL4A1 in small-vessel disease and hemorrhagic stroke. N Engl J Med 354: 1489-1496. [Crossref]
- Zhang X, Cao X, Xu X, Li A, Xu Y (2015) Correlation between the -1562C/T polymorphism in the matrix metalloproteinase-9 gene and hemorrhagic transformation of ischemic stroke. Exp Ther Med 9: 1043-1047. [Crossref]
- Mallolas J, Rodríguez R, Gubern C, Camós S, Serena J, et al. (2014) A polymorphism in the promoter region of the survivin gene is related to hemorrhagic transformation in patients with acute ischemic stroke. Neuromolecular Med 16: 856-861. [Crossref]
- Tong DC, Adami A, Moseley ME, Marks MP (2001) Prediction of hemorrhagic transformation following acute stroke: role of diffusion- and perfusion-weighted magnetic resonance imaging. Arch Neurol 58: 587-593. [Crossref]
- Campbell BCV, Christensen S, Parsons MW, Churilov L, Desmond PM, et al. (2013) Advanced imaging improves prediction of hemorrhage after stroke thrombolysis: Hemorrhage after rtPA. Ann Neurol 73: 510-519. [Crossref]
- Lin K, Zink WE, Tsiouris AJ, John M, Tekchandani L, et al. (2012) Risk assessment of hemorrhagic transformation of acute middle cerebral artery stroke using multimodal CT. J Neuroimaging 22: 160-166. [Crossref]
- Strbian D, Engelter S, Michel P, Meretoja A, Sekoranja L, et al. (2012) Symptomatic intracranial hemorrhage after stroke thrombolysis: the SEDAN score. Ann Neurol 71: 634-641. [Crossref]
- Zou M, Churilov L, He A, Campbell B, Davis SM, et al. (2013) Hyperdense middle cerebral artery sign is associated with increased risk of hemorrhagic transformation after intravenous thrombolysis for patients with acute ischaemic stroke. J Clin Neurosci 20: 984-987. [Crossref]
- Neumann-Haefelin T, Hoelig S, Berkefeld J, Fiehler J, Gass A, et al. (2006) Leukoaraiosis is a risk factor for symptomatic intracerebral hemorrhage after thrombolysis for acute stroke. Stroke 37: 2463-2466. [Crossref]
- Vo KD, Santiago F, Lin W, Hsu CY, Lee Y, et al. (2003) MR imaging enhancement patterns as predictors of hemorrhagic transformation in acute ischemic stroke. AJNR Am J Neuroradiol 24: 674-679. [Crossref]
- Aviv RI, d'Esterre CD, Murphy BD, Hopyan JJ, Buck B, et al. (2009) Hemorrhagic transformation of ischemic stroke: prediction with CT perfusion. Radiology 250: 867-877. [Crossref]
- Kidwell CS, Latour L, Saver JL, Alger JR, Starkman S, et al. (2008) Thrombolytic toxicity: blood brain barrier disruption in human ischemic stroke. Cerebrovasc Dis 25: 338-343. [Crossref]
- Bang OY, Saver JL, Kim SJ, Kim GM, Chung CS, et al. (2011) Collateral flow averts hemorrhagic transformation after endovascular therapy for acute ischemic stroke. Stroke 42: 2235-2239. [Crossref]
- Strbian D, Michel P, Seiffge DJ, Saver JL, Numminen H, et al. (2014) Symptomatic intracranial hemorrhage after stroke thrombolysis: comparison of prediction scores. Stroke 45: 752-758. [Crossref]
- De Bock M, Culot M, Wang N, Bol M, Decrock E, et al. (2011) Connexin channels provide a target to manipulate brain endothelial calcium dynamics and blood-brain barrier permeability. J Cereb Blood Flow Metab 31: 1942-1957. [Crossref]
- Jiao H, Wang Z, Liu Y, Wang P, Xue Y (2011) Specific role of tight junction proteins claudin-5, occludin, and ZO-1 of the blood–brain barrier in a focal cerebral ischemic insult. J Mol Neurosci 44: 130-139. [Crossref]
- Liu J, Jin X, Liu KJ, Liu W (2012) Matrix metalloproteinase-2-mediated occludin degradation and caveolin-1-mediated claudin-5 redistribution contribute to blood-brain barrier damage in early ischemic stroke stage. J Neurosci 32: 3044-3057. [Crossref]
- Jickling GC, Ander BP, Stamova B, Zhan X, Liu D, et al. (2013) RNA in blood is altered prior to hemorrhagic transformation in ischemic stroke. Ann Neurol 74: 232-240. [Crossref]
- Gliem M, Mausberg AK, Lee JI, Simiantonakis I, van Rooijen N, et al. (2012) Macrophages prevent hemorrhagic infarct transformation in murine stroke models. Ann Neurol 71: 743-752. [Crossref]
- Hafez S, Abdelsaid M1, El-Shafey S1, Johnson MH1, Fagan SC1, Ergul A (2016) Matrix metalloprotease 3 exacerbates hemorrhagic transformation and worsens functional outcomes in hyperglycemic stroke. Stroke 47: 843-851. [Crossref]
- Fujimoto M, Takagi Y, Aoki T, Hayase M, Marumo T, et al. (2008) Tissue inhibitor of metalloproteinases protect blood-brain barrier disruption in focal cerebral ischemia. J Cereb Blood Flow Metab 28: 1674-1685. [Crossref]
- Piccardi B, Palumbo V, Nesi M, Nencini P, Gori AM, et al. (2015) Unbalanced Metalloproteinase-9 and Tissue Inhibitors of Metalloproteinases Ratios Predict Hemorrhagic Transformation of Lesion in Ischemic Stroke Patients Treated with Thrombolysis: Results from the MAGIC Study. Front Neurol 6: 121. [Crossref]
- Gliem M, Mausberg AK, Lee JI, Simiantonakis I, van Rooijen N, et al. (2012) Macrophages prevent hemorrhagic infarct transformation in murine stroke models. Ann Neurol 71: 743-752. [Crossref]
- Mildner A, Mack M, Schmidt H, Brück W, Djukic M, et al. (2009) CCR2+Ly-6Chi monocytes are crucial for the effector phase of autoimmunity in the central nervous system. Brain 132: 2487-2500. [Crossref]
- Dimitrijevic OB, Stamatovic SM, Keep RF, Andjelkovic AV (2007) Absence of the chemokine receptor CCR2 protects against cerebral ischemia/reperfusion injury in mice. Stroke 38: 1345-1353. [Crossref]
- Urra X, Villamor N, Amaro S, Gómez-Choco M, Obach V, et al. (2009) Monocyte subtypes predict clinical course and prognosis in human stroke. J Cereb Blood Flow Metab 29: 994-1002. [Crossref]
- Gliem M, Klotz L, van Rooijen N, Hartung HP, Jander S (2015) Hyperglycemia and PPARγ antagonistically influence macrophage polarization and infarct healing after ischemic stroke. Stroke 46: 2935-2942. [Crossref]
- Zhao XR, Gonzales N, Aronowski J (2015) Pleiotropic role of PPARγ in intracerebral hemorrhage: an intricate system involving Nrf2, RXR, and NF-κB. CNS Neurosci Ther 21: 357-366. [Crossref]
- Frankowski JC, DeMars KM, Ahmad AS, Hawkins KE, Yang C, et al. (2015) Detrimental role of the EP1 prostanoid receptor in blood-brain barrier damage following experimental ischemic stroke. Sci Rep 5: 17956. [Crossref]
- Qiu J, Xu J, Zheng Y, Wei Y, Zhu X, et al. (2010) High-mobility group box 1 promotes metalloproteinase-9 upregulation through Toll-like receptor 4 after cerebral ischemia. Stroke 41: 2077-2082. [Crossref]
- Wang W, Li M, Chen Q, Wang J (2015) Hemorrhagic transformation after tissue plasminogen activator reperfusion therapy for ischemic stroke: Mechanisms, models, and biomarkers. Mol Neurobiol 52: 1572-1579. [Crossref]
- Fiehler J, Remmele C, Kucinski T, Rosenkranz M, Thomalla G, et al. (2005) Reperfusion after severe local perfusion deficit precedes hemorrhagic transformation: an MRI study in acute stroke patients. Cerebrovasc Dis 19: 117-124. [Crossref]
- Zhenjun Tan, Xinlan Li, Ryan C Turner, Aric F Logsdon, Brandon Lucke-Wold, et al. (2014) Combination treatment of R-rtPA and an optimized human apyrase reduces mortality rate and hemorrhagic transformation 6h after ischemic stroke in aged female rats. Eur J Pharmacol 738: 368-373. [Crossref]
- Yan P, Zhu A, Liao F, Xiao Q, Kraft AW, et al. (2015) Minocycline reduces spontaneous hemorrhage in mouse models of cerebral amyloid angiopathy. Stroke 46: 1633-1640. [Crossref]
- Murata Y, Rosell A, Scannevin RH, Rhodes KJ, Wang X, et al. (2008) Extension of the thrombolytic time window with minocycline in experimental stroke. Stroke 39: 3372-3377. [Crossref]
- Amiri-Nikpour MR, Nazarbaghi S, Hamdi-Holasou M, Rezaei Y (2015) An open-label evaluator-blinded clinical study of minocycline neuroprotection in ischemic stroke: gender-dependent effect. Acta Neurol Scand 131: 45-50. [Crossref]
- Padma Srivastava MV, Bhasin A, Bhatia R, Garg A, Gaikwad S, et al. (2012) Efficacy of minocycline in acute ischemic stroke: a single-blinded, placebo-controlled trial. Neurol India 60: 23-28. [Crossref]
- Wang Z, Lin Y, Liu Y, Chen Y, Wang B, et al. (2016) Serum uric acid levels and outcomes after acute ischemic stroke. Mol Neurobiol 53: 1753-1759. [Crossref]
- Chamorro A, Amaro S, Castellanos M, Segura T, Arenillas J, et al. (2014) Safety and efficacy of uric acid in patients with acute stroke (URICO-ICTUS): a randomised, double-blind phase 2b/3 trial. Lancet Neurol 13: 453-460. [Crossref]
- Llull L, Laredo C, Renú A, Pérez B, Vila E, et al. (2015) Uric acid therapy improves clinical outcome in women with acute ischemic stroke. Stroke 46: 2162-2167. [Crossref]
- Amaro S, Llull L, Renú A, Laredo C, Perez B, et al. (2015) Uric acid improves glucose-driven oxidative stress in human ischemic stroke. Ann Neurol 77: 775-783. [Crossref]
- Okamura K, Tsubokawa T, Johshita H, Miyazaki H, Shiokawa Y (2014) Edaravone, a free radical scavenger, attenuates cerebral infarction and hemorrhagic infarction in rats with hyperglycemia. Neurol Res 36: 65-69. [Crossref]
- Yagi K, Kitazato KT, Uno M, Tada Y, Kinouchi T, et al. (2009) Edaravone, a free radical scavenger, inhibits MMP-9-related brain hemorrhage in rats treated with tissue plasminogen activator. Stroke 40: 626-631. [Crossref]
- Isahaya K, Yamada K, Yamatoku M, Sakurai K, Takaishi S, et al. (2012) Effects of edaravone, a free radical scavenger, on serum levels of inflammatory biomarkers in acute brain infarction. J Stroke Cerebrovasc Dis 21: 102-107. [Crossref]
- Mishina M, Komaba Y, Kobayashi S, Kominami S, Fukuchi T, et al. (2008) Administration of free radical scavenger edaravone associated with higher frequency of hemorrhagic transformation in patients with cardiogenic embolism. Neurol Med Chir (Tokyo) 48: 292-297. [Crossref]
- Wada T, Yasunaga H, Inokuchi R, Horiguchi H, Fushimi K, et al. (2014) Effects of edaravone on early outcomes in acute ischemic stroke patients treated with recombinant tissue plasminogen activator. J Neurol Sci 345: 106-111. [Crossref]
- Li L, Zuo Z (2009) Isoflurane preconditioning improves short-term and long-term neurological outcome after focal brain ischemia in adult rats. Neuroscience 164: 497-506. [Crossref]
- Zhang HP, Sun YY, Chen XM, Yuan LB, Su BX, et al. (2014) The neuroprotective effects of isoflurane preconditioning in a murine transient global cerebral ischemia–reperfusion model: The role of the notch signaling pathway. Neuro Molecular Medicine 16: 191-204.
- Li G, Jia J, Fu J, Wang H, Ji K, et al. (2014) The effects of preconditioning and postconditioning with isoflurane on focal cerebral ischemi/reperfusion injury in rats. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 26: 431-435. [Crossref]
- Wang S, Yin J, Ge M, Dai Z, Li Y, et al. (2016) Transforming growth-beta 1 contributes to isoflurane postconditioning against cerebral ischemia-reperfusion injury by regulating the c-Jun N-terminal kinase signaling pathway. Biomed Pharmacother 78: 280-290. [Crossref]
- Lee JJ, Li L, Jung HH, Zuo Z (2008) Postconditioning with isoflurane reduced ischemia-induced brain injury in rats. Anesthesiology 108: 1055-1062. [Crossref]
- Kim EJ, Kim SY, Lee JH, Kim JM, Kim JS, et al. (2015) Effect of isoflurane post-treatment on tPA-exaggerated brain injury in a rat ischemic stroke model. Korean J Anesthesiol 68: 281-286. [Crossref]
- Esposito E, Mandeville ET, Lo EH (2013) Lower doses of isoflurane treatment has no beneficial effects in a rat model of intracerebral hemorrhage. BMC Neurosci 14: 129. [Crossref]
- Hu Q, Ma Q, Zhan Y, He Z, Tang J, et al. (2011) Isoflurane enhanced hemorrhagic transformation by impairing antioxidant enzymes in hyperglycemic rats with middle cerebral artery occlusion. Stroke 42: 1750-1756. [Crossref]
- Chen CH1, Manaenko A, Zhan Y, Liu WW, Ostrowki RP, et al. (2010) Hydrogen gas reduced acute hyperglycemia-enhanced hemorrhagic transformation in a focal ischemia rat model. Neuroscience 169: 402-414. [Crossref]
- Shuaib A, Lees KR, Lyden P, Grotta J, Davalos A, et al. (2007) NXY-059 for the treatment of acute ischemic stroke. N Engl J Med 357: 562-571. [Crossref]
- Lapchak PA, Chapman DF, Zivin JA (2001) Pharmacological effects of the spin trap agents N-T-Butyl-Phenylnitrone (PBN) and 2,2,6, 6-Tetramethylpiperidine-N-Oxyl (TEMPO) in a rabbit thromboembolic stroke model: combination studies with the thrombolytic tissue plasminogen activator. Stroke 32: 147-153. [Crossref]
- Cai Y, Liu X, Chen W, Wang Z, Xu G, et al. (2015) TGF-β1 prevents blood–brain barrier damage and hemorrhagic transformation after thrombolysis in rats. Exp Neurol 266: 120-126. [Crossref]
- Shih HM, Lin WC, Wang CH, Lin LC (2014) Hypertensive patients using thiazide diuretics as primary stroke prevention make better functional outcome after ischemic stroke. J Stroke Cerebrovasc Dis 23: 2414-2418. [Crossref]
- Fujii K, Weno BL, Baumbach GL, Heistad DD (1992) Effect of antihypertensive treatment on focal cerebral infarction. Hypertension 19: 713-716. [Crossref]
- Chen HS, Chen XM, Feng JH, et al. (2015) Peroxynitrite decomposition catalyst reduces delayed thrombolysis-induced hemorrhagic transformation in ischemia-reperfused rat brains. CNS Neurosci Ther 21: 585-590. [Crossref]
- Baulieu EE (1997) Neurosteroids: of the nervous system, by the nervous system, for the nervous system. Recent Prog Horm Res 52: 1-32. [Crossref]
- Stein DG, Wright DW, Kellermann AL (2008) Does progesterone have neuroprotective properties? Ann Emerg Med 51: 164-172. [Crossref]
- Ishrat T, Sayeed I, Atif F, Hua F, Stein DG (2010) Progesterone and allopregnanolone attenuate blood-brain barrier dysfunction following permanent focal ischemia by regulating the expression of matrix metalloproteinases. Exp Neurol 226: 183-190. [Crossref]
- Kumon Y, Kim SC, Tompkins P, Stevens A, Sakaki S, et al. (2000) Neuroprotective effect of postischemic administration of progesterone in spontaneously hypertensive rats with focal cerebral ischemia. J Neurosurg 92: 848-852. [Crossref]
- Jiang N, Chopp M, Stein D, Feit H (1996) Progesterone is neuroprotective after transient middle cerebral artery occlusion in male rats. Brain Res 735: 101-107. [Crossref]
- Won S, Lee JH, Wali B, Stein DG, Sayeed I (2014) Progesterone attenuates hemorrhagic transformation after delayed rtPA treatment in an experimental model of stroke in rats: involvement of the VEGF–MMP pathway. J Cereb Blood Flow Metab 34: 72-80. [Crossref]
- Kanazawa M, Igarashi H, Kawamura K, Takahashi T, Kakita A, et al. (2011) Inhibition of VEGF signaling pathway attenuates hemorrhage after rtPA treatment. J Cereb Blood Flow Metab 31: 1461-1474. [Crossref]
- Ishrat T, Sayeed I, Atif F, Hua F, Stein DG (2012) Progesterone is neuroprotective against ischemic brain injury through its effects on the phosphoinositide 3-kinase/protein kinase B signaling pathway. Neuroscience 210: 442-450. [Crossref]
- Hong SH, Khoutorova L, Bazan NG, Belayev L2 (2015) Docosahexaenoic acid improves behavior and attenuates blood-brain barrier injury induced by focal cerebral ischemia in rats. Exp Transl Stroke Med 7: 3. [Crossref]
- Marcheselli VL, Hong S, Lukiw WJ, Tian XH, Gronert K, et al. (2003) Novel docosanoids inhibit brain ischemia-reperfusion-mediated leukocyte infiltration and pro-inflammatory gene expression. J Biol Chem 278: 43807-43817. [Crossref]
- Lin Y, Xu M, Wan J, Wen S, Sun J, et al. (2015) Docosahexaenoic acid attenuates hyperglycemia-enhanced hemorrhagic transformation after transient focal cerebral ischemia in rats. Neuroscience 301: 471-479. [Crossref]
- Gautier S, Ouk T, Tagzirt M, Lefebvre C, Laprais M, et al. (2014) Impact of the neutrophil response to granulocyte colony-stimulating factor on the risk of hemorrhage when used in combination with tissue plasminogen activator during the acute phase of experimental stroke. J Neuroinflammation 11: 96. [Crossref]
- dela Peña IC, Yoo A, Tajiri N, Acosta SA, Ji X, et al. (2015) Granulocyte colony-stimulating factor attenuates delayed rtPA-induced hemorrhagic transformation in ischemic stroke rats by enhancing angiogenesis and vasculogenesis. J Cereb Blood Flow Metab 35: 338-346. [Crossref]
- Sprigg N, Bath PM, Zhao L, Willmot MR, Gray LJ, et al. (2006) Granulocyte-colony-stimulating factor mobilizes bone marrow stem cells in patients with subacute ischemic stroke: the Stem cell Trial of recovery EnhanceMent after Stroke (STEMS) pilot randomized, controlled trial (ISRCTN 16784092). Stroke 37: 2979-2983. [Crossref]
- Fan Y, Shen F, Frenzel T, Zhu W, Ye J, et al. (2010) Endothelial progenitor cell transplantation improves long-term stroke outcome in mice. Ann Neurol 67: 488-497. [Crossref]
- Fan ZZ, Cai HB, Ge ZM, Wang LQ, Zhang XD, et al. (2015) The efficacy and safety of granulocyte colony-stimulating factor for patients with stroke. J Stroke Cerebrovasc Dis 24: 1701-1708. [Crossref]
- Shyu WC, Lin SZ, Lee CC, Liu DD, Li H (2006) Granulocyte colony-stimulating factor for acute ischemic stroke: a randomized controlled trial. CMAJ 174: 927-933. [Crossref]
- Wang W, Li M, Wang Y, Li Q, Deng G, et al. (2015) GSK-3β Inhibitor TWS119 attenuates rrtPA-induced hemorrhagic transformation and activates the Wnt/β-Catenin signaling pathway after acute ischemic stroke in rats. Mol Neurobiol 53: 7028-7036. [Crossref]
- Liu L, Wan W, Xia S, Kalionis B, Li Y (2014) Dysfunctional Wnt/β -catenin signaling contributes to blood-brain barrier breakdown in Alzheimer's disease. Neurochem Int 75: 19-25. [Crossref]
- Ajmone-Cat MA, D'Urso MC, di Blasio G, Brignone MS, De Simone R, et al. (2016) Glycogen synthase kinase 3 is part of the molecular machinery regulating the adaptive response to LPS stimulation in microglial cells. Brain Behav Immun 55: 225-235. [Crossref]
- Qu B, Liu BR, DU YJ, Chen J, Cheng YQ, et al. Wnt/β‑catenin signaling pathway may regulate the expression of angiogenic growth factors in hepatocellular carcinoma. Oncol Lett 1175-1178. [Crossref]
- Kim I, Kim HG, So JN, Kim JH, Kwak HJ, et al. (2000) Angiopoietin-1 regulates endothelial cell survival through the phosphatidylinositol 3'-Kinase/Akt signal transduction pathway. Circ Res 86: 24-29. [Crossref]
- Gamble JR, Drew J, Trezise L, Underwood A, Parsons M, et al. (2000) Angiopoietin-1 is an antipermeability and anti-inflammatory agent in vitro and targets cell junctions. Circ Res 87: 603-607. [Crossref]
- Zhang ZG, Zhang L, Croll SD, Chopp M (2002) Angiopoietin-1 reduces cerebral blood vessel leakage and ischemic lesion volume after focal cerebral embolic ischemia in mice. Neuroscience 113: 683-687. [Crossref]
- Shin HY, Lee YJ, Kim HJ, Park CK, Kim JH, et al. (2010) Protective role of COMP-Ang1 in ischemic rat brain. J Neurosci Res 88: 1052-1063. [Crossref]
- Yu H, Wang P, An P, Xue Y (2012) Recombinant human angiopoietin-1 ameliorates the expressions of ZO-1, occludin, VE-cadherin, and PKCα signaling after focal cerebral ischemia/reperfusion in rats. J Mol Neurosci 46: 236-247. [Crossref]
- Baffert F, Le T, Thurston G, McDonald DM (2006) Angiopoietin-1 decreases plasma leakage by reducing number and size of endothelial gaps in venules. Am J Physiol Heart Circ Physiol 290: H107-118. [Crossref]
- Kawamura K, Takahashi T, Kanazawa M, Igarashi H, Nakada T, et al. (2014) Effects of angiopoietin-1 on hemorrhagic transformation and cerebral edema after tissue plasminogen activator treatment for ischemic stroke in rats. PLoS One 9: e98639. [Crossref]
- Zhenjun Tan, Xinlan Li, Ryan C Turner, Aric F Logsdon, Brandon Lucke-Wold, et al. (2014) Combination treatment of R-rtPA and an optimized human apyrase reduces mortality rate and hemorrhagic transformation 6h after ischemic stroke in aged female rats. Eur J Pharmacol 738: 368-373. [Crossref]
- Sugimoto S, Lin X, Lai J, Okazaki M, Das NA, et al. (2009) Apyrase treatment prevents ischemia-reperfusion injury in rat lung isografts. J Thorac Cardiovasc Surg 138: 752-759. [Crossref]
- Wang C (2010) APT 102 Protects the ischemically injured brain in rats. Oral Presentation Neuroscience Meeting.
- Pinsky DJ, Broekman MJ, Peschon JJ, Stocking KL, Fujita T, et al. (2002) Elucidation of the thromboregulatory role of CD39/ectoapyrase in the ischemic brain. J Clin Invest 109: 1031-1040. [Crossref]
- Cheng O, Ostrowski RP, Wu B, Liu W, Chen C, et al. (2011) Cyclooxygenase-2 mediates hyperbaric oxygen preconditioning in the rat model of transient global cerebral ischemia. Stroke 42: 484-490. [Crossref]
- Bian H, Hu Q, Liang X, Chen D, Li B, et al. (2015) Hyperbaric Oxygen Preconditioning Attenuates Hemorrhagic Transformation through Increasing PPAR? in Hyperglycemic MCAO Rats. Exp Neurol 265: 22-29. [Crossref]
- Min LJ, Mogi M, Shudou M, Jing F, Tsukuda K, et al. (2012) Peroxisome proliferator-activated receptor- activation with angiotensin II type 1 receptor blockade is pivotal for the prevention of blood-brain barrier impairment and cognitive decline in type 2 diabetic mice. Hypertension 59: 1079-1088. [Crossref]
- Zeng Y, Xie K, Dong H, Zhang H, Wang F, et al. (2012) Hyperbaric oxygen preconditioning protects cortical neurons against oxygen-glucose deprivation injury: role of peroxisome proliferator-activated receptor-gamma. Brain Res 1452: 140-150. [Crossref]
- Zhao X, Zhang Y, Strong R, Grotta JC, Aronowski J (2006) 15d-Prostaglandin J2 activates peroxisome proliferator-activated receptor-gamma, promotes expression of catalase, and reduces inflammation, behavioral dysfunction, and neuronal loss after intracerebral hemorrhage in rats. J Cereb Blood Flow Metab 26: 811-820. [Crossref]
- Lin TN, Cheung WM, Wu JS, Chen JJ, Lin H, et al. (2006) 15d-prostaglandin J2 protects brain from ischemia-reperfusion injury. Arterioscler Thromb Vasc Biol 26: 481-487. [Crossref]
- Guo ZN, Xu L, Hu Q, Matei N, Yang P, et al. (2016) Hyperbaric oxygen preconditioning attenuates hemorrhagic transformation through reactive oxygen species/thioredoxin-interacting protein/nod-like receptor protein 3 pathway in hyperglycemic middle cerebral artery occlusion rats. Crit Care Med 44: e403-411. [Crossref]
- Ma Q, Chen S, Hu Q, Feng H, Zhang JH, et al. (2014) NLRP3 inflammasome contributes to inflammation after intracerebral hemorrhage. Ann Neurol 75: 209-219. [Crossref]
- Yang F, Wang Z, Wei X, Han H, Meng X, et al. (2014) NLRP3 deficiency ameliorates neurovascular damage in experimental ischemic stroke. J Cereb Blood Flow Metab 34: 660-667. [Crossref]
- Martinon F (2010) Signaling by ROS drives inflammasome activation. Eur J Immunol 40: 616-619. [Crossref]
- Miyazaki T, Kimura Y, Ohata H, Hashimoto T, Shibata K, et al. (2011) Distinct effects of tissue-type plasminogen activator and SMTP-7 on cerebrovascular inflammation following thrombolytic reperfusion. Stroke 42: 1097-1104. [Crossref]
- Ito A, Niizuma K, Shimizu H, Fujimura M, Hasumi K, et al. (2014) SMTP-7, a new thrombolytic agent, decreases hemorrhagic transformation after transient middle cerebral artery occlusion under warfarin anticoagulation in mice. Brain Res 1578: 38-48. [Crossref]
- Sawada H, Nishimura N, Suzuki E, Zhuang J, Hasegawa K, et al. (2014) SMTP-7, a novel small-molecule thrombolytic for ischemic stroke: a study in rodents and primates. J Cereb Blood Flow Metab 34: 235-241. [Crossref]
- Tureyen K, Kapadia R, Bowen KK, Satriotomo I, Liang J, et al. (2007) Peroxisome proliferator-activated receptor-gamma agonists induce neuroprotection following transient focal ischemia in normotensive, normoglycemic as well as hypertensive and type-2 diabetic rodents. J Neurochem 101: 41-56. [Crossref]
- Sundararajan S, Gamboa JL, Victor NA, Wanderi EW, Lust WD, Landreth GE (2005) Peroxisome proliferator-activated receptor-gamma ligands reduce inflammation and infarction size in transient focal ischemia. Neuroscience 130: 685-696. [Crossref]
- Allahtavakoli M, Moloudi R, Arababadi MK, Shamsizadeh A, Javanmardi K (2009) Delayed post ischemic treatment with Rosiglitazone attenuates infarct volume, neurological deficits and neutrophilia after embolic stroke in rat. Brain Res 1271: 121-127. [Crossref]
- Zhang FH, Lin YH, Huang HG, Sun JZ, Wen SQ, et al. (2013) Rosiglitazone attenuates hyperglycemia-enhanced hemorrhagic transformation after transient focal ischemia in rats. Neuroscience 250: 651-657. [Crossref]
- Abdul Muneer PM, Alikunju S, Szlachetka AM, Murrin LC, Haorah J (2011) Impairment of brain endothelial glucose transporter by methamphetamine causes blood-brain barrier dysfunction. Mol Neurodegener 6: 23. [Crossref]
- Arboix A, García-Eroles L, Oliveres M, Targa C, Balcells M, Massons J (2010) Pretreatment with statins improves early outcome in patients with first-ever ischaemic stroke: A pleiotropic effect of statins or a beneficial effect of hypercholesterolemia? BMC Neurol 10: 47. [Crossref]
- Ní Chróinín D, Asplund K, Asberg S, Callaly E, Cuadrado-Godia E, et al. (2013) Statin therapy and outcome after ischemic stroke: systematic review and meta-analysis of observational studies and randomized trials. Stroke 44: 448-456. [Crossref]
- Wang TY, Newby LK, Chen AY, Mulgund J, Roe MT, et al. (2009) Hypercholesterolemia paradox in relation to mortality in acute coronary syndrome. Clin Cardiol 32: E22-28. [Crossref]
- Nardi K, Leys D, Eusebi P, Cordonnier C, Gautier S, et al. (2011) Influence of lipid profiles on the risk of hemorrhagic transformation after ischemic stroke: systematic review. Cerebrovasc Dis Extra 1: 130-141. [Crossref]
- Wilhelmsson U, Andersson D, de Pablo Y, Pekny R, Stahlberg A, et al. (2017) Injury leads to the appearance of cells with characteristics of both microglia and astrocytes in mouse and human brain. Cereb Cortex 27: 3360-3377. [Crossref]
- Skrobot OA, Attems J, Esiri M, Hortobágyi T, Ironside JW, et al. (2016) Vascular cognitive impairment neuropathology guidelines – a multi-centre study of the contribution of cerebrovascular disease to cognitive impairment. Brain 139: 2957-2969. [Crossref]
- McAleese KE, Alafuzoff I, Charidimou A, De Reuck J, Grinberg LT, et al. (2016) Post-mortem assessment in vascular dementia: advances and aspirations. BMC Med 14: 129. [Crossref]
- Szepesi R, Csokonay Á, Murnyák B, Kouhsari MC, Hofgárt G, et al. (2016) Haemorrhagic transformation in ischaemic stroke is more frequent than clinically suspected - A neuropathological study. J Neurol Sci 368: 4-10. [Crossref]
- Szepesi R, Széll IK, Hortobágyi T, Kardos L, Nagy K, et al. (2015) New prognostic score for the prediction of 30-day outcome in spontaneous supratentorial cerebral haemorrhage. Biomed Research International.
- Alafuzoff I, Gelpi E, Al-Sarraj S, Arzberger T, Attems J, et al. (2012) The need to unify neuropathological assessments of vascular alterations in the ageing brain: multicentre survey by the BrainNet Europe consortium. Exp Gerontol 47: 825-833. [Crossref]
- Kirvell SL, Elliott MS, Kalaria RN, Hortobágyi T, Ballard CG, et al. (2010) Vesicular glutamate transporter and cognition in stroke: a case-control autopsy study. Neurology 75: 1803-1809. [Crossref]
- Elliott MS, Ballard CG, Kalaria RN, Perry R, Hortobágyi T, Francis PT (2009) increased binding to 5-HT1A and 5-HT2A receptors in vascular dementia and stroke is related to infarct injury and cognition. Brain 132: 1858-1865. [Crossref]
- Sharp SI, Francis PT, Elliott MS, Kalaria RN, Bajic N, et al. (2009) Choline acetyltransferase activity in vascular dementia and stroke. Dement Geriatr Cogn Disord 28: 233-238. [Crossref]
- Mulugeta E, Molina-Holgado F, Elliott MS, Hortobagyi T, Perry R, et al. (2008) Inflammatory mediators in the frontal lobe of patients with mixed and vascular dementia. Dement Geriatr Cogn Disord 25: 278-286. [Crossref]
- Zádor Z, Benyó Z, Lacza Z, Hortobágyi T sr, Harkany T, Hortobágyi T (2004) Neuroprotection in brain ischemia – doubts and hopes. Clinical Neuroscience/Ideggyógyászati Szemle 57: 81-93.
- Zádor Z, Lacza Z, Benyó Z, Harkany T, Hortobágyi T (2001) Apoptosis in focal brain ischemia. Clinical Neuroscience/Ideggyógyászati Szemle 56: 216-228.
- Lacza Z, Hermán P, Görlach C, Hortobágyi T, Sándor P, et al. (2001) NO synthase blockade induces chaotic cerebral vasomotion via activation of thromboxane receptors. Stroke 32: 2609-2614. [Crossref]
- Wahl M, Görlach C, Hortobágyi T, Benyó Z (1999) Effects of bradykinin in the cerebral circulation. Acta Physiol Hung 86: 155-160. [Crossref]
- Bum Joon Kim, Hyun Goo Kang, Hye-Jin Kim, Sung-Ho Ahn, Na Young Kim, et al. (2014) Magnetic resonance imaging in acute ischemic stroke treatment. J Stroke 16: 131-145. [Crossref]