Marked improvement in the accuracy and sensitivity of current diagnostic techniques for cardiac diseases have contributed to the identification of haematologic and oncologic diseases as a possible aetiology of cardiomyopathy (CM). If undiagnosed, the CM may progress to HF and eventually death. This type of CM is exceedingly rare and a precise understanding of its natural cause, pathophysiology, diagnosis and management is undermined by the lack of supporting clinical evidence. Cancer as a direct cause of CM remains controversial. Differentiating cancer-associated CM from chemotherapy-induced CM is difficult since diagnosis of CM is nearly always made after the initiation chemotherapy. Hematologic diseases on the other hand have been classified under restrictive cardiomyopathy (RCM) or dilated cardiomyopathy (DCM) yet they are a distinct clinical entity uniquely characterized by cardiac chamber (atrial and/or ventricular) dilation, diastolic dysfunction and preserved systolic function. Oncologic or haematologic CM has an ominous prognosis although diagnosis is incidental or delayed making management difficult. There is a need to better understand the role of oncologic and/or haematologic diseases in the pathogenesis of CM aimed to improve diagnosis and development of aetiology-specific therapy. Similarly, this review provides an overview of CM in oncology and haematology patients including pathophysiology, presentation, diagnosis and treatment.
hematologic cardiomyopathy, neoplastic cardiomyopathy, leukaemia, myeloma, sickle-cell anaemia, henoch-schonlein purpura, neoplasm
Heart diseases and cancer are responsible for almost half of the deaths in developed countries [1]. The intersection between cancer and heart diseases has been primarily about advances made in anti-cancer therapy that has not only increased survival but has also increased both the risk and burden of cardiovascular (CV) complications. The increased risk has undermined the efficacy of the current validated long-term oncology (anti-cancer) therapy [2]. Hematologic malignancies are among the major forms of cancer that have increased the risk of cardiac diseases. As early as the late 1970s, several studies implicated long-standing oncology therapy to the development of cardiomyopathies (CMs) in cancer survivors [3,4]. Recently, both the European Society of Cardiology (ESC) and the American Heart Association (AHA) scientific statements on the classification of CMs have also recognized oncology treatment as one of the rare causes of therapy-related CMs in clinical settings [5-8].
Studies on haematology and oncology patients report also suggest CMs in cancer survivors may be the direct consequence of the cancer itself. A systematic evaluation of older adults with cancer found they have an increased risk of thromboembolic events (heart attack) months prior to cancer diagnosis [9]. In a more specific evidence, an echocardiographic study among patients with acute leukaemia (AL) and lymphoma reported nearly half had clinical evidence of left ventricular (LV) dysfunction prior to the initiation of chemotherapy later diagnosed as non-infiltrative CM unrelated to chemotherapy. Another speckle tracking echocardiographic (STE) study on AL patients reported a significant decrease in LV global longitudinal strain (GLS) compared to age-matched breast cancer and non-cancer controls suggesting AL itself may be involved in the pathological changes in the myocardium [11]. The ESC 2007 position statement on classification of CMs lists carcinoid heart disease and metastatic cancers as one of the causes of an acquired form of restrictive cardiomyopathy (RCM) [8].
Despite accumulating evidence of hematologic and oncologic involvement in CM, they remain an under-appreciated aetiology of CM both in academia and in clinical practice possibly due to the difficulty in differentiating CMs due to cancer itself and due to aggressive chemotherapy. In most cases, diagnosis of CM in cancer survivors is during or after aggressive chemotherapy, which obscures the involvement of cancer itself in the pathogenesis of CMs. Nevertheless, increased recognition of cancer-related CM is clinically relevant because the disease has an unfavourable prognosis and the disease does not appear to be reversible like that due to chemotherapy but patients surviving longer than nine months may show some improvement in LV function [10]. Increased recognition may also help to improve diagnosis and management of CM in cancer patients. Cardio-oncology clinicians need to appreciate CMs in cancer patients are not only due to chemotherapy but also may be due to the cancer itself. They need to identify patients at risk of cancer-related CMs and give them priority to lessen CV risks including their deleterious effect on the heart. Similarly, the present paper provides a focused overview of CMs in the setting of hematologic and oncologic disorders with an emphasis on its epidemiology, pathological features, diagnosis and clinical management.
Haematologic cardiomyopathy
Haematology is the sub-speciality of internal medicine concerned with the study of the cause, prognosis, treatment and prevention of diseases related to blood. Thus, haematologic disorders are pathological conditions primarily affecting the blood and blood-producing organs, which include but not limited to various types of anaemia, blood cancers and haemorrhagic conditions [12,13]. Blood is a vital medium consisting of plasma, blood cells and platelets. Platelets are responsible for clotting, white blood cells (WBC) for inflammation and red blood cells (RBC) for carrying oxygen and nutrients to all tissues of the body and waste products from the organs. Any abnormality of these blood components may result in haematologic disorders. Whereas disorders involving platelets and coagulation resulting in thrombosis and/or excessive haemorrhaging remain the primary concern for most cardiologists, disorders involving RBCs and platelets can also affect blood flow and viscosity to vital organs including the heart with attendant deleterious effect [12,14]. Common haematologic disorders implicated as a cause of dilated cardiomyopathy (DCM) include oncological disorders such as leukaemia and multiple myeloma, and non-oncological disorders such as sickle-cell anaemia, anaemia, and Henoch-Schonlein Purpura.
Leukaemia: Types and prevalence of leukaemia by definition, leukaemia is “a progressive malignant disease of the blood-forming organs characterized by distorted proliferation and development of leukocyte and their precursors in the blood and bone marrow” [15]. Leukaemia can be categorised based on disease onset (acute or chronic) or on the type of leukocyte affected (lymphoblast, myeloblasts, lymphocytes or myelocytes) [16,17]. The acute type results from the proliferation of immature WBCs or blasts whereas the chronic types results from the proliferation of mature cells [16]. Table 1 provides the basic classification criteria of leukaemia.
Table 1. Classification of leukaemia
Onset/Progression |
Clinical Name |
Leukocyte Affected |
Acute leukaemia (due to proliferation of immature blast cells) |
Acute lymphoblastic leukaemia |
Lymphoblasts |
Acute myeloid leukaemia |
Myeloblasts |
Chronic leukaemia (due to proliferation of mature cells) |
Chronic lymphocytic leukaemia |
Lymphocytes |
Chronic myeloid leukaemia |
Myelocytes (neutrophils, eosinophils, basophils) |
*Large granular lymphocytic leukaemia, hairy cell leukaemia |
|
*Less common chronic leukaemia
The most prevalent acute types are acute lymphoblastic leukaemia (ALL) and acute myeloid leukaemia (AML) [17]. In the U.K., there are about 8,000 new diagnoses of leukaemia each year, and in 2010, the reported leukaemia-associated deaths stood at 4,504 [16]. ALL has an overall incidence of 1 to 1.5 per 100,000 persons and is particularly common in children aged between 4 and 5 years with an incidence of 4 to 5 per 100,000 persons, and in adults aged 50 years, the incidence is 2 per 100,000 persons. ALL accounts for 80% of acute leukaemia in children and 20% in adults [18]. AML is the most common type of leukaemia in the U.S., with the number of cases ranging from 3.8 to 17.9 per 100,000 persons with a male to female ratio of 3:2 [19]. The cause of ALL is largely idiopathic but there are suggestions of a possible genetic predisposition and certain viral infections (human T-cell lymphotropic virus type 1, Epstein-Barr virus, and Human Immunodeficiency Virus) [18]. On the other hand, AML is associated with the exposure to ionising radiation, benzene (from chronic exposure to cigarette smoking) and cytotoxic chemotherapy [17].
Evidence of leukaemic CM: The direct effect of leukaemia on the cardiac structure and function has not been well described, and at most, remains an academic interest. At present, chemotherapy-induced CM has attracted the attention of oncology although recognizing a direct involvement of leukaemia in the pathogenesis of CM may improve cardio-oncological management of these patients beyond the current management strategies targeting dose-adjustment or alternative chemotherapeutic drugs with lower cardiotoxic effects. Despite the lack of evidence of leukaemia-associated CM, there are increasing reports of CM and HF in patients diagnosed with leukaemia prior to the initiation of chemotherapy. Two clinical and echocardiographic studies [10,11] and one population-based matched cohort study [20], as well as a handful of case reports [21-23] have suggested a direct involvement of leukaemia itself in myocardial dysfunction not related to the cytotoxic effect of chemotherapeutic drugs.
The first clinical and echocardiographic study performed by Mir [10] in 1978 described association between AL or lymphoma and non-infiltrative CM. The basis of the study was previous observations of HF in young leukaemic patients who had not received cytotoxic drugs and had no history of heart disease. The study enrolled 38 patients with AL and lymphoma, and 17 healthy controls. Nearly half of the patients (47%) had echocardiographic evidence of LV dysfunction before chemotherapy demonstrated by significantly increased (p < 0.05) LV systolic dimensions and decreased fractional shortening. Twelve (12) patients developed HF, seven of them prior to daunorubicin therapy while five after 3 to 24 weeks of therapy. Necropsy performed in four patients found leukaemic infiltration of the myocardium. In the first six months, sequential echocardiograms revealed deteriorating LV function but which steadily improved in patients living longer than nine months. The study concluded that 47% of AL and lymphoma patients had non-infiltrative CM not related to exposure to chemotherapeutic drugs and a significant proportion of these patients went on to develop HF. This leukaemic CM appears irreversible but patients living longer than nine months may show improvement in LV function.
The second and more recent echocardiographic study by Assuncao et al. (2017) [11] evaluated whether AL is associated with LV dysfunction prior to chemotherapy. The basis of the study were reports that AL patients developed higher rates of HF than patients with other cancer types, suggesting it may be a consequence of high cytokine release or direct leukaemic cell infiltration of the heart. The study enrolled 76 AL patients and 76 non-cancer patients with no known CM or valvular heart disease matched for age, gender, hypertension and the presence of diabetes. Twenty-eight (28) patients from each group were subsequently matched with breast cancer patients (non-haematological malignancy). Prior to chemotherapy, LV function was preserved in both AL and controls with no significant differences (63±5% vs. 62±6%; p=0.34). However, patients with AL had significantly higher LV mass and significantly lower GLS (-19±3% vs -21±2%, p<0.001) compared to controls. AL patients also had significantly lower GLS than breast cancer patients (-19.4 ±3.0% vs -21.2 ±2.2%; p=0.023). There was no difference in GLS between breast cancer and non-cancer patients. Risk factors for impaired GLS in AL patients were body mass index, LVEF and absolute lymphocyte count. The findings suggest a direct effect of leukaemia may result in myocardial dysfunction and subsequently CM.
The third population-based study by Chellapandian et al. [20] in 2019 aimed to describe the incidence and risk factors of HF in 2,053 children (≤18 years) diagnosed with ALL and AML. At ten years, the cumulative incidence of HF was higher in AML (7.5%) than in AML (1.7%). Risk factors for the development of HF in ALL children were female gender, age at diagnosis < 1 year, irradiation and cumulative anthracycline dose ≥250 mg/m2. In AML children, irradiation was the only risk factor. Of 23 patients with HF during active therapy, only one developed HF after treatment completion. The study suggests that in AL patients, risk factors other than irradiation (chemotherapy) may result in cardiac dysfunction and HF. However, additional clinical trials examining children with ALL is warranted to clarify the pathogenic role of cancer itself in the development of CM in paediatric patients.
Case reports have also suggested the involvement of leukaemia itself in the development of myocardial dysfunction [21-23]. The first case report is that of myelodysplastic syndrome with extensive myocardial infiltration by haemopoietic precursors associated with myocardial necrosis in the LV and interventricular septum [21]. Cardiac dysfunction was the result of RCM due to leukaemic infiltration, concomitant anaemia, cardiac dilatation and conduction blocks and myocardial necrosis. Marked rapid reduction in LVEF (66% to 33%) suggests the role of ischemia since leukaemic infiltration is not expected to cause this degree of systolic dysfunction over a 24-hour period. Differential diagnosis did not consider haemopoietic cell infiltration, which may have potentially contributed to death [21]. Madhavan et al. [22] reported a case of a 53-year old male diagnosed with plasma cell leukaemia who developed RCM due to plasma cell infiltration in the myocardium. After a high dose of chemotherapy, the patient became asymptomatic and echocardiographic evidence of significant improvement and almost normal physiology supporting the diagnosis that plasma cell infiltration caused RCM [22]. Van Haelst et al. [22] reported two cases of AML as a cause of ischemic heart disease.
Despite some evidence suggesting leukaemic CM, it is a rare clinical entity such that its pathogenesis, diagnosis and management remain unknown. However, three case reports [21-23] have highlighted three possible pathogenic mechanisms contributing to cardiac dysfunction in leukaemic patients. (i) The presence of leukaemic thrombi or leucostasis in major arteries; (ii) leukaemic infiltration into the myocardium or pericardium,; (iii) disorders of coagulation secondary to leukaemia causing emboli due to a hypercoaguable state or haemorrhage; and (iv) anti-leukaemic therapies [21-23]. These case reports also suggest the importance of including differential diagnosis of leukaemic myocardial infiltration (CM) in patients diagnosed with AML, myelodysplasia who exhibit signs and symptoms of cardiac dysfunction.
Multiple myeloma: Overview and prevalence multiple myeloma (MM) is a plasma cell malignant abnormality characterized by uncontrolled clonal plasma cell proliferation in the bone marrow, production of monoclonal protein in the blood and/or urine and associated organ dysfunction [24,25]. It is the second most prevalent haematological malignant abnormality with an annual age-adjusted incidence of 6.3 per 100,000 persons in the U.S. [24] and accounts for approximately 10% of all haematological cancers [26-28]. Anaemia is a common complication observed in about two-thirds of MM patients [29]. A triad of chronic kidney disease, HF and anaemia has been described in clinical practice [29,30]. The risk of CV and thrombotic events is an import consideration during treatment of MM patients because CV risk factors and CVD are prevalent in the MM patient population, and CV and thrombotic complications are associated with MM itself. MM typically affects older individuals with median age of diagnosis at about 70 years. In a retrospective analysis of 32,193 newly diagnosed or relapsed cases of MM, about two thirds had CV disease at baseline or during follow up – ischemic heart disease (IHD), HF, CM, arrhythmias and conduction disorders [31]. In a related analysis of 3,107 newly diagnosed MM cases in the U.K. between 1980 and 2002, myocardial infarction or cerebrovascular accident accounted for 8% of deaths within 60 days of trial entry [32]. With improved survival over the past decade due to the introduction of novel therapies such as immunomodulatory drugs and proteasome inhibitors, there is the need to recognize MM-associated CV to improve treatment efficacy as well as survival in MM patients [25].
Pathophysiology: Physiologically, MM manifests with restrictive cardiac dysfunction, conduction abnormalities and infiltrative CM. The exact pathophysiology of MM-associated CM remains unclear, but the association of MM with CV complications may provide valuable insights. In MM patients, rapidly proliferating malignant B-cells secrete large quantities of immunoglobulins or immunoglobulin fragments into the bloodstream, which may accumulate in end organs including the heart, liver and kidneys [33]. The accumulation of amyloid light chain immunoglobulin is believed to lead to clinical amyloidosis in about 12% to 15% of MM patients during the natural course of the disease and up to 30% of those with MM have subclinical amyloid deposits [33]. Myocardial involvement is estimated to be present in half of the amyloid light chain amyloidosis cases, which may subsequently result into restrictive physiology and CM [30,34]. These findings support the observation made in several case reports of the relationship between MM and amyloidosis in the development of CM [29,30]. MM has also been associated with a specific set of clinical manifestations often referred to as CRAB (elevated Calcium levels, Renal insufficiency, Anaemia and Bone lesions), which may increase the risk of CV comorbidities [35]. For instance, hypercalcaemia is associated with the development of arrhythmias, renal insufficiency with the development of uraemic CM, while anaemia may contribute to arrhythmias, CM, and high-output HF [25,36].
Manifestation/Treatment: Clinical manifestation and diagnostic features vary widely across case reports. Typical signs and symptoms of MM-induced CM at presentation include a history (3-months) of progressive exertional dyspnoea and orthopnoea, marked weight loss and lower extremity oedema [28,30]. On clinical examination, MM patients with CM may exhibit bilateral crackles on lung auscultation, bilateral lower extremity oedema and jugular venous distention [30]. Initial bone marrow biopsy may reveals 40-60% of plasma cell infiltration with Congo-red stain positive for amyloid light chains [28]. Some patients may exhibit elevated serum level of BNP possibly due to elevated ventricular filling pressure resulting from direct damage to cardiomyocytes caused by the accumulation of large quantities of immunoglobulin [33]. ECG is non-specific, often revealing a normal sinus rhythm, no sign of LV hypertrophy and low voltage complexes while chest x-ray may reveal cardiomegaly and pulmonary oedema [30]. On echocardiogram, depressed or mildly depressed LVEF (35-40%) with restrictive LV filling pattern and a sparkling appearance of the myocardium is a common observation [28]. Cardiac MRI may reveal bi-ventricular hypertrophy with diffuse sub-endocardial late gadolinium enhancement (LGE) and thickening of the intra-atrial septum, consistent with myocardial amyloidosis [28,30].
Due to the possible association of MM and amyloidosis in the pathogenesis of CM, cardiac screening in all patients with MM should include ECG and complete cardiac sonography. Conversely, in patients with cardiac amyloidosis, diagnosis for MM should be sought because of its ominous prognosis. Despite the lack of a single non-invasive test that can accurately diagnose MM and cardiac amyloidosis, a combination of HF signs and symptoms, sonography findings and low-voltage complexes on ECG are highly suggestive of CM secondary to MM. There is no specific treatment for CM due to MM. However, current strategies target the underlying disease and cardiac dysfunction. Case series suggest treating MM itself and for patients who have developed signs and symptoms of HF, the conventional HF mediations may be prescribed according to the current HF guidelines. Clinical trials specifically enrolling patients with CM secondary to MM should help us to understand the pathogenesis and the natural cause of the disease, which may in turn help to improve diagnosis and management.
Anaemia: Anaemia is one of the hematologic disorders of a non-oncological origin that is capable of causing several CV diseases including CM. Anaemia is a common condition in which the number of RBCs (and consequently their oxygen-carrying capacity) is insufficient to meet the body’s physiological needs. According to World Health Organization (WHO), one third of the global population is anaemic due to nutritional imbalance [37]. Anaemia has been classified based on various RBCs defects: (i) aplastic – production defect; (ii) megalobastic – maturation defect; (iii) iron deficiency – defects in haemoglobin synthesis; (iv) thalassaemia – defects of haemoglobin maturation; and (v) haemoglobinopathies, sickle cell anaemia and thalassaemia due to synthesis of abnormal haemoglobin [38,39]. Anaemia (defined as haemoglobin <13 g/dL in men and <12 g/dL in women or serum ferritin level <12 ng/mL) is prevalent in HF patients, occurring in approximately 30% of stable HF and 50% of hospitalized HF patients compared to < 10% in the general population [40]. Anaemia is frequently associated with chronic diseases and is an independent risk factor for the development of CV complications. One (1) g/dL decrease in haemoglobin level is an independent risk factor for cardiac morbidity and mortality in anaemic patients [41,42].
Pathophysiology: The general relationship between anaemia and HF is well-recognized, nearly always associated with high output HF syndrome. In CM patients, the most common type of anaemia implicated in the pathogenesis is iron-deficiency anaemia caused by defects in haemoglobin synthesis [43-45]. Initially, in patients with severe anaemia (haemoglobin 4-6 g/dL) and normal LV function [46,47], reduced oxygen-carrying capacity induces haemodynamic and non-haemodynamic compensatory mechanisms [48]. Low numbers of circulating RBCs reduces systemic vascular resistance by decreasing whole-blood viscosity and associated tissue hypoxia, while low-haemoglobin increases nitric oxide (NO) mediated vasodilation [47,49]. Reduced arterial blood pressure induces baroreceptor-mediated neurohormonal activation (increased sympathetic and renin angiotensin activity) leading to decreased renal blood flow and glomerular filtration rate, resulting in increased renal retention of salt and water, and the expansion of extracellular and plasma volumes [40]. These mechanisms results in high output HF and the correction of anaemia results in a rapid and complete reversal of high output HF [46].
Prolonged and severe anaemia results in cardiac dilatation and ventricular hypertrophy. It leads to the development of CV decompensation after a period of high output HF [13]. If anaemia is not corrected, the myocardium is unable to withstand the high workload leading to contraction and fatigue, myocardial remodelling and expansion of ventricles, normal or thin wall thickness, and eventually HF [50]. Induced iron deficiency anaemia in mice revealed increased cardiac output and sympathetic activation resulting in LV hypertrophy [51]. Anaemia may also induce hypoxia-linked inhibition of the mitochondrial respiratory chain or potassium channels leading to intracellular calcium accumulation, which in turn may lead to cardiomyocyte dysfunction. Iron is also an essential element in the synthesis of collagen – an import component of the CV system responsible for supporting and sustaining the hardness of the vascular wall. In iron deficiency anaemia, the content of collagen in the cardiac tissue is decreased, resulting in decreased elasticity of the myocardium and vascular wall as well as marked changes in the normal pressure/volume relationship [52,53].
Diagnosis: Clinical signs and symptoms of anaemia-associated CM are non-specific but patients may often experience fatigue, dizziness, headache, palpitations, shortness of breath and increased heart rate [13,14]. Prolonged anaemia may lead to the development of hypertrophy and cardiac chamber enlargement, eventually leading to HF [54]. Various auxiliary examinations (mainly ECG and echocardiography) are commonly used to detect changes in the anaemic myocardium – ECG abnormalities include prolonged P-wave duration and dispersion, T-wave and tachycardia [55,56], and significant decrease in QRS duration and increased QTc (especially in pregnant women with iron deficiency anaemia) [57]. Various echocardiographic modalities can also detect myocardial changes associated with prolonged anaemia. Two-dimensional (2D) echocardiography reveals significantly elevated LV end-diastolic and end-systolic dimensions, LV posterior wall thickness, left atrial volumes index and LV mass index with normal or mildly elevated LVEF [58]. Doppler echocardiography reveals significantly increased E/A ratio, transmitral Doppler early filling velocity to tissue Doppler early diastolic mitral annular velocity ratio (E/E’ ratio), stroke volume, and cardiac index but with no significant differences in deceleration time (DT) [58]. Speckle tracking echocardiography (STE) reveals impairments in the global longitudinal strain (GLS), global area strain, global radial strain (GRS) and global circumferential strain (GCS), suggesting LV remodelling and LV systolic dysfunction in patients with haemoglobin levels 6-9 g/dL [59]. In a 2D-STE study that evaluated patients with iron deficiency anaemia, LA functional dimensions – global peak atrial longitudinal strain, and strain rate of the systolic LV and the early and late diastolic LV strain rate curves of the LA show better potential for accurate assessment of LA dysfunction [60]. Recently, myocardial perfusion single-photon emission computed tomography (SPECT) reveal an inverse association between haemoglobin levels and myocardial washout rate of thalium-201 (TI-201) in patients with normal myocardial perfusion [61]. Anaemic patients may have significantly increased levels of troponin I secondary to upper gastrointestinal (GI) bleeding in the early course of myocardial damage [62].
Treatment: Cardiomyopathy secondary to anaemia (especially CM resulting from iron deficiency) may be completely reversible prior to the development of HF [63]. Chest ECG and radiographic images of a patient with chronic anaemia due to long-term bloodletting with cardiac hypertrophy reveal three months of iron supplementation therapy caused marked improvement in cardiac hypertrophy [64]. Low haemoglobin has been associated with enlarged cardiac chambers, increased LV mass and higher LV filling pressure [58]. Appropriate correction of the cause of anaemic results in a decrease in LV mass, LA volume and E/E’. Furthermore, ferric carboxyl-maltose may alleviate iron deficiency in patients for whom oral iron supplementation is ineffective [65]. A systematic review of 13 small uncontrolled or randomized placebo-controlled studies published between 2000 and 2010 associate treatment by erythropoietin-stimulating agents (ESA) to an increase in haemoglobin levels accompanied by symptomatic improvement [40].
Among anaemic patients who have developed HF, it is important to consider whether treatment of anaemia might improve outcomes. Unfortunately, few options are available to increase haemoglobin in anaemic HF patients. Packed RBC transfusion as a short-term therapy has many risks and provide only temporary benefit [40]. In a large public discharge database, about 600,000 patients admitted for HF, 27% had anaemia. Untreated anaemia was associated with approximately 10% increase in adjusted risk of mortality and approximately 70% higher in patients who had received transfusions [66]. However, the study did not consider confounders such as the severity and reason for transfusion, which may require large prospective RCTs to clarify the harmful effects of transfusion on anaemic patients with HF. The Transfusion Requirements in Cardiac Surgery (TRICS) III trial also reported RBC transfusion in HF patients with anaemia is not necessarily beneficial and may even be associated with poor outcomes [67]. Thus, routine blood transfusion in asymptomatic patients with non-acute anaemia is not always recommended [68]. Careful consideration of clinical and individual patient factors such as age, comorbidities and the need for surgical intervention is advisable when determining clinical indications for transfusion in HF patients.
Sickle cell anaemia: Sickle cell anaemia (SCA) refers to a group of inherited hematologic disorders caused by a point mutation in the beta-subunit of haemoglobin. The abnormal haemoglobin polymerizes after releasing oxygen to the tissues to produce long filaments that contort the erythrocyte into a characteristic sickle-cell conformation [69-71]. Over the past few decades, improvement in the medical care of SCA patients have led to a significant decrease in childhood mortality, especially in developed countries [72]. With improved survival into adulthood, the cumulative burden of acute and chronic organ damage has become an important determinant of quality of life, morbidity and life-expectancy, which appear not to have improved in the last 15 years while adult SCA-related mortality appear to have increased [73,74]. Cardiopulmonary complications including heart chamber dilation, diastolic dysfunction and elevated pulmonary arterial pressure (PAPs) are common findings in SCA patients although the mechanisms and pathophysiology that unify these factors to the cardiac phenotype of SCA and its underlying unexplained complications such as arrhythmias and sudden death are not well-recognized [69,75,76]. Sudden unexplained death has been reported in 25% to 30% of SCA patients associated with cardiopulmonary complications [77,78].
Pathophysiology: Pathophysiologic mechanisms involved in SCA-CM is multi-factorial and mainly includes anaemia-related hyperdynamic features and restrictive physiology [76]. Hyperdynamic features appear at the onset of myocardial dysfunction resulting from various adaptive CV mechanisms associated with anaemia. Altered blood viscosity, tissue hypoxia, and increased sympathetic tone appear to drive hyper-dynamic features [79]. Prolonged anaemia leads to arteriolar dilation and reduced afterload. Reduced venous tone increases preload, and together with increased sympathetic tone, results in increased stroke volume and cardiac output and eventually a state of volume overload [80-82]. Overtime, volume overload and increased cardiac load leads to cardiac enlargement and LVH [83]. Typical features of volume overload in SCA include increased stroke volume, cardiac output, LV end-diastolic dimensions and eccentric LVH [69]. LV dilatation in SCA has increased or preserved LVEF and increased stroke volume, which differs from LV dilatation of failing ventricles typical in DCM that is associated with LV systolic dysfunction [70]. Although hyperdynamic features associated with anaemia are prevalent in SCA patients, it is difficult to distinguish the effect of anaemia itself from that of pathological processes due to SCA including vaso-occlusion and inflammation in the heart. The involvement of anaemic-related hyperdynamic features to other pathologic cardiac features such as diastolic dysfunction, pulmonary hypertension, and elevated tricuspid regurgitant jet velocity (TRV) has yet to be established [40].
In addition to hyperdynamic features, patients with SCA exhibit a unique form of CM with restrictive physiology. This form of CM may provide an explanation for typical pathologic cardiac features in SCA patients, especially mild pulmonary hypertension, elevated TRV, diastolic dysfunction, LA dilation, and LV dilation with normal systolic function [69]. Restrictive physiology often manifests as stiffened myocardium that leads to increases in ventricular pressures with only mild increases in volume [84]. It is primarily a disease of the myocardium characterized by reduced myocardial compliance and subsequently diastolic dysfunction leading to increases in ventricular filling pressure, LA pressure and restricted filling [84]. RCM can be idiopathic or in the setting of infiltrative diseases, radiation or chemotherapy [85]. Primary pathophysiologic mechanism underlying RCM is progressive fibrosis of the myocardium resulting in impaired ventricular relaxation and progressive diastolic function [86].
Generally, RCM has a poor prognosis without heart transplantation. Age and LA size are the strongest independent risk factors for mortality in patients with RCM [87] while ischemia and arrhythmias, which are possible consequences of fibrosis encasing the conducting pathways, are frequent causes of death in RCM patients, which occurs in about 30% of RCM patients [86,88]. This restrictive physiology hypothesis of SCA-CM has been supported by a meta-analysis study that included 134 SCA patients, which reported primary pathologic features are diastolic dysfunction, LA dilation and LV enlargement with preserved systolic function [69]. The main difference between idiopathic RCM and SCA-CM is LV enlargement, which is absent in idiopathic RCM. Indeed, the formal criteria of RCM exclude enlarged ventricles but this distinction is made to differentiate RCM from DCM. Thus, LV dilatation is one of the hyperdynamic features that coexist with the features of restrictive physiology in the hearts of SCA patients [70].
Autopsy and cardiac MRI (CMRI) studies provide supporting data on the pathophysiology of SCA-CM. Autopsy studies in SCA patients provide an insight into cardiac histopathology. In particular, important autopsy findings include cardiac chamber enlargement and increased heart weight, pulmonary vascular changes and myocardial fibrosis [89,90-92]. Autopsy findings also reveal different patterns of myocardial fibrosis – transmural fibrosis (scarring) with no evidence of atherosclerosis, patchy fibrosis, diffuse fibrosis and fibrotic foci involving the conduction system, which predisposes SCA patients to arrhythmias [89,93]. Recent CMRI-LGE studies show the modality is useful for detecting scar tissue or focal macroscopic fibrosis based on the difference in the volumes of distribution of extracellular contrast agent (gadolinium). However, LGE may vary significantly in SCA patients because it is based on detecting differences in enhancement between the affected area and surrounding myocardial tissue and may not detect diffuse myocardial fibrosis observed in autopsy of some SCA patients [89]. Autopsy and CMRI studies suggest that fibrosis is an important but overlooked pathology contributing to cardiac dysfunction in SCA. Ongoing studies evaluating novel CMRI techniques indicate diffuse myocardial fibrosis is a common finding in SCA patients [94].
Diagnosis: Diagnosis of SCA-CM remains controversial. The 2014 National Heart, Lung and Blood Institute (NHLBI) expert panel report on evidence-based management of SCA-CM did not find any demonstrable evidence to support echocardiographic screening in asymptomatic patients [95]. In contrast, the American Thoracic Society Clinical Practice guidelines recommend performing echocardiography every 1 to 3 years and increasing frequency depending on the presence of adverse risk factors such as high TRV and elevated plasms NT-pro-BNP or confirmed pulmonary hypertension [96]. However, so far, these expert consensus reports are not based on strong evidence. While it remains unclear at what point screening for SCA-CM should begin, Nis et al. [70] suggests ECG and echocardiographic screening on asymptomatic SCA starting between 15 and 18 years. Screening should look for chamber enlargement especially of the LA, systolic function and diastolic abnormalities using mitral and Doppler imaging. If cardiac abnormalities are detected, the need for imaging and referral to a cardiologist should be determined based on individual patient presentation.
Treatment: Same to diagnosis, treatment of SCA-CM remains controversial and challenging. At present, there is no proven treatment for cardiac dysfunction or pulmonary hypertension (PH) for SCA patients. Observational studies suggest a potential effect of therapy directed towards pulmonary arterial hypertension (PAH) in SCA patients with PH but small-randomized controlled trials (RCTs) do not show these benefits [97-99]. Small RCTs using endothelin receptor antagonist bosentan in SCA patients with PH [20] or comparing sildenafil to placebo in SCA patients with elevated TRV [100] were discontinued due to slow enrolment and adverse effects respectively. The role of disease-modifying therapy (targeting SCA) in SCA-CM remains undetermined. Going forward, understanding the pathophysiology of SCA-CM and different forms of PH in SCA patients may be important to identify directed therapies to slow or even reverse cardiac dysfunction in SCA patients. Until then, optimizing general SCA care using disease modifying therapy such as hydroxyurea or chronic transfusion remain the only therapeutic option with established benefits for SCD patients [70].
Henoch-schonlein purpura: Henoch-Schonlein purpura (HSP) is non-oncologic haematologic disorder defined as vasculitis (inflammation of blood vessels) characterized by the deposition of immune complexes mainly polymeric immunoglobulin a (IgA) and C3 in the small blood vessel mainly in the skin, intestines and kidneys [101-103]. Its aetiology remains unknown but it is often associated with infectious agents. Its course is often self-limiting but may manifest long-term renal morbidity [101]. HSP can affect all age groups although by far the most commonly affected are children aged 2-6 years [104,105]. The incidence in children is approximately 10.5 to 20.4 per 100,000 children per year [105,106]. The incidence is highest in the 4 to 6 years age group, with up to 70.3 per 100,000 children per year with a slight male preponderance of 1.2 to 1 ratio to female [105]. The dominant clinical characteristics of HSP are non-thrombocytopenic palpable purpura (100%), arthritis (82%), abdominal pain (63%), gastrointestinal bleeding (33%) and nephritis (40%) [107,108].
Pathophysiology: Kidney manifestations is a frequent occurrence in HSP patients causing haematuria and IgA type nephropathy. Extra renal involvement is rare but can be life threatening if diagnosis is missed. Direct evidence of HSP related CM are lacking but case reports of cardiopulmonary involvement [109-112] and cardiac involvement [113,114] suggest CM of an ischaemic origin in HSP patients. Lecutier [113] described a case report of an 11-year old boy who died one month after the onset of HSP. A throat culture detected streptococcus pyogenes as the infectious aetiology. Autopsy examination revealed several myocardial areas of necrosis with calcification but without the involvement of coronary arteries or arterioles perfusing the myocardium. The study suggested that antigen antibody reaction might affect the myocardium in the same way it produces the vascular lesions [113]. Imai and Matsumoto [114] reported a case of a 14-year old girl with HSP who developed HF two-weeks after the onset of the disease. On ECG, the patient had myocardial injury compatible with acute carditis and anti-streptococcal titre was markedly raise [114].
Case reports of cardiopulmonary involvement in HSP patients support the hypothesis of an underlying ischaemic process in the development of HSP-related CM. In a case report of HSP, Hayakawa et al. [111] described two patients who developed transient myocardial ischemia in the course of the disease. On ECG, ischaemic changes returned to normal within 4 to 5 days and the level of creatinine phosphokinase was not elevated with no evidence of acute myocardial infarction. In one patient, acute pericarditis was excluded because elevation of ST segments was confined to pericardial limbs on ECG and echocardiography revealed no pericardial effusion. The study concluded that coronary spasm was the likely cause of transient myocardial ischaemia in the two patients [111]. Tan [109] reports a case of adult onset of HSP in a 60-year old woman who developed ST elevation myocardial infarction (STEMI) and pulmonary haemorrhage in the course of the disease. Prompt normalization of ST segment change suggested transient ischemic of the myocardium possible due to coronary artery spasm precipitated by vasculitis process, which causes injury to coronary vessels endothelium and releases vasoconstrictive substances from those cells (endothelin-1) that is known to possess potent vasoconstrictive actions [115]. Another suggested mechanism of myocardial ischemia could be related to acute arterial thrombosis associated with the presence of antiphospholipid antibodies sometimes detected in HSP patients [116].
Diagnosis/Treatment: Diagnosis of HSP-CM rests on demonstrable clinical evidence of HSP and cardiac involvement. In 2006, the European League against Rheumatism and Paediatric Rheumatology European Society (EULAR/PReS) published a new classification of childhood vasculitis, which updated the 1990 American College of Rheumatology classification of HSP [117]. The criteria support diagnosis of HSP in the presence of palpable purpura, diffuse abdominal pain, biopsy showing predominant IgA, acute arthritis/arthralgia or renal involvement (haematuria/proteinuria). Ancillary tests such as ECG and non-invasive imaging are useful in the diagnosis of HSP complications including CM [117]. Although non-specific, cardiac involvement in HSP patients can manifest in the form of chest pain, bradycardia and hypotension. Some patients may exhibit coronary events manifesting as T-wave inversion and myocardial infarction as well as bundle branch block and sub-endocardial leucocytoclastic vasculitis. Pericarditis and subsequent pericardia effusion is very unusual [118]. Differential diagnosis of cutaneous purpura and myocardial infarction is challenging but the addition of other organ involvement such as kidney, lung and joints should suspect a systemic cause such as vasculitis of small to medium-sized vessels. Although HSP has a paediatric preponderance, it can also affect adults and should not be overlooked in adults presenting with typical signs of HSP and myocardial dysfunction [109]. Nevertheless, studies specific to HSP-CM are needed to clarify its diagnostic features as current evidence includes features general to cardiac dysfunction or HF.
Treatment for HSP-CM is often supportive. Unlike CM due to hematologic malignancies such as anaemia and SCA-CM, HSP-CM lacks disease-modifying therapy since the natural course of HSP is usually one of complete resolution often without the need for therapeutic intervention [101]. For severe cases, treatment is usually symptomatic and supportive. Unlike other vasculitis, corticosteroids (anti-inflammation treatment) have not been shown to change the course of the disease although it may reduce the length of severe abdominal pain. In the case of severe cardiac involvement, treatment usually targets symptoms of cardiac (HF medication), gastrointestinal and renal dysfunction based on current guidelines [109].
Neoplastic cardiomyopathy
The human body comprises of trillions of cells that grow, divide and die in an orderly fashion in a process tightly regulated by the DNA elements within the cell. The term neoplasm refers to an abnormal growth of cells that exceeds, and is uncoordinated with, that of the normal cells and persists in the same excessive manner even after the cessation of the stimuli that evoked the change. As the excessive growth of these neoplastic cells persist, a tumour (neoplasm) that lacks any purpose or function in the body may develop [119]. The neoplasm may be benign (non-cancerous) such as uterine fibroids or malignant (neoplasm that have become cancerous) characterized by abnormal cell growth, capacity of invading other tissues, and capacity to spread to distant organs via blood vessels or lymphatic channels (metastasis). Thus, neoplastic diseases define neoplasm that can cause both benign and malignant tumour growth [119,120]. Primary neoplasm (tumour restricted to the original organ) or metastatic (tumour that have spread to other distant organs) can potentially lead to CM in cancer patients.
Primary neoplasm: Cardiac tumours are a reclusive sub-division of oncology classified as primary (benign or malignant) or metastatic (secondary) but lacks a definitive classification based on aetiology, histopathology or a system of staging [121]. Cardiac myxoma represents the most common type of primary cardiac neoplasm regarded as benign. It can occur in almost any age although the usual patient age-range is between 30 and 60 years with a female predilection [122]. Patients with myxoma may present with a variety of symptoms including obstructive cardiac, embolic and constitutional symptoms. Obstructive cardiac signs include dyspnoea, thoracic pain, cough, dizziness and HF due to tumour prolapse into the mitral orifice mimicking HF symptoms [123,124]. Embolic manifestations may occur in any organ and usually accompanied with peripheral or pulmonary emboli or stroke [122]. Constitutional symptoms include arthralgia, myalgia, fever, rash, weight loss, cachexia and fatigue believed to be due to IL-6 production by tumour cells [125]. In a pooled autopsy series, primary cardiac neoplasm are rare, occurring in approximately 0.02% (about 200 tumours in 1 million autopsies) [126]. Histologically, about 75% of primary neoplasm are benign and almost half are myxoma with the remainder being lipomas, papillary fibroelastomas and rhabdomyomas. Whether benign or malignant, a majority of primary cardiac neoplasm manifest intracavitary and preferentially develop in the left atrium leading to LV inflow obstruction [127].
Evidence of myocardial involvement: Reports of CM in the setting of primary cardiac neoplasm are very rare, with only a handful of case reports describing this clinical entity. In a case report published in 1992, Kanemoto et al. [128] was the first to associate atrial myxoma with HCM in an English-language medical literature in a 67-year old male who presented with exertional dyspnoea. An association between HCM and LA myxoma has also been observed in patients with progressive cardiomyopathic lentiginosis (LEOPARD syndrome), which is a rare autosomal dominant multi-organ disease [129]. In three subsequent case reports of patients with HCM and atrial myxoma, no evidence of LEOPARD disease was discovered [130-132]. Abdou et al. [130] described a case of incidental diagnosis of atrial myxoma in a 71-year old female with HCM. Transthoracic and transesophageal echocardiogram revealed asymmetric hypertrophy of the basal and mid-anteroseptal wall (consistent with HCM) later confirmed by CMRI. Kałuzna‑Oleksy et al. [131] also described an incidental echocardiography diagnosis of LA myxoma and HCM who did not have LEOPARD syndrome. Finally, Mani et al. [132] described a 5-year boy with ventricular myxoma and HCM in which diagnosis was incidental on echocardiography and confirmed by CMRI.
Two case reports have also described DCM in patients diagnosed with atrial myxoma [133,134]. Adachi et al. [133] described a 54-year old female who had echocardiographic and angiographic evidence of markedly reduced LV wall motion and a large LA mass. The histological findings of the biopsied RV specimens were consistent with DCM. Muthiah [134] described a rare case of LV myxoma originating from the apical interventricular septum and projecting into LV cavity on 2D transthoracic echocardiography in a 29-year old male. The patient presented with myxoma with symptoms mimicking infectious hepatitis and DCM with depressed LVEF (20%). Cases of LV or bi-ventricular failure in patients with atrial myxoma have also been described suggesting HF may be the first manifestation of a giant myxoma in patients with normal coronary arteries [135,136]. Thus, timely echocardiographic evaluation and surgical removal of tumour is recommended to prevent severe cardiac complications.
Pathophysiology: The association between cardiac myxoma and HCM is very rare such that its pathophysiologic relevance and clinical course remain unclear. However, its pathophysiology is believed to be multifactorial based on evidence associating primary cardiac myxoma and HF. Left atrium myxoma may result in atrial and annular dilatation with mitral insufficiency from inadequate coaptation of valve leaflets in systole and mitral stenosis from obstruction of LV filling during diastole [137,138]. In some patients with LA myxoma, LV dilatation is likely related to co-occurring DCM, which may also be responsible for LV systolic dysfunction. A rapid onset of atrial fibrillation associated with ventricular tachycardia may possibly reduce the diastolic time, which further impairs LV function and aggravates latent chronic heart failure. Atrial myxoma may also produce a direct cardiodepressive effect, which may result in the release of cytokines such as interleukin (IL) 6 and IL-8 that may cause myocardial tissues inflammation and bi-ventricular hypertrophy and dilation with global ventricular dysfunction [135]. Over-production of IL-6 and IL-8 may induce thrombopoiesis, neutrophil migration and myxoma cell-fragment adhesion to coronary artery endothelium, leading to myocardial ischemia and infarction [139-142].
Diagnosis/Treatment: Diagnosis of primary neoplasm CM has not been elucidated in medical literature. Case reports suggest diagnosis is often incidental especially when examining for other more common causes of cardiac dysfunction, CM or HF. In all the four case reports on myxoma and HCM or DCM [130-133], common diagnostic test was transthoracic or transesophageal echocardiography used for the detection of myxoma mass and ventricular hypertrophy and/or dilation. Echocardiogram can detect asymmetric hypertrophy of the basal and mid-anteroseptal wall (consistent with HCM) while CMRI may reveal asymmetric LVH with a thickened interventricular septum and a mass in the LA along the inter-atrial septum [130]. Echocardiography is the best first-lime imaging modality because of its simplicity, wide availability and cost-effectiveness, and detect tumour structure and location as well as hemodynamic alterations induced by the tumour. Cardiac MRI is a complementary technique that is not associated with radiation risk and provides information on tumour vasculature and tissue appearance [133,134]. The management of asymptomatic cardiac myxoma remain a topic of debate because there are no reports clearly favouring conservative or surgical intervention. However, tumour excision (surgically or by radiotherapy) has been associated with improvement in cardiac function especially in patients with primary cardiac myxoma and familial HCM, which increases the risk of complications [130,133,134]. To improve diagnosis and management, autopsy studies or surgically excised myxoma should be histologically examined for diagnosis as well as the structure should be evaluated by histochemical analysis for cardio-depressant factors particularly in patients with ventricular dysfunction.
Metastatic neoplasms: Metastatic cardiac neoplasm refers to tumours that have travelled from other organs of the body to the heart preferentially from melanoma, primary mediastinal, breast and colon cancers [122]. These tumours are among the least known and highly debated issues in oncology with a few systematic studies devoted to this topic. Metastatic cardiac neoplasm reach the heart through hematogenous route entering the small coronary vessels from which they invade the heart or direct extension from intrathoracic cancer or by retrograde flow through mediastinal and tracheobronchial lymphatic channels [144]. The incidence of cardiac metastases is on the rise because of improved survival of cancer patients due to improved anti-cancer treatment and increased sensitivity of diagnostic modalities. The incidence of metastatic cardiac neoplasm reported in literature varies widely between 2.3% to 18.3% in case reports [143] and up to 25% in autopsy series of patient who had died of malignancies [127]. Although no malignant tumours are known that diffuse preferentially to the heart, some tumours do involve the heart more than others do such as melanoma and mediastinal primary tumours [143-145]. Thus, for every other site, the ability of the tumour to spread to the heart depends on several factors including the characteristics of the primary tumour and the specific histological and functional characteristics of the heart [146].
Evidence of myocardial involvement: Evidence on the involvement of the myocardium in metastatic cardiac neoplasm is limited to autopsy series and a handful of case reports possibly due to the rarity of the disorder as well as heterogeneity of cardiac and extra cardiac signs and symptoms rendering a definitive diagnosis challenging. Thus, the evidence of tumours metastasizing to the heart are largely based on diagnosis during autopsy series [147-149]. The most common primary neoplasm metastasizing to the heart are lung (31.6%), lymphoma (15.8%), breast (12.8%), leukaemia (13.7%), stomach (5.3%), and liver, melanoma and colon (3.2%) [147]. The myocardium is the most common cardiac site (53.9%), followed by the pericardium (28.4%), the epicardium/pericardium (13.7%) and finally the endocardium (3.9%) [147]. Although neuroendocrine and thyroid cancers rarely lead to cardiac metastases, few case reports have described patients with these unusual forms of cardiac metastases leading to CM [146,147].
Myocardial metastases directly from neuroendocrine carcinoma have been described in two case reports. Robinson et al. [150] initially reported an unusual case of a 92-year old male patient presenting with clinical signs of acute myocardial infarction. The patient had elevated jugular venous pressure, pulmonary crackles, ST changes and a minor degree of right bundle branch block (RBBB) on ECG but with normal levels of serum creatinine phosphokinase. The patient died due to cardiogenic shock and autopsy analysis revealed carcinoma of the pancreas and metastasis in the myocardium. Recently, Kondo et al. [151] described an autopsy case of non-functioning pancreatic neuro-endocrine carcinoma metastasizing to the myocardium in a 63-year old Japanese male patient who died of HF. The patient presented with dyspnoea and echocardiography revealed marked LVH and diffuse myocardial thickening with pericardial effusion. Autopsy analysis revealed the entire pancreas was occupied with neuroendocrine differentiation, diagnosed on histopathology as non-functioning pancreatic neuroendocrine carcinoma. Immunohistochemical analysis revealed the heart had numerous metastatic nodules and diffuse myocardial thickening mimicking HCM [151].
Myocardial metastases of thyroid cancer are very rare. In an analysis of 4,124 autopsies [148] and 3,314 autopsies [147], no myocardial metastases of thyroid cancer were found and in 3,249 autopsies, cases of metastases of thyroid to the myocardium was 0.18% [149]. However, case reports have described 13 cases of cardiac metastases of thyroid tumours including two cases of metastatic papillary thyroid carcinomas [152-158]. In a recent case of a 62-year old male with a history of surgical therapy for thyroid carcinoma, Fukuda et al. [159] report myocardial metastases of papillary thyroid carcinoma in both ventricles with a mobile RV pedunculated tumour. The patient was admitted because of chest pain, exertional dyspnoea, pretibial oedema, and febrile. ECG revealed ST segment elevation and low voltage. A large solid tumour was demonstrated in both ventricles with pericardial effusion by echocardiography, thoracic CT scan and angiography. Autopsy analysis revealed widespread metastases to all layers of the heart – myocardium, endocardium and pericardium. Thus, metastatic cardiac tumours should be considered in patients who have undergone thyroidectomy especially when new cardiac symptoms or ECG changes occur. Recently, a case of metastases of hepatocellular carcinoma was misdiagnosed as isolated HCM [160].
Diagnosis/Treatment: Diagnosis and treatment for metastatic neoplasm is a clinical challenge since the disease is rare and it lacks pathognomonic diagnostic signs and symptoms, and usually presents with extra-cardiac signs and symptoms. Autopsy series and case reports suggest clinical manifestation of metastatic cardiac tumour varies widely. Diagnosis is incidental in clinical settings but in a greater majority of cases, diagnosis is made during autopsy. Since diagnosis is usually delayed, surgical intervention of the primary tumour may have little therapeutic benefit to the patient because the tumour has already spread to other organs. Further studies are warranted to evaluate cardiac features that would raise clinical suspicion of CM in the setting of metastatic cardiac neoplasm since the disorder has an ominous prognosis when detected late.
Meta-analysis of SCA-CM echocardiographic diagnosis
The understanding of the natural course, clinical presentation, diagnosis and management of oncologic CM has lagged far behind that of other forms of CM possibly due to the rarity of the disease as well as the involvement of both cardiac and extra-cardiac manifestations making the development of a generalized diagnosis and treatment impossible. Moreover, the rarity of the disease has been associated with the paucity of research despite the disease having an ominous prognosis. On the other hand, non-oncologic hematologic causes of CM such as sickle-cell anaemia has received a greater proportion of research. However, diagnosis and management remain unclear with current treatment targeting the management of HF symptoms and cardiac complications [95,96]. To date, only two review articles [69,70] have specifically discussed SCA-CM, with a majority of evidence based on general cardiac dysfunction and serum changes in the levels of natriuretic peptides in SCA-patients. This meta-analysis goes further to evaluate echocardiographic-based evidence of cardiac changes in SCD patients in comparison to healthy controls matched to body surface area (for infants and children), age and sex.
Electronic search for studies was performed in PubMed and Google Scholar using the following search terms: sickle cell anaemia, sickle cell disease or sickle cell combined with each of the following terms: cardiomyopathy, haemodynamic, heart failure, cardiac dysfunction, pulmonary hypertension or echocardiography. Studies that enrolled SCA patients and evaluated different modalities of echocardiography (conventional, 2D, 3D, M-mode, Doppler or speckle tracking) were retrieved for a full review. Studies that did not compare SCA patients with age- and sex-matched healthy controls or did not provide raw quantitative data (mean, standard deviation or number of observations) were excluded from the final data set. Categorical data was expressed as frequency and percentage, continuous data as mean and standard deviation, and dichotomous data as weighted mean difference (WMD) and 95% confidence interval (CI). The inconsistency parameter (I2) was used to estimate the amount of heterogeneity as well as determine the basis of random effect approach (either fixed or random effect model) used for analysis. Differences were considered statistical significant at p < 0.05 (Table 2).
Table 2. Summary of patient characteristics and summary of echocardiographic findings
1st Author [Ref #]
|
Year
|
Sample
(SCA/ Ctrl)
|
Male (n)
|
Mean Age (yrs.)
|
Summary of Echocardiographic Findings
|
Rees [161] |
1978 |
44/28 |
27 |
8.6±0.4 |
LV dysfunction is prevalent in a significant proportion of SCA children but unrelated to the degree of anaemia or percentage of foetal haemoglobin. |
Lester [162] |
1990 |
64/99 |
29 |
7.37±0.71 |
Cardiac abnormalities in children are related to volume overload with no evidence of SCA-CM or cardiac dysfunction |
Lewis [163] |
1991 |
30/30 |
15 |
29(19-39) |
LV diastolic filling patterns are altered in SCA patients, which may be present even in the absent of HF symptoms suggesting intrinsic myocardial abnormality. |
Martins [164] |
1999 |
25/25 |
NR |
26.56±9.19 |
Chamber dilation, eccentric hypertrophy and systolic dysfunction characterize SCA-CM, which is significantly increased compared to controls. |
Seliem [165] |
2002 |
26/20 |
19 |
10.2±2.6 |
LV systolic performance is preserved in SCD patient in early childhood but with LVH and diastolic filling abnormalities by Doppler that do not fit specific cardiomyopathic pattern - diastolic dysfunction - restrictive physiology with delayed relaxation |
Raman [166] |
2006 |
22/11 |
8 |
28.5±8.18 |
A subset of adult SCA patients may have myocardial ischemia in the absence of myocardium infarct/iron overload or CAD. RV dysfunction may be present in stable patients despite normal resting pulmonary artery pressure suggesting under-recognized mechanisms. |
Aleem [167] |
2007 |
65/58 |
28 |
24.5±9.2 |
PH is the most common cardiac abnormality in SCD patients, who exhibit significantly increased LV dimensions and volume compared to controls. |
Sachdev [168] |
2007 |
235/41 |
94 |
35±11 |
Diastolic dysfunction and PH exert an independent effect on mortality in SCD patients with extremely poor prognosis supporting the need for echo screening to stratify at risk population |
Westwood [169] |
2007 |
47/40 |
27 |
33±13 |
LV changes in SCD are partly due to chronic anaemia but there appears to be e secondary mechanism, which is not myocardial iron loading or fibrosis |
Aliyu [170] |
2008 |
208/94 |
0 |
22±8 |
Incidence of PH in SCD patients is higher in sub-Saharan Africa compared to age-matched US cohort. |
Arslankoylu [171] |
2010 |
32/30 |
17 |
10.4±4.2 |
Doppler myocardial performance index may be a useful non-invasive and sensitive tool for assessing sub-clinical cardiac LV and RV dysfunction in SCA patients |
Colombatti [172] |
2010 |
37/8 |
20 |
6.25(3.07-15.68) |
SCA patients have significantly higher LV dimensions compared to healthy controls |
Knight-Perry [173] |
2011 |
53/33 |
28 |
34±11 |
SCD adults exhibit LV and RV chamber enlargement and higher LV and RV filling pressure compared to controls |
Abdul-Mohsen [174] |
2012 |
45/45 |
28 |
25.73±6.79 |
SCD patients have significantly increased ventricular dimensions and volume and diastolic dysfunction compared to age and sex-matched controls |
Ahmad [175] |
2012 |
28/28 |
12 |
33±7 |
3D STE confirms diastolic dysfunction in some SCA patients but does not reveal any changes in LV systolic function |
Ghaderian [176] |
2012 |
64/50 |
|
11.7±5.5 |
TDI indices is a sensitive indicator for the cardiac function in chronic diseases and the RV function in some disorders such as SCA |
Desai [177] |
2014 |
38/13 |
17 |
32(27-38) |
SCA-CM is characterized by 4-chamber dilation, and in some patients, myocardial fibrosis, abnormal perfusion reserve, diastolic dysfunction and rarely myocardial iron overload. |
CAD: Coronary Artery Disease; CM: Cardiomyopathy; HF: Heart Failure; LV: Left Ventricular; LVH: PH: Pulmonary Hypertension; SCA: Sickle Cell Anaemia; SCD: Sickle Cell Disease; STE: Speckle Tracking Echocardiography; TDI: Tissue Doppler Imaging; RV: Right Ventricular
The electronic search yielded 374 studies, of which only 17 were included in the final analysis [161-177]. The 17 studies enrolled a combined population of 1,716 consisting of 1,063 SCA patients and 653 controls matched for sex, age and body surface areas (for children). Patients had an equal gender representation (male = 51%; female = 49%) and were relatively young (mean = 22.22±10.63 years) consisting of children in six studies [161,162,165,171,172,176] and young adults in the remaining 11 studies [163,164,166-170,173-175,177]. The most common cardiac structural and functional parameters assessed were LV systolic function (LVEF and cardiac index), LV structure (LVEDD, LVESD, LVM, LA and IVRT), and diastolic function (E/A ratio, deceleration time [DT]) and global longitudinal strain (GLS).
Pooled analysis of echocardiographic data from 14 studies [161,162,164-171,173-176] revealed that SCA patients have a preserved LV systolic function (LVEF) similar to that of age- and sex-matched controls. Weighted mean for LVEF at baseline was 62.19% (95% CI: 60.12% to 64.25%). Weighted mean difference (WMD) of LVEF between SCA and control patients was -1.95% (95% CI: -4.253 to 0.359) in favour of control patients although the difference was not statistically significant (p = 0.098; Figure 1). Heterogeneity among the studies was high (I2 = 99.29%), which could not be explained by differences in patient and study characteristics, and thus a random fixed model was used. Cardiac index was also significantly higher in SCA patients (WMD: 2.30; 95% CI: 0.73 to 3.87; p = 0.004) [161,162,164]. Despite a preserved systolic function, LV dimensions and size differed between SCA and control patients. In eight studies [163,164,167,168,170-172,174], SCA had significantly higher LVEDD (WMD: 4.267 mm; 95% CI: 3.15 to 5.38; p = 0.000; Figure 2). In six studies [164,168,170-172,174], LVESD was significantly higher in SCA patients then in controls (WMD: 3.569 mm; 95% CI: 2.93 to 4.21; p = 0.000; Figure 3). In five studies, LVMI was significantly higher in SCA patients (WMD: 28.40; 95% CI: 22.87 to 33.98; p = 0.000; Figure 4). Left atrial diameter (LAD) was also significantly increased in SCA patients (WMD: 3.44 mm; 95% CI: 2.19 to 4.69; p = 0.000) [163,167]. Finally, there was a tendency towards diastolic dysfunction indicated by a slightly lower E/A ratio in SCA patients (WMD: -0.01; 95% CI: -0.12 to 0.099; p = 0.854; Figure 5) [165,168,170-174] and slightly higher deceleration time (DT: WMD: 1.34; -9.04 to 11.72; p= 0.801; Figure 6) [165,168,170,171,173-175].
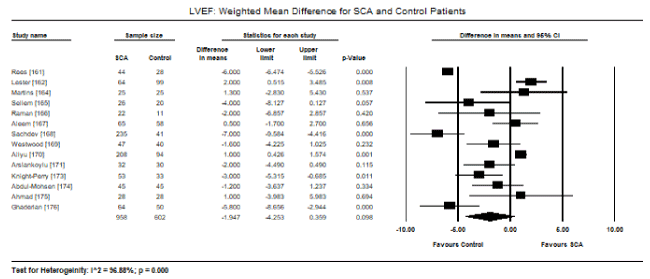
Figure 1. LVEF weighted mean difference and 95% CI for SCA and control patients
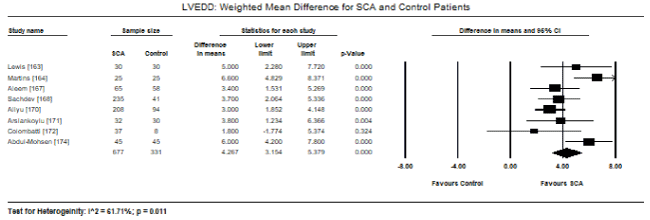
Figure 2. LVEDD weighted mean difference and 95% CI for SCA and control patients
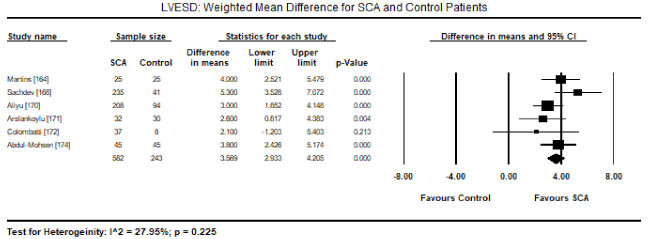
Figure 3. LVESD weighted mean difference and 95% CI for SCA and control patients
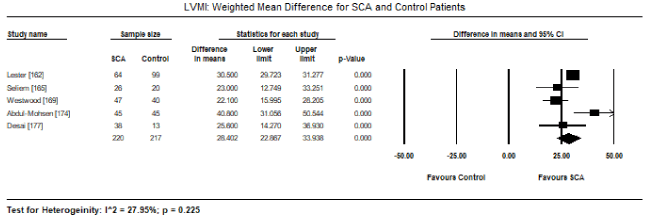
Figure 4. LVMI weighted mean difference and 95% CI for SCA and control patients
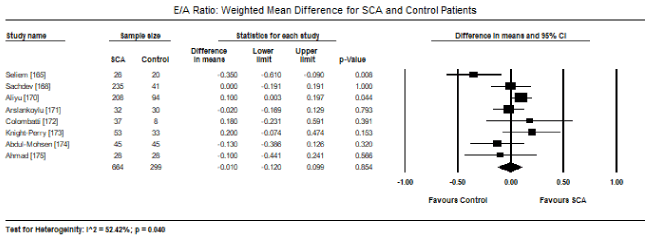
Figure 5. E/A ratio weighted mean difference and 95% CI for SCA and control patients
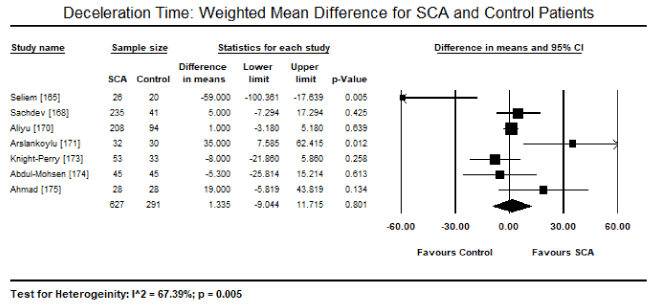
Figure 6. DT weighted mean difference and 95% CI for SCA and control patients
The present meta-analysis involving 1,716 SCA and healthy (control) patients evaluated cardiac changes in SCA-CM. The findings reveal that SCA patients have a preserved systolic function, but with significantly increased LV dimensions (LVEDD and LVESD), left atrial diameter and cardiac index compared to healthy controls. There was also a tendency towards diastolic dysfunction indicated by a slight non-significant decrease in E/A ratio and increase in DT in SCA patients. The findings suggest common cardiac changes in SCA-CM are preserved systolic function, dilated LV, LA enlargement and diastolic dysfunction. LV dilation (hypertrophy) and other detectable abnormalities in the diastolic filling patterns by Doppler suggest a specific cardiomyopathic pattern due to impaired LV relaxation and characterized by abnormally delayed early diastolic filling (deceleration time > 166 msec) or a decreased rate of decline of row velocity in early diastole (restrictive physiology and delayed relaxation).
The present findings support the hypothesis that SCA-CM patients have a restrictive physiology mimicking that of RCM although it is uniquely different from RCM. The defining features of RCM are preserved systolic function, diastolic dysfunction and left atrial enlargement and the standard definition of RCM usually does not include ventricular dilation [5-8]. Thus, the SCA-CM does not fit the classical definition of RCM since ventricular dilation is one of its key diagnostic feature. The present findings support the also hypothesis that the pathophysiologic mechanisms of SCA-CM is a restrictive physiology super-imposed on anaemia-related hyperdynamic features [80-83]. Since SCA-CM has ventricular dilation and preserved systolic function, it still differs from DCM, which is characterized by depressed systolic function [6,7].
The presence of diastolic dysfunction in SCA-CM has been identified as an independent risk factor for early death and an important feature for differential diagnosis of SCA-CM from RCM [168,178]. Paediatric studies reveal LA dilation is more prevalent in young SCA patients than the enlargement of other chambers suggesting it precedes the enlargement of other cardiac chambers [172,179-181]. Moreover, LA dilation and LVH are adaptive processes to anaemia-related hyperdynamic features, which worsen overtime [82,162,182]. In a review by cardiac dysfunction in SCA patients, Nis et al. [69] finds LV end-diastolic and LVH are more noticeable in older patients compared to a younger cohort of SCA patients. In a recent study, adults with SCA exhibit 4-chamber dilation, normal systolic function and diastolic dysfunction associating increasing age with worsening LV dilation and LVH progressing into a 4-chamber dilatation [177]. A related review study on systolic function in SCA patients, LV systolic function was preserved although the patients had progressive LV dilation over time [183]. The presence of LA dilation preceding chamber enlargement is a characteristic feature that allows for the identification of SCA-CM with restrictive physiology that would otherwise have been masked by dilated ventricles in older patients [69,70].
The present findings have clinical relevance for the diagnosis of SCA-CM. Currently, the use and application of adult diastolic HF grading guidelines have not been validated in paediatric patients [184]. Many patients with overt RCM were missed when the current guidelines were used to classify diastolic function coupled with high discordance among different parameters and poor inter-observer agreement [185,186]. Thus, different criteria may be needed to diagnose and grade diastolic dysfunction in paediatric patients with SCA. Grade III/IV or restrictive diastolic dysfunction are not common in some studies [69] but are prevalent in the present meta-analysis accompanied by LA dilation and normal systolic function. Precise understanding of the pathophysiology of diastolic dysfunction would be beneficial for clinical management of SCA-CM patients. At present, the cause of diastolic dysfunction in SCA patients is not well-established. Although the presence of myocardial fibrosis has been suggested as a possible underlying mechanism for restrictive physiology in SCA, cardiac MRI studies have been inconsistent in detecting the extent of fibrosis in the SCA myocardium while microscopic fibrosis has not been well studied. Other studies suggest that ventricular stiffness secondary to fibrosis is involved in the development of SCA-CM with restrictive physiology [187-190].
In summary, the present findings suggest that SCA patients develop a unique form of CM presenting with restrictive physiology super-imposed on anaemic-related hyperdynamic state. The disease is progressive, leading to the development of LV dilatation and diastolic dysfunction. Presently, diastolic dysfunction remains an independent risk factor for morbidity and mortality. Determining the pathogenic mechanisms leading to SCA-CM could help identify therapies that decrease cardiopulmonary morbidity and mortality in these patients. Besides SCA-CM, there is also need for additional studies specifically focussing on CM due to other haematologic and oncologic aetiologies to improve diagnosis and identify therapies that would improve disease management and survival for patients with CMs in general.
Cardiomyopathy (CM) is a rare but important cause of HF and death in patients with primary or metastatic cardiac neoplasm, or haematological disorders especially leukaemia, multiple myeloma, anaemia, sickle-cell anaemia and Henoch-Schonlein Purpura. The pathophysiology of oncologic/haematologic CM remains unclear but appears to have restrictive physiology although a few isolated cases of DCM and HCM phenotypes have been described in some cancer patients. In particular, CM secondary to sickle cell anaemia (SCA) consists of restrictive physiology superimposed on anaemia-related hyperdynamic state, which may explain the prevalence of diastolic dysfunction, cardiac chamber (LA/LV) dilation and normal systolic function in SCA-CM patients. Dilated LA/LV differentiates SCA-CM from RCM while preserved systolic function differentiates SCA-CM from DCM. Oncologic/haematologic CM lacks specific diagnostic tests. Diagnosis rests on the patient’s history and physical examination supported by ancillary tests such as ECG, non-invasive cardiac tests and endomyocardial biopsy. Management of oncologic/haematologic CM is challenging due to a wide heterogeneity of clinical signs and symptoms, and varying extent of myocardial and extra cardiac involvement. Current therapy rests on optimizing general care for the underlying oncologic or haematologic disease causing the CM, and in the case of severe cardiac dysfunction, the standard HF therapy based on the current guidelines.
- Hamo CE, Bloom MW (2017) Cancer and heart failure: understanding the intersection. Card Fail Rev 3: 66-70. [Crossref]
- Barlogis V, Auquier P, Bertrand Y, Chastagner P, Plantaz D et al. (2015) Late cardiomyopathy in childhood acute myeloid leukemia survivors: a study from the LEA program. Haematologica 100: e186-e189. [Crossref]
- Kasper EK, Agema WR, Hutchins GM, Deckers JW, Hare JM, et al. (1994) The causes of dilated cardiomyopathy: a clinicopathologic review of 673 consecutive patients. J Am Coll Cardiol 23: 586-590. [Crossref]
- Lantz B, Adolfsson J, Lagerlöf B, Reizenstein P (1979) Cardiomyopathy in leukemia, with reference to rubidomycin cardiotoxicity. Cancer Chemother Pharmacol 2: 95-99. [Crossref]
- Bozkurt B, Colvin M, Cook J, Cooper LT, Deswal A et al. (2016) Current diagnostic and treatment strategies for specific dilated cardiomyopathies: a scientific statement from the American Heart Association. Circulation 134: e579-e646. [Crossref]
- Pinto YM, Elliott PM, Arbustini E, Adler Y, Anastasakis A et al. (2016) Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J 37: 1850-1858. [Crossref]
- Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D et al. (2006) Contemporary definitions and classification of the cardiomyopathies: an American Heart Association scientific statement from the council on clinical cardiology, heart failure and transplantation committee; quality of care and outcomes research and functional genomics and translational biology interdisciplinary working groups; and council on epidemiology and prevention. Circulation 113: 1807-1816. [Crossref]
- Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F et al. (2007) Classification of the cardiomyopathies: a position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 29: 270-276. [Crossref]
- Navi BB, Reiner AS, Kamel H, Iadecola C, Okin PM et al. (2019) Arterial thromboembolic events preceding the diagnosis of cancer in older persons. Blood 133: 781-789. [Crossref]
- Mir MA (1978) Evidence for non-infiltrative cardiomyopathy in acute leukaemia and lymphoma. A clinical and echocardiographic study. Br Heart J 40: 725-733. [Crossref]
- Assuncao BM, Handschumacher MD, Brunner AM, Yucel E, Bartko PE et al. (2017) Acute leukaemia is associated with cardiac alterations before chemotherapy. J Am Soc Echocardiogr 30: 1111-1118. [Crossref]
- Xu W, Wang TY, Becker RC (2011) Hematologic diseases: from within the heart. Rev Esp Cardiol 64: 606-613. [Crossref]
- Phillips MD, Willerson JT (2007) Hematologic Disease and Heart Disease. In Willerson JT, Wellens HJJ, Cohn JN, Holmes DR (Eds.) Cardiovascular Medicine. Springer, London, 2409-2421.
- Mozos I (2015) Mechanisms linking red blood cell disorders and cardiovascular diseases. Biomed Res Int 2015: 682054. [Crossref]
- Burgun A, Bodenreider O, Jacquelinet C (2005) Issues in the classification of disease instances with ontologies. Stud Health Technol Inform 116: 695. [Crossref]
- Grigoropoulos NF, Petter R, Van‘t Veer MB, Scott MA, Follows GA (2013) Leukaemia update. Part 1: diagnosis and management. BMJ 346: f1660. [Crossref]
- Blundell R, Xuereb J (2007) Acute Leukaemia. International Journal of Molecular Medicine and Advance Sciences 3: 138-144.
- Jabbour EJ, Faderl S, Kantarjian HM (2005) Adult acute lymphoblastic leukaemia. Mayo Clin. Proc 80: 1517-1527. [Crossref]
- Jabbour EJ, Estey F, Kantarjian HM (2006) Adult acute myeloid leukaemia. Mayo Clin Proc 81: 247-260. [Crossref]
- Chellapandian D, Pole JD, Nathan PC, Sung L (2019) Congestive heart failure among children with acute leukemia: a population-based matched cohort study. Leuk Lymphoma 60: 385. [Crossref]
- Mateen FJ, Harding SR, Saxena A (2006) Extensive myocardial infiltration by hemopoietic precursors in a patient with myelodysplastic syndrome. BMC Blood Disord 6: 4. [Crossref]
- Madhavan S, Sasidharan PK, Krishnan R (2004) Restrictive cardiomyopathy due to primary plasma cell leukaemia. J Assoc Physicians India.52: 826-827. [Crossref]
- Van Haelst PL, Schot B, Hoendermis ES, van den Berg MP (2006) Acute myeloid leukaemia as a cause of acute ischaemic heart disease. Neth Heart J 14: 62-65. [Crossref]
- Li W, Garcia D, Cornell RF, Gailani D, Laubach J et al. (2017) Cardiovascular and thrombotic complications of novel multiple myeloma therapies: a review. JAMA Oncol 3: 980-988. [Crossref]
- Plummer C, Driessen C, Szabo Z, Mateos MV (2019) Management of cardiovascular risk in patients with multiple myeloma. Blood Cancer J 9: 26. [Crossref]
- Kocoglu M, Badros A (2016) The role of immunotherapy in multiple myeloma. Pharmaceuticals (Basel) 2016; 9 [Crossref]
- Lub S, Maes K, Menu E, De Bruyne E, Vanderkerken K et al. (2016) Novel strategies to target the ubiquitin proteasome system in multiple myeloma. Oncotarget 7: 6521–6537. [Crossref]
- Beg R, Hurwitz B, Cendan I, Alvarez D, Rosario L et al. (2016) Infiltrative Cardiomyopathy In Multiple Myeloma: Delayed Treatment, Devastating Consequences. Critical Care Case Reports: Cardiovascular Disease and Hemodynamics II A6963-A6963.
- Dahraoui S, Uwingabiye J, Belarj B, Biaz A, Rachid A et al. (2018) Unexpected discovery of multiple myeloma following cardiomyopathy. Clin Case Rep 6: 86-90. [Crossref]
- Arous S, Bensahi I, Noureddine M, Habbal R (2015) Typical case of a multiple myeloma revealed by cardiac amyloidosis. Angiology 3: 1000163.
- Kristin D. Kistler KR, Faich G, Lanes S (2012) Cardiac event rates in patients with newly diagnosed and relapsed multiple myeloma in US clinical practice. Paper presented at: 54th American Society of Hematology Annual Meeting; December 8-11, 2012; Atlanta, GA.
- Augustson BM, Begum G, Dunn JA, et al. (2005) Early mortality after diagnosis of multiple myeloma: analysis of patients entered onto the united kingdom Medical Research Council trials between 1980 and 2002: Medical Research Council Adult Leukaemia Working Party. J Clin Oncol 23: 9219-9226. [Crossref]
- Bahlis NJ, Lazarus HM (2006) Multiple myeloma-associated AL amyloidosis: is a distinctive therapeutic approach warranted? Bone Marrow Transplant. 38: 7-15. [Crossref]
- Hassan W, Al-Sergani H, Mourad W, Tabbaa R (2005) Amyloid heart disease. New frontiers and insights in pathophysiology, diagnosis, and management. Tex Heart Inst J 32: 178-184. [Crossref]
- International Myeloma Working Group (2015) International Myeloma Working Group (IMWG) criteria for the diagnosis of multiple myeloma 2015. Available from: http://imwg.myeloma.org/international-myelomaworking-group-imwg-criteria-for-the-diagnosis-of-multiple-myeloma/.
- Hegde N, Rich MW, Gayomali C (2006) The cardiomyopathy of iron deficiency. Tex Heart Inst J 33: 340-344. [Crossref]
- McLean E, Cogswell M, Egli I, Wojdyla D, De Benoist B (2009) Worldwide prevalence of anaemia, WHO vitamin and mineral nutrition information system, 1993–2005. Public Health Nutr 12: 444-454. [Crossref]
- Mukherjee KL, Ghosh S (2012) Medical laboratory technology. Procedure Manual for Routine Diagnostic Tests Vol I, 263-266.
- Soundarya N, Suganthi P (2017) A review on anaemia–types, causes, symptoms and their treatments. Journal of Science and Technology Investigation 10-17
- Anand IS, Gupta P (2018) Anemia and iron deficiency in heart failure: current concepts and emerging therapies. Circulation 138: 80-98. [Crossref]
- Anderson J, Glynn LG, Newell J, Iglesias AA, Reddan D et al. (2009) The impact of renal insufficiency and anaemia on survival in patients with cardiovascular disease: a cohort study. BMC Cardiovascular Disorders 9: 51. [Crossref]
- Eckardt KU (1999) Cardiovascular consequences of renal anaemia and erythropoietin therapy. Nephrology Dialysis Transplantation 14: 1317-1344. [Crossref]
- Hegde N, Rich MW, Gayomali C (2006) The cardiomyopathy of iron deficiency. Tex Heart Inst J 33: 340. [Crossref]
- Van Veldhuisen DJ, Anker SD, Ponikowski P, Macdougall IC (2011) Anemia and iron deficiency in heart failure: mechanisms and therapeutic approaches Nat Rev Cardiol.8: 485. [Crossref]
- Sohn IS, Jin ES, Cho JM, Kim CJ, Bae JH et al. (2008) Bloodletting-induced cardiomyopathy: reversible cardiac hypertrophy in severe chronic anaemia from long-term bloodletting with cupping. Eur J Echocardiogr. 9: 585-586. [Crossref]
- Anand IS, Chandrashekhar Y, Ferrari R, Poole-Wilson PA, Harris PC (1993) Pathogenesis of oedema in chronic severe anaemia: studies of body water and sodium, renal function, haemodynamic variables, and plasma hormones. Br Heart J 70: 357-362. [Crossref]
- Anand IS, Chandrashekhar Y, Wander GS, Chawla LS (1995) Endothelium derived relaxing factor is important in mediating the high output state in chronic severe anaemia. J Am Coll Cardiol 25: 1402-1407. [Crossref]
- Anand IS (2008) Anaemia and chronic heart failure implications and treatment options. J Am Coll Cardiol 52: 501-511. [Crossref]
- Ni Z, Morcos S, Vaziri ND (1997) Up-regulation of renal and vascular nitric oxide synthase in iron-deficiency anaemia. Kidney Int 52: 195-201. [Crossref]
- Kalra PR, Anagnostopoulos C, Bolger AP, Coats AJ, Anker SD (2002) The regulation and measurement of plasma volume in heart failure. J Am Coll Cardiol 39: 1901-1908. [Crossref]
- Turner LR, Premo DA, Gibbs BJ, Hearthway ML, Motsko M et al. Adaptations to iron deficiency: Cardiac functional responsiveness to norepinephrine, arterial remodeling, and the effect of beta-blockade on cardiac hypertrophy. BMC Physiol 2: 1. [Crossref]
- Chvapil M, Hurych J, Ehrlichova E (1968) The effect of iron deficiency on the synthesis of collagenous and non-collagenous proteins in wound granulation tissue and in the heart of rats. Exp Med Surg 26: 52-60. [Crossref]
- Gow AJ, Payson AP, Bonaventura J (2005) Invertebrate haemoglobins and nitric oxide: How heme pocket structure controls reactivity. J Inorg Biochem 99: 903-911. [Crossref]
- Naito Y, Tsujino T, Matsumoto M, Sakoda T, Ohyanagi M et al. (2009) Adaptive response of the heart to long-term anaemia induced by iron deficiency. Am J Physiol Heart Circ Physiol 296: H585-H593. [Crossref]
- Simsek H, Gunes Y, Demir C, Sahin M, Gumrukcuoglu HA et al. (2010) The effects of iron deficiency anaemia on p-wave duration and dispersion. Clinics (Sao Paulo) 65: 1067-1071. [Crossref]
- Khode VH, Kammar KF (2012) QTc changes in non-pregnant females with severe iron deficiency anaemia. J Clin Diagn Res 6: 777-779.
- Tangeda PR, Patil S, Shastri N, Noorali SN (2016) Maternal myocardial performance in second trimester of pregnancy with iron deficiency anaemia. J Clin Diagn Res 10: Cc16-18. [Crossref]
- Cho IJ, Mun YC, Kwon KH, Shin GJ (2014) Effect of anaemia correction on left ventricular structure and filling pressure in anaemic patients without overt heart disease. Korean J Intern Med 29: 445-453. [Crossref]
- Zhou Q, Shen J, Liu Y, Luo R, Tan B et al. (2017) Assessment of left ventricular systolic function in patients with iron deficiency anaemia by three-dimensional speckle tracking echocardiography. Anatol J Cardiol 18: 194-199. [Crossref]
- Shen J, Zhou Q, Liu Y, Luo R, Tan B, Li G et al. (2016) Evaluation of left atrial function in patients with iron-deficiency anaemia by two-dimensional speckle tracking echocardiography. Cardiovasc Ultrasound 14: 34. [Crossref]
- Kurisu S, Sumimoto Y, Ikenaga H, Watanabe N, Ishibashi K et al. (2017) Effects of haemoglobin level on myocardial washout rate of thallium-201 in patients with normal myocardial perfusion assessed by single-photon emission computed tomography. Heart Vessels 32: 1062-1066. [Crossref]
- Bellotto F, Fagiuoli S, Pavei A, Gregory SA, Cati A et al. (2005) Anemia and ischemia: Myocardial injury in patients with gastrointestinal bleeding. Am J Med 118: 548-551. [Crossref]
- Hegde N, Rich MW, Gayomali C (2006) The cardiomyopathy of iron deficiency. Tex Heart Inst J 33: 340-344. [Crossref]
- Sohn IS, Jin ES, Cho JM, Kim CJ, Bae JH et al. (2008) Bloodletting-induced cardiomyopathy: Reversible cardiac hypertrophy in severe chronic anaemia from long-term bloodletting with cupping. Eur J Echocardiogr 9: 585-586. [Crossref]
- Bregman DB, Goodnough LT (2014) Experience with intravenous ferric carboxymaltose in patients with iron deficiency anaemia. Ther Adv Hematol 5: 48-60. [Crossref]
- Kao DP, Kreso E, Fonarow GC, Krantz MJ (2001) Characteristics and outcomes among heart failure patients with anemia and renal insufficiency with and without blood transfusions (public discharge data from California 2000–2006). Am J Cardiol 107:69-73. [Crossref]
- Mazer CD, Whitlock RP, Fergusson DA, Hall J, Belley-Cote E et al. (2017) TRICS Investigators and Perioperative Anesthesia Clinical Trials Group. Restrictive or liberal red-cell transfusion for cardiac surgery. N Engl J Med 377: 2133-2144. [Crossref]
- Kidney Disease Improving Global Outcomes) (KDIGO) Anaemia Work Group (2012) KDIGO clinical practice guideline for anaemia in chronic kidney disease. Kidney Int Suppl 2: 279-335.
- Niss O, Quinn CT, Lane A, Daily J, Khoury PR et al. (2016) Cardiomyopathy with restrictive physiology in sickle cell disease. JACC: Cardiovasc Imaging 9: 243-252. [Crossref]
- Niss O, Quinn CT (2016) The Cardiomyopathy of Sickle Cell Disease. IntechOpen 5: 1-10.
- Wood JC (2016) The heart in sickle cell disease, a model for heart failure with preserved ejection fraction. Proc Natl Acad Sci U S A 113: 9670-9672. [Crossref]
- Quinn CT, Rogers ZR, McCavit TL, Buchanan GR (2010) Improved survival of children and adolescents with sickle cell disease. Blood 115: 3447-3452. [Crossref]
- Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O et al. (1994) Mortality in sickle cell disease. Life expectancy and risk factors for early death. New Eng J Med 330: 1639-1644. [Crossref]
- Lanzkron S, Carroll CP, Haywood C (2013) Mortality rates and age at death from sickle cell disease: U.S., 1979–2005. Public Health Reports 128: 110-116. [Crossref]
- Caughey MC, Ataga KI, Hinderliter AL (2016) Sickle Cardiomyopathy: The Missing Forest in the Trees. JACC. Cardiovasc Imaging 9: 253. [Crossref]
- Bakeer N, James J, Roy S, Wansapura J, Shanmukhappa S et al. (2016) Sickle cell anaemia mice develop a unique cardiomyopathy with restrictive physiology. Proc Natl Acad Sci U S A 113: E5182. [Crossref]
- Fitzhugh CD, Lauder N, Jonassaint JC, Telen MJ, Zhao X et al. (2010) Cardiopulmonary complications leading to premature deaths in adult patients with sickle cell disease. Am J Hematol 85: 36-40. [Crossref]
- Manci EA, Culberson DE, Yang YM, Gardner TM, Powell R et al. (2003) Causes of death in sickle cell disease: An autopsy study. Br J Haematol 123: 359-365. [Crossref]
- Metivier F, Marchais SJ, Guerin AP, Pannier B, London GM (2000) Pathophysiology of anaemia: Focus on the heart and blood vessels. Nephrol Dial Transplant 15: 14-18. [Crossref]
- London GM, Guerin AP, Marchais SJ, Pannier B, Safar ME et al. (1996) Cardiac and arterial interactions in end-stage renal disease. Kidney International 50: 600-608. [Crossref]
- Muller R, Steffen HM, Brunner R, Saric J, Pollok M et al. (1991) Changes in the alpha adrenergic system and increase in blood pressure with recombinant human erythropoietin (rhuepo) therapy for renal anaemia. Clin Invest Med 14: 614-622. [Crossref]
- Varat MA, Adolph RJ, Fowler NO (1972) Cardiovascular effects of anaemia. Am Heart J 83: 415-426. [Crossref]
- Lester LA, Sodt PC, Hutcheon N, Arcilla RA (1990) Cardiac abnormalities in children with sickle cell anaemia. Chest 98:1169-1174. [Crossref]
- Kushwaha SS, Fallon JT, Fuster V (1997) Restrictive cardiomyopathy. New Eng J Med 336: 267-276. [Crossref]
- Nihoyannopoulos P, Dawson D (2009) Restrictive cardiomyopathies. Eur J Echocardiogr 10: iii23–iii33. [Crossref]
- Webber SA, Lipshultz SE, Sleeper LA, Lu M, Wilkinson JD et al. (2012) Outcomes of restrictive cardiomyopathy in childhood and the influence of phenotype: A report from the paediatric cardiomyopathy registry. Circulation 126: 1237-1244. [Crossref]
- Rivenes SM, Kearney DL, Smith EO, Towbin JA, Denfield SW (2000) Sudden death and cardiovascular collapse in children with restrictive cardiomyopathy. Circulation 102: 876-882. [Crossref]
- Walsh MA, Grenier MA, Jefferies JL, Towbin JA, Lorts A et al. (2012) Conduction abnormalities in paediatric patients with restrictive cardiomyopathy. Circ Heart Fail 5: 267-273. [Crossref]
- Desai AA, Patel AR, Ahmad H, Groth JV, Thiruvoipati T et al. (2014) Mechanistic insights and characterization of sickle cell disease-associated cardiomyopathy. Circ Cardiovasc Imaging 7: 430-437. [Crossref]
- Haque AK, Gokhale S, Rampy BA, Adegboyega P, Duarte A et al. (2002) Pulmonary hypertension in sickle cell hemoglobinopathy: A clinicopathologic study of 20 cases. Human Pathol 33: 1037-1043. [Crossref]
- Martin CR, Johnson CS, Cobb C, Tatter D, Haywood LJ (1996) Myocardial infarction in sickle cell disease. J Natl Med Assoc 88: 428-432. [Crossref]
- Darbari DS, Kple‐Faget P, Kwagyan J, Rana S, Gordeuk VR et al. (2006) Circumstances of death in adult sickle cell disease patients. Am J Hematol 81: 858-863. [Crossref]
- James, TN, Riddick, L, Massing, GK (1994) Sickle cells and sudden death: Morphologic abnormalities of the cardiac conduction system. J Lab Clin Med 124: 507-520. [Crossref]
- Niss, O, Taylor, MD, Moore, R, Fleck, R, Malik, P et al. (2015) 2015 Aspho abstracts. Pediatric Blood & Cancer 62: S21-S119.
- Yawn BP, Buchanan GR, Afenyi-Annan AN, Ballas SK, Hassell KL et al. (2014) Management of sickle cell disease: Summary of the 2014 evidence-based report by expert panel members. The Journal of the American Medical Association 312: 1033-1048. [Crossref]
- Klings ES, Machado RF, Barst RJ, Morris CR, Mubarak KK et al. (2014) An official american thoracic society clinical practice guideline: Diagnosis, risk stratification, and management of pulmonary hypertension of sickle cell disease. American Journal of Respiratory and Critical Care Medicine 189: 727-740. [Crossref]
- Machado RF, Martyr S, Kato GJ, Barst RJ, Anthi A et al. (2005) Sildenafil therapy in patients with sickle cell disease and pulmonary hypertension. Br J Haematol 130:445- 453. [Crossref]
- Minniti CP, Machado RF, Coles WA, Sachdev V, Gladwin MT et al. (2009) Endothelin receptor antagonists for pulmonary hypertension in adult patients with sickle cell disease. Br J Haematol 147: 737-743. [Crossref]
- Morris CR (2006) New strategies for the treatment of pulmonary hypertension in sickle cell disease : The rationale for arginine therapy. Treat Respir Med.5:31-45. [Crossref]
- Machado RF, Barst RJ, Yovetich NA, Hassell KL, Kato GJ et al. (2011) Hospitalization for pain in patients with sickle cell disease treated with sildenafil for elevated TRV and low exercise capacity. Blood 118: 855-864. [Crossref]
- McCarthy HJ, Tizard EJ (2010) Clinical practice: Diagnosis and management of Henoch–Schönlein purpura. Eur J Pediatr 169: 643-650. [Crossref]
- Punnoose AR, Lynm C, Golub RM (2012) Henoch-Schonlein Purpura. JAMA 307: 742. [Crossref]
- Yang YH, Yu HH, Chiang BL (2014) The diagnosis and classification of Henoch–Schonlein purpura: an updated review. Autoimmunity Reviews 13: 355-358. [Crossref]
- Aalberse J, Dolman K, Ramnath G et al (2007) Henoch Schonlein purpura in children: an epidemiological study among Dutch paediatricians on incidence and diagnostic criteria. Ann Rheum Dis 66: 1648-1650. [Crossref]
- Gardner-Medwin JM, Dolezalova P, Cummins C, Southwood TR (2002) Incidence of Henoch–Schonlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet 360: 1197-1202. [Crossref]
- Chang WL, Yang YH, Lin YT, Chiang BL (2004) Gastrointestinal manifestations in Henoch–Schonlein purpura: a review of 261 patients. Acta Paediatr 93: 1427-1431. [Crossref]
- Saulsbury FT (1999) Henoch–Schonlein purpura in children. Report of 100 patients and review of the literature. Medicine 78: 395-409. [Crossref]
- Patrignelli R, Sheikh SH, Shaw-Stiffel TA (1995) Henoch–Schonlein purpura. A multisystem disease also seen in adults. Postgrad Med 97: 123-124. [Crossref]
- Tan GM (2015) A case of Henoch-Schonlein purpura with cardiopulmonary involvement. Rheumatology Reports 7.
- Bando K, Maeba H, Shiojima I (2018) IgA Vasculitis with Simultaneous Cardiopulmonary Involvement. Internal Medicine 57: 829-834. [Crossref]
- Hayakawa K, Shiohara T (2003) Two cases of Henoch-Schonlein purpura with transient myocardial ischaemia. Acta dermato-venereologica 83: 393. [Crossref]
- Carmichael P, Brun E, Jayawardene S, Abdulkadir A, O'Donnell PJ (2002) A fatal case of bowel and cardiac involvement in Henoch–Schonlein purpura. Nephrol Dial Transplant 17: 497-499. [Crossref]
- Lecutier MA (1952) A case of the Schonlein-Henoch syndrome with myocardial necrosis. J Clin Pathol 5: 336-338. [Crossref]
- Imai T, Matsumoto S (1970) Anaphylactoid purpura with cardiac involvement. Arch Dis Child 45: 727-729. [Crossref]
- Soloukides A, Moutzouris DA, Metaxatos G, Hadjiconstantinou V (2006) Pulmonary involvement in Henoch-Schonlein purpura. Emerg Med J 23: 886. [Crossref]
- Shin JI, Lee JS (2007) Severe coronary artery disease in Henoch-Schoenlein purpura: superimposition of antiphospholipid or polyangitis overlap syndrome? Int J Cardiol 122: 182-183. [Crossref]
- Ozen S, Ruperto N, Dillon MJ, Bagga A, Barron K et al (2006) EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis 65: 936–941. [Crossref]
- Sundriyal D, Gupta BB, Sharma B, Chawla MP (2013) Cardiac involvement in Henoch-Schönlein purpura. JIACM 14: 173-174.
- Joshi SK, Jadon G (2012) Introduction to neoplasm: Tumour classification a review article. Int J Adv Res Pharm Biosci 1: 227-263.
- Hulikal N, Ray S, Thomas J, Fernandes DJ (2012) Second primary malignant neoplasms: A clinicopathological analysis from a cancer centre in India. Asian Pac J Cancer Prev 13: 6087-6091. [Crossref]
- Makhija Z, Deshpande R, Desai J (2009) Unusual tumours of the heart: diagnostic and prognostic implications. J Cardiothorac Surg 4: 4. [Crossref]
- Siminelakis S, Kakourou A, Batistatou A, Sismanidis S, Ntoulia A et al. (2014) Thirteen years follow-up of heart myxoma operated patients: what is the appropriate surgical technique? J Thorac Dis 6: S32. [Crossref]
- Yuan SM (2012) Mitral valve myxoma: clinical features, current diagnostic approaches, and surgical management. Cardiol J 19: 105-109. [Crossref]
- Ojji DB, Mamven MH, Omonua O, Habib Z, Osaze H et al. (2012) Left atrial myxoma mimicking mitral stenosis. Clin Med Insights Case Rep 5: 111-114. [Crossref]
- Pinede L, Duhaut P, Loire R (2001) Clinical presentation of left atrial cardiac myxoma. A series of 112 consecutive cases. Medicine (Baltimore) 80: 159-172. [Crossref]
- Reynen K (1996) Frequency of primary tumours of the heart. Am J Cardiol 77: 107. [Crossref]
- Reynen K, Kockeritz U, Strasser RH (2004) Metastases to the heart. Ann Oncol 15: 375-381. [Crossref]
- Kanemoto N, Nishiumi N, Inoue H, Koide S, Kawada S et al. (1992) Combined apical hypertrophic cardiomyopathy and left atrial myxoma. Chest 101: 1149-1150. [Crossref]
- Coppin BD, Temple IK (1997) Multiple lentigines syndrome (LEOPARD syndrome or progressive cardiomyopathic lentiginosis). J Med Genet 34: 582‑586. [Crossref]
- Abdou M, Hayek S, Williams III BR (2013) Atrial Myxoma in a patient with hypertrophic cardiomyopathy. Tex Heart Inst J 40: 462-464. [Crossref]
- Kałużna‑Oleksy M, Stefaniak S, Oko‑Sarnowska Z, Janus M, Straburzyńska‑Migaj E (2014) Hypertrophic cardiomyopathy and left atrial myxoma. Pol Arch Med Wewn 124: 336-337. [Crossref]
- Mani A, Gopalakrishnan A, Ayyappan A, Valaparambil A (2019). Image of the month: Ventricular myxoma mimicking hypertrophic cardiomyopathy. Clin Med (Lond) 19: 131-132. [Crossref]
- Adachi H, Ohshima S, Hayashi R, Yamamoto S, Hasegawa A et al. (1987) A case of dilated cardiomyopathy associated with a left atrial myxoma. Kitakanto Medical Journal. 36: 505-510.
- Muthiah R (2016) Left Ventricular Myxoma: A Case Report. Case Reports in Clinical Medicine 5 :88.
- Chiariello GA, Bruno P, Colizzi C, Pavone N, Nesta M et al. (2018) Acute heart failure related to a large left atrial myxoma. Proc (Bayl Univ Med Cent) 31: 331-333. [Crossref]
- Raut M, Maheshwari A, Swain B (2018) Unsolved enigma of atrial myxoma with biventricular dysfunction. Ann Card Anaesth 21: 105-106. [Crossref]
- Keeling IM, Oberwalder P, Anelli-Monti M, et al. (2002) Cardiac myxomas: 24 years of experience in 49 patients. Eur J Cardiothorac Surg 22: 971-977. [Crossref]
- Goswami KC, Shrivastava S, Bahl VK, Saxena A, Manchanda SC et al. (1998) Cardiac myxomas: clinical and echocardiographic profile. Int J Cardiol 63: 251-259. [Crossref]
- Chockalingam A, Jaganathan V, Gnanavelu G, Dorairajan S, Chockalingam V (2006) Severe left ventricular dysfunction in left atrial myxoma: report of 2 cases. Angiology 57: 119-122. [Crossref]
- Currey HL, Mathews JA, Robinson J (1967) Right atrial myxoma mimicking a rheumatic disorder. Br Med J 1: 547–548. [Crossref]
- Kanda T, Takahashi T (2004) Interleukin-6 and cardiovascular diseases. Jpn Heart J 45:183-193. [Crossref]
- Marta L, Peres M, Alves M, Da Silva GS (2012) Giant left atrial myxoma presenting as acute myocardial infarction. Rev Port Cardiol 31:815-819. [Crossref]
- Bussani R, De-Giorgio F, Abbate A, Silvestri F (2007) Cardiac metastases. J Clin Pathol 60: 27-34. [Crossref]
- Hanfling SM (1960) Metastatic cancer to the heart: review of the literature and report of 127 cases. Circulation 22: 474-483. [Crossref]
- Sakai T, Tsushima T, Kimura D, Hatanaka R, Sawada M et al. (2017) Primary Lung Cancer Associated with Dilated Phase of Hypertrophic Cardiomyopathy; Report of a Case. Kyobu Geka 70: 147-150. [Crossref]
- Prichard RW (1951) Tumours of the heart: review of the subject and report of one hundred thirty-seven cases. Arch Pathol 51: 98-128. [Crossref]
- Abraham KP, Reddy VI, Gattuso P (1990) Neoplasms metastatic to the heart: review of 3314 consecutive autopsies. Am J Cardiovasc Pathol 3: 195-198. [Crossref]
- Gassman HS, Meadows R, Baker LA (1955) Metastatic tumours of the heart. Am J Med 19: 357-365. [Crossref]
- McAllister Jr HA. Fenoglio JJ Jr (1978) Tumors of the cardiovascular system. Atlas of Tumour Pathology, Fascicle 15: 1-20.
- Robinson BW, Lewis RR (1982) Myocardial metastasis from carcinoma of pancreas presenting as acute myocardial infarction. J R Soc Med 75:560-562. [Crossref]
- Kondo T, Kitazawa R, Kawata E, Mori K, Kitazawa S (2009) Diffuse cardiac lymphatic involvement by metastatic neuroendocrine carcinoma mimicking hypertrophic cardiomyopathy: a case report. Cases J 2: 9127. [Crossref]
- Chiewvit S, Pusuwan P, Chiewvit P, Pleehachinda R, Attanatho V et al. (1998) Metastatic follicular carcinoma of thyroid to pericardium. J Med Assoc Thai 81: 799-802. [Crossref]
- Clare-Salzler MJ, Van AH, Varki NM, Tillisch J (1991) Endocardial metastases of follicular thyroid carcinoma: a case report and review of the literature. Eur J Surg Oncol 17: 219-223. [Crossref]
- Hara K, Ohno M, Takenaga M, Tsuneyoshi H, Takeuchi H et al. (1986) Metastatic thyroid cancer to the right ventricle causing obstruction of the right ventricular outflow tract and associated with disseminated intravascular coagulopathy: A case report. J Cardiol 16: 765-773. [Crossref]
- Kasprzak JD, Religa W, Krzemiñska-Pakula M, Marszal-Marciniak M, Zaslonka J et al. (1996) Right ventricular outflow tract obstruction by cardiac metastasis as the first manifestation of follicular thyroid carcinoma. J Am Eoc Echocardiogr 9: 733-735. [Crossref]
- Murata S, Kobayashi A, Nakajima H (1991) Surgical resection of cardiac metastasis of thyroid cancer--a case report. Nihon Kyobu Geka Gakkai 39: 219-223. [Crossref]
- Larsimont D, Renard N, Andry G (1998) Death from myocardial metastasis of thyroid cancers (insular, papillary with undifferentiated foyers). Annales de chirurgie 52: 465-468. [Crossref]
- Mark G (1981) Papillary carcinoma of the thyroid with cardiac metastasis. Br J Clin Pract 35: 52-53.
- Fukuda A, Saito T, Imai M, Ishii K, Miwa K (2000) Metastatic cardiac papillary carcinoma originating from the thyroid in both ventricles with a mobile right ventricular pedunculated tumour. Jpn Circ J 64: 890-892. [Crossref]
- Greco A, De Masi R, Orlando S, Metrangolo A, Zecca V et al. (2017) Metastases of Hepatocellular Carcinoma Misdiagnosed as Isolated Hypertrophic Cardiomyopathy. Annals of Hepatology 16: 966-969. [Crossref]
- Rees AH, Stefadouros MA, Strong WB, Miller MD, Gilman P et al. (1978) Left ventricular performance in children with homozygous sickle cell anaemia. Br Heart J 40: 690-696. [Crossref]
- Lester LA, Sodt PC, Hutcheon N and Arcilla RA (1990) Cardiac abnormalities in children with sickle cell anaemia. Chest 98: 1169-1174. [Crossref]
- Lewis JF, Maron BJ, Castro O and Moosa YA (1991) Left ventricular diastolic filling abnormalities identified by Doppler echocardiography in asymptomatic patients with sickle cell anaemia. J Am Coll Cardiol 17: 1473-1478. [Crossref]
- Martins W, Mesquita ET, Cunha DM, Pinheiro LA, Romeo Filho LJ et al. (1999) Doppler echocardiographic study in adolescents and young adults with sickle cell anaemia. Arq Bras Cardiol 73: 463-474. [Crossref]
- Seliem MA, Al-Saad HI, Bou-Holaigah IH, Khan MN, Palileo MR (2002) Left ventricular diastolic dysfunction in congenital chronic anaemias during childhood as determined by comprehensive echocardiographic imaging including acoustic quantification. Eur J Echocardiogr 3: 103-110. [Crossref]
- Raman SV, Simonetti OP, Cataland SR, Kraut EH (2006) Myocardial ischemia and right ventricular dysfunction in adult patients with sickle cell disease. Haematologica 91: 1329-1335. [Crossref]
- Aleem A, Jehangir A, Owais M, Al-Momen A, Al-Diab A et al. (2007) Echocardiographic abnormalities in adolescent and adult Saudi patients with sickle cell disease. Saudi Med J. 28: 1072-1075. [Crossref]
- Sachdev V, Machado RF, Shizukuda Y, Rao YN, Sidenko S et al. (2007) Diastolic dysfunction is an independent risk factor for death in patients with sickle cell disease. J Am Coll Cardiol. 49: 472-479. [Crossref]
- Westwood MA, Shah F, Anderson LJ, Strange JW, Tanner MA et al. (2007) Myocardial tissue characterization and the role of chronic anaemia in sickle cell cardiomyopathy. J Magn Reson Imaging 26: 564-568. [Crossref]
- Aliyu ZY, Gordeuk V, Sachdev V, Babadoko A, Mamman AI et al. (2008) Prevalence and risk factors for pulmonary artery systolic hypertension among sickle cell disease patients in Nigeria. Am J Hematol 83: 485-490. [Crossref]
- Arslankoylu AE, Hallioglu O, Yilgor E, Duzovali O (2010) Assessment of cardiac functions in sickle cell anaemia with Doppler myocardial performance index. J Trop Pediatr 56: 195-197. [Crossref]
- Colombatti R, Maschietto N, Varotto E, Grison A, Grazzina N et al. (2010) Pulmonary hypertension in sickle cell disease children under 10 years of age. Br J Haematol 150: 601-609. [Crossref]
- Knight-Perry JE, de las Fuentes L, Waggoner AD, Hoffmann RG, Blinder MA et al. (2011) Abnormalities in Cardiac Structure and Function in Adults with Sickle Cell Disease are not Associated with Pulmonary Hypertension. J Am Soc Echocardiogr 24: 1285-1290. [Crossref]
- Abdul-Mohsen MF (2012) Echocardiographic evaluation of left ventricular diastolic and systolic function in Saudi patients with sickle cell disease. J Saudi Heart Assoc 24: 217-224. [Crossref]
- Ahmad H, Gayat E, Yodwut C, Abduch MC, Patel AR et al. (2012) Evaluation of myocardial deformation in patients with sickle cell disease and preserved ejection fraction using three‐dimensional speckle tracking echocardiography. Echocardiography 29: 962-969. [Crossref]
- Ghaderian M, Keikhaei B, Heidari M, Salehi Z, Azizi MR (2012) Tissue Doppler echocardiographic findings of left ventricle in children with sickle-cell anaemia. J Tehran Heart Cent 7: 106-110. [Crossref]
- Desai AA, Patel AR, Ahmad H, Groth JV, Thiruvoipati T et al. (2014) Mechanistic insights and characterization of sickle cell disease-associated cardiomyopathy. Circ Cardiovasc Imaging 7: 430-437. [Crossref]
- Cabrita IZ, Mohammed A, Layton M, et al (2013) The association between tricuspid regurgitation velocity and 5-year survival in a North West London population of patients with sickle cell disease in the United Kingdom. Br J Haematol 162: 400-408. [Crossref]
- Blanc J, Stos B, de Montalembert M, Bonnet D, Boudjemline Y (2012) Right ventricular systolic strain is altered in children with sickle cell disease. J Am Soc Echocardiogr 25:511-517. [Crossref]
- Caldas MC, Meira ZA, Barbosa MM (2008) Evaluation of 107 patients with sickle cell anaemia through tissue Doppler and myocardial performance index. J Am Soc Echocardiogr 21: 1163-1167. [Crossref]
- Simmons BE, Santhanam V, Castaner A, Rao KR, Sachdev N et al. (1988) Sickle cell heart disease: two-dimensional echo and Doppler ultrasonographic findings in the hearts of adult patients with sickle cell anaemia. Arch Intern Med 148: 1526-1528. [Crossref]
- Covitz W, Espeland M, Gallagher D, Hellenbrand W, Leff S et al. (1995) The heart in sickle cell anaemia: the Cooperative Study of Sickle Cell Disease (CSSCD). Chest 108: 1214-1219. [Crossref]
- Poludasu S, Ramkissoon K, Salciccioli L, Kamran H, Lazar JM (2013) Left ventricular systolic function in sickle cell anaemia: a meta-analysis. J Card Fail 19: 333-341. [Crossref]
- Nagueh SF, Appleton CP, Gillebert TC, et al. (2009) Recommendations for the evaluation of left ventricular diastolic function by echocardiography. J Am Soc Echocardiogr 22: 10733. [Crossref]
- Dragulescu A, Mertens L, Friedberg MK (2013) Interpretation of left ventricular diastolic dysfunction in children with cardiomyopathy by echocardiography: problems and limitations. Circ Cardiovasc Imaging 6: 254-261. [Crossref]
- Unzek S, Popovic ZB, Marwick TH (2011) Diastolic Guidelines Concordance Investigators. Effect of recommendations on inter-observer consistency of diastolic function evaluation. J Am Coll Cardiol Img 4: 460-467. [Crossref]
- George A, Pushkaran S, Konstantinidis DG, et al. (2013) Erythrocyte NADPH oxidase activity modulated by Rac GTPases, PKC, and plasma cytokines contributes to oxidative stress in sickle cell disease. Blood 121: 2099-2107. [Crossref]
- Mohtat D, Thomas R, Du Z, et al. (2011) Urinary transforming growth factor beta-1 as a marker of renal dysfunction in sickle cell disease. Pediatr Nephrol 26: 275-280. [Crossref]
- Tharaux PL, Hagege I, Placier S, et al. (2005) Urinary endothelin-1 as a marker of renal damage in sickle cell disease. Nephrol Dial Transplant 20: 2408-2413. [Crossref]
- Fertrin KY, Costa FF (2010) Genomic polymorphisms in sickle cell disease: implications for clinical diversity and treatment. Expert Rev Hematol 3: 443-458. [Crossref]