Abstract
55-year-old Caucasian male presented with fevers (39.4C), resting tremors, and influenza- like symptoms. Two months prior he had a febrile illness, presumed to be viral, which persisted for two weeks. On admission, the patient was found to encephalopathy, asterixis and fever. Laboratory studies demonstrated pancytopenia, renal and hepatic failure. No specific cause of hepatic failure was identified despite extensive evaluation. An extensive immunological, metabolic and infectious disease work-up was negative. Abdominal ultrasound and CAT scan and MRI of the chest and abdomen revealed mild ascites with pleural effusion and hepatosplenomegaly. Subsequent liver biopsy demonstrated nonspecific acute hepatitis. The clinical picture then raised suspicion for HLH. A bone marrow biopsy demonstrated histiocytes without hemophagocytes with concomitant B-cell lymphoma in brain biopsy.
Key words
Hemophagocytic Lymphohistiocytosis (HLH), B-cell lymphoma, fever of unknown origin.
Introduction
HLH is a rare lethal disease caused by mutations in genes crucial to the cytolytic secretory pathway, resulting in induced apoptosis of target cells by perforin and granzymes [1]. This results in dysfunction in natural killer and cytotoxic T-cells [2,3]. Few patients worldwide are reported to have HLH associated with B-cell lymphoma [4,5].
Case presentation
A 55-year-old Caucasian male presented with fevers (39.4C), fine resting tremors, and influenza- like symptoms. Two months prior he had a febrile illness presumed to be viral that was followed by fatigue, malaise and wasting, which persisted for two weeks. On admission, the patient was found to have encephalopathy, asterixis and fever. Laboratory studies demonstrated pancytopenia; white blood cell of 1.1 thou/cmm, haemoglobin 9.9 g/dL, platelets 35 thou/cmm. Acute renal failure; creatinine 5.23 mg/dL, BUN 101 mg/dL. Furthermore, the patient acute liver failure manifested with encephalopathy and coagulopathy. His AST 800 U/L, ALT 510 U/L, Alkaline Phosphatase 608 U/L, Bilirubin 14.5 mg/dL, INR 3.9. Moreover, his ferritin >40000 ng/mL, Fibrinogen 51 mg/dL, triglycerides 955 ng/mL. An Extensive infectious work up including Pan-Cultures, viral hepatitis, HIV and tuberculosis was negative. Hepatotoxic drug, autoimmune and hemochromatosis were also excluded. Abdominal ultrasound and CAT scan and MRI/MRCP of the chest and abdomen revealed mild ascites with pleural effusion and hepatosplenomegaly. Subsequent liver biopsy demonstrated acute nonspecific hepatitis. The clinical picture then raised suspicion for HLH. A bone marrow biopsy demonstrated histiocytes without hemophagocytes with concomitant B-cell lymphoma in brain biopsy. The fever, pancytopenia, and multiple organs dysfunction then raised suspicion for HLH. A bone marrow biopsy demonstrated histiocytes without hemophagocytes. The patient was transferred to a tertiary center, where a brain biopsy demonstrated B-cell lymphoma. Unfortunately, the patient died shortly after the diagnosis (Figure 1).
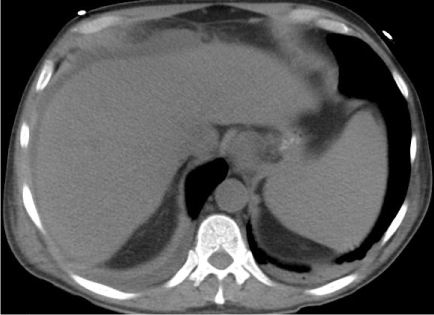
Figure 1. Hepatosplenomegaly on CT scan
Discussion
We present a case of acute liver failure as a part of multi organ failure due to secondary HLH. The causes of secondary HLH are infection, rheumatologic disorders and neoplasms with marked regional variations in cause [2,3]. HLH is diagnosed if patients fulfil one of the two following criteria; molecular diagnosis of HLH or the presence of five of these eight sub-criteria: fever, splenomegaly, pancytopenia, hypertriglyceridemia >3 time’s normal, hypofibrinogenemia, high ferritin, soluble CD 25 >2,400 U/mL and hemophagocytosis in BM, spleen, or lymph nodes [3,6] (Table 1). Our patient met seven out of the eight sub-criteria. A recent review article pooled several case series’ together and found that hepatic involvement was seen in 67% of patients with HLH. Another study demonstrated that almost one third of pediatric patients presenting with hepatic failure of unknown origin had features of HLH [2,3]. HLH should be considered in all patients with idiopathic hepatic failure3. It is crucial to start treatment with corticosteroids, cyclosporine A and etoposide6 once clinical suspicion HLH has been raised and prior to fulfilling the complete diagnostic criteria as early diagnosis and subsequent early treatment significantly reduce mortality [3]. Other findings supporting a diagnosis of HLH include neurological symptoms, cerebrospinal fluid pleocytosis, elevated D-dimer. Certain malignancies have association with HLH [7], most commonly seen in different types of T-cell lymphomas [3]. Studies have shown that it may occur close to 20% of the time in certain type of lymphomas. The association between HLH and B-cell lymphoma is more common in Asians [8], however among Caucasians is extremely rarely [9,10].
Table 1. Molecular diagnosis consistent with HLH OR?(B) Fulfillment of 5/8 criteria
Fever?Temperature > 38.5 C for > 7 days |
Splenomegaly?Spleen tip palpated > 3 cm below left costal margin |
Cytopenias involving 2 or more lines ?Hb < 9 g/dL, or Plt < 100 000/mL, or ANC <1000/mL |
Hypertriglyceridemia and/or hypofibrinogenemia?Fasting triglycerides > 2 mmol/L, or >3SD above age-adjusted ?normal; fibrinogen < 1.5 g/L, or < 3SD below age-adjusted normal |
Hemophagocytosis in bone marrow, spleen, or lymph nodes |
Low or absent NK-cell activity ?determined by 51-Cr release assay |
Ferritin > 500 mg/L (84% sensitivity) ?>10 000 (90% sensitivity, 96% specificity) |
Soluble CD25 (IL-2 receptor) > 2400 U/mL ?or >2SD above the age-adjusted mean |
The absence of hemophagocytosis from a bone marrow biopsy does not exclude the diagnosis of HLH. This case demonstrates that B-cell lymphoma should be investigated even after the diagnosis of HLH has been made.
Conclusion
gastroenterologist should carry a high index of suspicion for HLH in patient presenting with acute liver failure. This becomes especially important in patients presenting with fever, pancytopenia and idiopathic fulminant hepatic failure after ruling out other more common differential diagnosis.
References
- Tothova Z, Berliner N2 (2015) Hemophagocytic syndrome and critical illness: new insights into diagnosis and management. J Intensive Care Med 30: 401-412. [Crossref]
- Ramos-Casals M, Brito-Zeron P, Lopez-Guillermo A (2014) Adult haemophagocytic syndrome. Lancet 383: 1503-16.
- Weitzman S1 (2011) Approach to hemophagocytic syndromes. Hematology Am Soc Hematol Educ Program 2011: 178-183. [Crossref]
- Falini B, Pileri S, De Solas I, Martelli MF, Mason DY, et al. (1990) Peripheral T-cell lymphoma associated with hemophagocytic syndrome. Blood 75: 434-444. [Crossref]
- Miyahara M, Sano M, Shibata K, Matsuzaki M, Ibaraki K, et al. (2000) B-cell lymphoma-associated hemophagocytic syndrome: clinicopathological characteristics. Ann Hematol 79: 378-388. [Crossref]
- Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, et al. (2007) HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 48: 124-131. [Crossref]
- Machaczka M (2013) Hemophagocytic lymphohistiocytosis in adults. Ups J Med Sci 118: 201-203. [Crossref]
- Li F, Li P, Zhang R (2014) Identification of clinical features of lymphoma-associated hemophagocytic syndrome (LAHS): an analysis of 69 patients with hemophagocytic syndrome from a single-center in central region of China. Med Oncol 31: 902.
- Fung KM, Chakrabarty JH, Kern WF, Magharyous H, Gehrs BC, et al. (2012) Intravascular large B-cell lymphoma with hemophagocytic syndrome (Asian variant) in a Caucasian patient. Int J Clin Exp Pathol 5: 448-454. [Crossref]
- Shimazaki C, Inaba T, Shimura K, Okamoto A, Takahashi R, et al. (1999) B-cell lymphoma associated with haemophagocytic syndrome: a clinical, immunological and cytogenetic study. Br J Haematol 104: 672-679. [Crossref]