Hemophagocytic lymphohistiocytosis is a rare life-threatening syndrome caused by an excessive chaotic immune activation. It can be primary or sporadic and thus secondary to a trigger such as an auto-immune disease, an infectious process or a malignancy. It is usually associated with hematological malignancies and less frequently with solid tumors. This medical emergency has variable presentations lacking pathognomonic clinical and laboratory findings. The prognosis is usually dismal and is associated with high mortality rates. Here we report the case of a 45-year-old male patient previously healthy who presented with alteration of general status and high-grade fever. The work-up revealed a pancytopenia with biological and pathological findings confirming the diagnosis of hemophagocytic lymphohistiocytosis (HLH). After excluding the most common causes of HLH, the investigations concluded for a newly diagnosed gastric adenocarcinoma, initially presenting with bone marrow metastasis leading to an excessive immune dysfunction. There were no other associated metastatic sites as usually seen in generalized gastric cancers. The patient clinical condition deteriorated despite the urgent initiation of a standard treatment of HLH and passed away after multi-organ failure.
Hemophagocytic lymphohistiocytosis (HLH) and its related syndromes are life threatening medical diagnoses with serious outcomes secondary to excessive and inadequate immune activation [1]. Macrophage activation syndrome (MAS) shares many clinical similarities with HLH, thus many clinicians use the same diagnostic guidelines for the diagnosis [2]. However, the macrophage activation syndrome is commonly used to describe the HLH secondary to auto-immune disorders, especially juvenile rheumatoid arthritis [3]. It is a syndrome of excessive immune stimulation triggered by a severe infection, inflammation or malignancy that lead to a cytokine storm and a continuous macrophage activation with persistent phagocytosis [1].
Our case is about a 45-year-old male patient, previously healthy, referred to our center for worsening fatigue, alteration of general status and high grade fever. History goes back to 2 weeks prior to his presentation, when he started complaining of low grade fever and chills. Then, the fever became high grade and he started suffering from asthenia, anorexia, undocumented weight loss and diffuse bone pain. On the review of system, he didn’t report any respiratory, gastro-intestinal or urinary symptoms. Physical examination revealed an icteric patient, with 39.2 ͦC fever without any hemodynamic instability. There were no palpable lymphadenopathies, no hepato-splenomegaly, and no palpable mass in the abdomen. Laboratory investigations showed a pancytopenia, with grade IV neutropenia, grade II normocytic anemia and grade III thrombocytopenia. The peripheral smear didn’t reveal any abnormal immature cells, no blasts, no schistocytes, or evidence of leucoerythroblastic circulating cells. The fibrinogen was 90 mg/dL (normal value 150-400 mg/dL). The bilirubin was increased to 4.5 mg/dL (normal value < 1.2 mg/dL) with a grade III elevation of transaminases and a grade II elevation of cholestatic liver enzymes. The LDH was 10 times more than the upper limit of normal (ULN). The patient was admitted to the intensive care unit. We did pan-cultures, asked for a chest abdomen pelvis CT scan, and completed the laboratory work-up with bone marrow aspirate and biopsy, viral serologies, lipid panel, auto-immune profile, erythrocyte sedimentation rate (ESR) and ferritin. Our differential diagnoses were a bone marrow failure either due to a primary bone marrow malignant infiltrative process (leukemia or lymphoma), bone marrow necrosis, hemophagocytic lymphohistiocytosis or secondary bone marrow aplasia. The chest abdomen pelvis CT scan described a gastric wall thickening at the level of the fundus with borderline splenomegaly, but there were no other suspicious bone, lung, pancreatic, kidney or liver lesions. There was no dilatation of intra or extra-hepatic biliary tree. The ferritin was 12598 ng/mL (normal value < 200 ng/mL) and there was a hypertriglyceridemia of 750 mg/dL (normal value < 150 mg/dL). The viral serologies including hepatitis viruses, herpes viridae panels, human immunodeficiency virus (HIV), as well as the salmonella and brucella titers were all negative. Additionally, the auto-immune work-up was also negative. The bone marrow aspirate revealed a hypo-cellular bone marrow, with large non hematopoietic cells grouped in islets and a fulminant picture of hemophagocytosis: “macrophages ingesting the host cells’’. There were no blasts, no plasma cells, nor atypical lymphocytes. The bone marrow biopsy confirmed the diagnosis of macrophage activation syndrome with the co-existence of carcinoma cells (Figure 1). The immuno-histochemistry was in favor of adenocarcinoma cells compatible with a gastric cancer primary. We did an FDG-PET CT scan showing diffuse heterogenic bone avidity, without lytic or sclerotic lesions and a hypermetabolic gastric fundus thickening associated with hypermetabolic perigastric infracentimetric lymphadenopathies not reported on the CT scan. The gastroscopy showed an erythematous mucosa with a 2 cm ulcerated lesion at the greater curvature of the stomach. Biopsies were taken, and the pathology confirmed the same adenocarcinoma cells seen in the bone marrow. We stabilized the patient with platelet and packed red blood cells (PRBCs) transfusions. The carcino-embryonic antigen (CEA) was elevated at 36.8 ng/mL (normal < 5 ng/mL). The rest of the tumor markers CA19.9 and CA125 were within normal limits. He was started on high dose dexamethasone of 10 mg/m2 intravenous (iv) daily and he received Etoposide 150 mg/m2 iv on day 1 and day 4. Unfortunately, the liver enzymes kept worsening, and the ferritin kept rising. The patient developed multi-organ failure with acute renal failure, disseminated intravascular coagulopathy and acute lung injury. He was intubated and 24 hours later he had cardiac arrest, not successfully resuscitated.
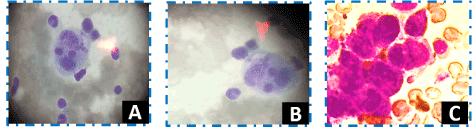
Figure 1. Hematoxylin and eosin (H/E) staining (x100): Bone marrow examination revealing the hemophagocytic lymphohistiocytosis picture with the macrophages ingesting the host cells (A and B): erythrophagocytosis. The trigger of the HLH is the metastatic carcinoma cells in the bone marrow (C)
This case describes a hemophagocytic lymphohistiocytosis occurring as an unusual initial presentation of gastric adenocarcinoma, with perigastric lymph nodes and exclusive bone marrow metastasis. The disease outcome was quite poor as the patient presented with this excessive immune dysfunction triggered by the adenocarcinoma cells massively infiltrating the bone marrow.
HLH is not a malignant disorder. It is a severe inflammatory syndrome associated with a major immune dysfunction defined by the absence of down-regulation of abnormally activated lymphocytes leading to tissue destruction by phagocytosis. The tissue damage will result in further cytokine release that keeps the vicious circle of macrophage activation and paralyzes the cytotoxic T cells and NK cells. There is a high level of interferon gamma (IFN ϒ), tumor necrosis factor alpha (TNF α), soluble receptor of Interleukin (IL) 2 or CD25, IL-6 and IL-10. The pathophysiology is resumed in the Figure 2 [4-6].
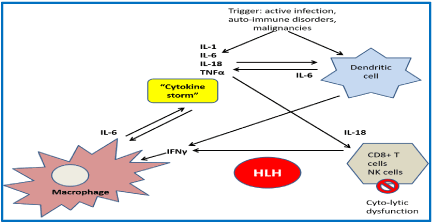
Figure 2. Pathophysiology of hemophagocytic lymphohistiocytosis. IL: interleukin; NK: natural killer; TNFα: tumor necrosis factor alpha; IFNϒ: interferon gamma; HLH: hemophagocytic lymphohistiocytosis
Primary HLH is mainly caused by familial disorders and other immunodeficiencies. Controversially, secondary HLH are mainly related to infectious etiologies, malignancies and auto-immune disorders. Approximately 50% of adult HLH are malignancy-associated HLH as shown by a large study including 2,197 adults. Most of the HLH cases were triggered by hematological neoplasms (93.7%) and rarely solid tumors and non-specified neoplasms account for 3.1% and 3.2%, respectively. As seen in the literature, solid malignancies associated with HLH are rare and mainly reported in case series. Solid tumors may play the role of trigger to HLH, such as colon carcinoma, gastric cancer, small cell lung carcinoma and others. They are usually highly aggressive associated with a very poor prognosis [7-9].
Patients present with an acute onset severe illness mimicking a clinical picture of severe sepsis. They usually suffer from a rapid clinical deterioration. The clinical and biological findings are the following: high grade fever, chills, hepatomegaly, splenomegaly, diffuse lymphadenopathies, pancytopenia (secondary to hemophagocytosis and bone marrow failure due to the macrophages engulfing the three lineages), high ferritin (secondary to liver injury and erythrophagocytosis), hypertriglyceridemia (secondary to inhibition of lipoprotein lipase by IL1 and TNF α), low fibrinogen and disturbed liver function tests. To evaluate the pancytopenia, bone marrow examination is mandatory, not only to exclude lymphoproliferative disorders and leukemia, but also to confirm the infiltration by hemophagocytes that is pathognomonic of HLH. There is an imminent, active phagocytosis of the hematopoietic cells in the bone marrow and a tissue destruction by a similar mechanism in the liver, lymph nodes and the spleen [5,6,10]. So, the diagnosis depends on both clinical and pathological investigations. These define the HLH-2004 criteria, and five of the eight criteria reported in the Figure 3 are required to make the diagnosis of HLH [11].

Figure 3. The eight diagnostic criteria according to HLH-2004 trial: 5 criteria out of 8, must be at least present to make the diagnosis of HLH [11].
HLH is usually of poor prognosis, and treatment must be started once the diagnosis is established, because an earlier initiation of the induction therapy is associated with a better outcome. In clinical practice, we rely on the HLH-94 based treatment protocol to treat the patients. In the cases of HLH related to solid tumors as a trigger of the uncontrolled immune dysregulation, the patients may be severely ill and they won’t be able to receive the adequate anti-neoplastic cytotoxic treatment for their malignancies. The management of secondary sporadic, non-primary/hereditary HLH is resumed in the Figure 4 [12].
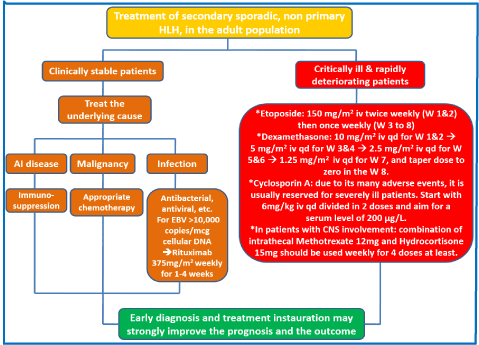
Figure 4. Algorithm describing the management of the secondary hemophagocytic lymphohistiocytosis in the adult population. HLH: hemophagocytic lymphohistiocytosis; AI: auto-immune; mcg: micrograms; iv: intravenous; W: week; qd: daily
Besides the early instauration of the treatment, the outcome also depends on the availability of an adequate supportive care that consists of transfusion management, broad spectrum antibiotics as well as appropriate prophylactic anti-viral and antifungal therapies. The response to initial induction treatment is a major determinant prognostic factor, and mainly depends on clinical and biological assessment: the evolution of the ferritin and other cytokines which are disease specific markers. HLH is usually fatal and associated with 50 % mortality despite the administration of the HLH-94-based treatment [13].
Hemophagocytic lymphohistiocytosis is an aggressive syndrome characterized by a multi-organ failure secondary to a widespread uncontrolled inflammation. Clinicians must be aware of this rare entity. Due to its irregular clinical presentation, it is often diagnosed later in a patient's clinical course, leading to poor survival outcomes. Nonetheless, it may be the initial presentation of the main triggering disorder and thus the prognosis will be worse. The treatment consists of stopping the inflammatory cascade via the HLH treatment protocol followed by the appropriate treatment of the underlying trigger.
The authors declare no conflicts of interests.
- EI Hachem G (2017) Hemophagocytic Lymphohistiocytosis, A Syndrome of Excessive Immune Activation: Review of the Literature. J Hematol Mult Myeloma 1: 1002.
- Henter JI, Elinder G, Ost A (1991) Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL Study Group of the Histiocyte Society. Semin Oncol 18: 29-33.
- Janka GE (2007) Hemophagocytic syndromes. Blood Rev 21: 245-253. [Crossref]
- Filipovich A, McClain K, Grom A (2010) Histiocytic disorders: recent insights into pathophysiology and practical guidelines. Biol Blood Marrow Transplant 16: S82.
- Pachlopnik Schmid J, Côte M, Ménager MM, Burgess A, Nehme N, et al. (2010) Inherited defects in lymphocyte cytotoxic activity. Immunol Rev 235: 10-23. [Crossref]
- Risma K, Jordan MB (2012) Hemophagocytic lymphohistiocytosis: updates and evolving concepts. Curr Opin Pediatr 24: 9-15. [Crossref]
- Michot JM, Hié M, Galicier L, Lambotte O, Michel M, et al. (2013) [Hemophagocytic lymphohistiocytosis]. Rev Med Interne 34: 85-93. [Crossref]
- Ramos-Casals M, Brito-Zerón P, López-Guillermo A, Khamashta MA, Bosch X (2014) Adult haemophagocytic syndrome. Lancet 383: 1503-1516. [Crossref]
- Machaczka M, Vaktnäs J, Klimkowska M, Hägglund H (2011) Malignancy associated hemophagocytic lymphohistiocytosis in adults: a retrospective population based analysis from a single center. Leuk Lymphoma 52: 613-619. [Crossref]
- Gars E, Purington N, Scott G, Chisholm K, Gratzinger D, et al. (2018) Bone marrow histomorphologic criteria can accurately diagnose hemophagocytic lymphohistiocytosis. Haematologica. Epub ahead of print.
- Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, et al. (2007) HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 48: 124-131. [Crossref]
- Henter JI, Aricò M, Egeler RM, Elinder G, Favara BE, et al. (1997) HLH-94: A treatment protocol for hemophagocytic lymphohistiocytosis. HLH study Group of the Histiocyte Society. Med Pediatr Oncol 28: 342. [Crossref]
- Rivière S, Galicier L, Coppo P, Marzac C, Aumont C, et al. (2014) Reactive hemophagocytic syndrome in adults: a retrospective analysis of 162 patients. Am J Med 127: 1118-1125. [Crossref]
2021 Copyright OAT. All rights reserv