Coronary artery disease accounts for one-third of all deaths in the world and it is estimated that 50% of the American population will have a cardiac event in a normal lifespan. Prevention of CAD has been successful, particularly for secondary prevention. Determining who is at risk for primary prevention based on traditional risk factors (TRFs) is inadequate because the TRFs are often not present until the 6th or 7th decade. Sensitivity based on TRFs is further compromised by the recent observation that early primary prevention, based on results of Mendelian Randomization Studies, is threefold more effective. Genetic risk stratification has been assessed in multiple studies involving over one million participants and found to be superior and complementary to methods based on TRS. The upper 20% of the genetic risk score exhibits one to threefold increased risk of CAD. Stratification based on the 10-year risk, as determined by the Pooled Cohort Equation in the current guidelines, would recommend only about 45% of these individuals to receive statin therapy. Results of randomized clinical trials show genetic risk is markedly reduced by statin therapy, lifestyle changes, and physical activity. Genetic risk score has a major advantage over risk stratification based on TRFs, since it is independent of age and provides the same risk at birth as anytime later in life since one’s DNA does not change in one’s lifetime. The test, performed on blood, saliva, or tissue, is inexpensive and is available throughout the world. Statin (those off-patent), the number one drug for prevention, is inexpensive and available worldwide. Genetic risk stratification will enable the implementation of primary prevention of CAD early in life throughout the world.
Genetic risk, CAD
Coronary Artery Disease (CAD) accounts for one-third of all of the deaths in the world, which includes high, low, and middle-income countries. Despite CAD being in large part preventable, it has continued to increase and is now pandemic, affecting not only low income but high-income countries. Over 60% of the global burden of CAD occurs in low and middle-income countries [1]. Application of prevention, particularly secondary, has been very successful in the western world with over 50% reduction in cardiac mortality in the past 30 years [2]. In the meantime, CAD has increased in the developing world in large part because of living longer, adoption of a western-style diet, and improved the treatment of communicable diseases such as malaria. The reduced incidence in western countries has been mediated in large by reducing the risk associated with Traditional Risk Factors (TRF): age, obesity, hypertension, diabetes, smoking, sedentary way of life, and plasma hypercholesterolemia.
Secondary prevention, by definition, follows a cardiac event that triggers the need for modification of the TRFs to reduce future cardiac events. Primary prevention, though paramount to halting the spread of CAD, is silent and difficult to detect. This review summarizes recent data showing the risk of CAD from plasma LDL-C is accumulative and the benefit from lowering of plasma LDL-C early in life is also accumulative with a threefold greater effect than intervening later in life. The core problem is how to select asymptomatic individuals early in life that is at risk since only 50% will experience a cardiac event in a normal lifespan [2]. The clinical guidelines based on TRS are age-dependent, making them less than adequate for primary prevention. Results of recent genetic risk stratification will be discussed as a means complementary to the TRFs for the detection of those at high risk for CAD. The genetic risk score is accessible and inexpensive. A statin off-patent is inexpensive making this approach applicable to high, middle, and low-income countries.
Coronary atherosclerosis is known to initiate early in life and gradually progresses to plaque formation with possible clinical manifestation. The young men, averaging 22 years of age, who died in the Korean War [3] or Vietnam War [4], had fatty streaks which are thought to be the forerunners of coronary atheroma. In the Pathological Determinants of Atherosclerosis, in Youth Biopsies [5] performed on 2,876 individuals 15 to 34 years of age who died from non-vascular causes, had fatty streaks and more advanced lesions which increased with age and were associated with increased plasma cholesterol [6,7]. This study showed slow progression until 29 years of age, then increased twofold in women and threefold in men from 30-34 years [5]. Pencina et al. [8] in a longitudinal Framingham study observed that most individuals who have high plasma cholesterol during adolescence tend to sustain high plasma cholesterol into their 50s or 60s unless they undergo therapeutic intervention. Navar-Boggan et al. [9], also based on longitudinal Framingham studies, observed increased duration of exposure to plasma LDL-C increased the risk of CAD. This was approximated by the relationship showing CAD risk doubled for every additional 10 years of exposure. A similar log-linear relationship was observed with respect to clinical cardiac events. At age 40 the incidence of myocardial infarction is about 1%, at age 50 it is 2%, at age 60 it is 4%, at age 70 it is 8%, and at age 80 it is 16% [10,11].
The accumulative risk for CAD associated with increased exposure to LDL-C prompted Ference et al. [12] to ask the question of whether the benefit from the early lowering of plasma LDL-C would also be accumulative. To answer this question, they employed the Mendelian Randomization method utilizing several DNA variants known to be associated with decreased plasma LDL-C and a lower risk for CAD. These variants are randomly assorted at the time of conception and are not confounded by other factors affecting CAD. Individuals with these genetic variants have been exposed consistently throughout their lifetime to a lower risk for CAD. The effect of these DNA variants on cardiac events can be compared to individuals without these variants. A meta-analysis was performed for nine DNA variants associated with lower plasma LDL-C and a lower risk for CAD. The results of the long-term exposure to lower plasma LDL-C resulted in a 54% reduction in the risk of CAD for each 39 mg/dl reduction in LDL-C [12]. These results were confirmed by two other Mendelian Randomization studies showing a 57% reduction of risk for CAD per 39 mg/dL reduction in plasma LDL-C using 13 DNA variants associated with reduced LDL-C [13]. Another Mendelian Randomization study utilized 12 DNA variants associated with reduced LDL-C showed a 53% reduction in the risk of CAD for each 39 mg/dl reduction of LDL-C [14]. These results confirmed reducing plasma LDL-C early in life reduces the risk of CAD by about threefold more than observed when plasma LDL-C is reduced later in life. The lesser benefit from starting later in life is confirmed in a recent meta-analysis of 170,000 individuals involving 27 randomized clinical trials. The average exposure during these clinical trials was three to five years, and therapy was initiated in the 6th and 7th decades in a population with the mean age of 63 years. The Cholesterol Treatment Trialists’ (CTT) [15,16] collaboration concluded that reduction in plasma LDL-C by any means was associated with a 22% reduction in CAD morbidity and mortality per 39 mg/dL reduction in plasma LDL-C. This is about one-third of the benefit observed per one unit of lowering of plasma LDL-C when initiated early in life.
Ference et al. from the results of the Mendelian Randomization Studies concluded the benefit from the early lowering of plasma LDL-C on CAD risk is accumulative and follows a log-linear relationship [17,18]. This might be expected as a corollary to the accumulative increase in risk for CAD associated with increased exposure to plasma LDL-C. The earlier in life one initiates primary prevention, the greater the reduction in risk for CAD. These results offer greater hope indicating that lowering plasma LDL-C earlier in life by 77 mg/dl could potentially lower the risk of CAD by 80%. One may also conclude that to have a similar reduction later in life would require a threefold further reduction in plasma LDL-C. In the female, since CAD is delayed during the premenopausal period [19], primary prevention may be effective even if initiated in the 40s, but for males, such benefit might require initiation of therapy in the 20s or 30s.
Today, the prevention and treatment of CAD have been codified into the Current Practice Guidelines (CPG) sponsored by AHA and ACC [20]. These guidelines recommend who should receive lifestyle changes and specific therapies for primary prevention based on their risk from traditional risk factors. They have had a remarkable influence on how we practice cardiology and have contributed greatly to the 50% reduction in morbidity and mortality from CAD over the past 30 years [21,22]. These CPGs for cardiology are targeted for the age group between 40 and 75-years. The TRFs for CAD are age-dependent and are mostly manifested from the sixth decade onwards in men and a decade later in women. To identify those at highest risk from TRFs the CPG recommends using the Pooled Cohort Equations to calculate the 10-year risk of a cardiac event and if ≥ 7.5, primary prevention is appropriate including statins. To have a 10-year risk of 7.5 at middle age usually requires at least 2 risk factors. This is illustrated by the following case: A 40-year-old male or female with a plasma LDL-C of 180 mg/dL, and no other traditional risk factors, has a ten-year risk of 2.0%, which would put them into the category of no specific treatment. Today some would argue this is a missed opportunity to prevent the development of CAD and its clinical sequelae.
The recommendations developed by the CPG committee were developed thoughtfully in an attempt to maximize efficacy and safety based on the known observation that preventive measures are most effective for those at the highest risk. The average plasma LDL-C in a 40-year-old American male is 147 mg/dL, and in a female, it is 121 mg/dL [23], which is roughly twice the current recommended plasma LDL-C of ≤ 70 mg/dL. Thus, if plasma cholesterol is worthy of treatment as a sole risk factor for CAD, then everyone, of middle age or older, at least in the U.S. and probably most of the world, would be a candidate for the treatment of statin therapy if diet alone is inadequate. However, if everyone with plasma LDL-C > 70 mg/dl were treated which is every male and female over 30 years, 50% would get therapy unnecessarily since only 50% of the population is expected to experience a cardiac event during a normal life span in the U.S [2]. To implement primary prevention for those who would benefit most could be an opportunity for risk stratification using the genetic risk score as discussed in subsequent sections.
The genetic predisposition for common, chronic, polygenic disorders, such as CAD, has been claimed for decades by epidemiologists to account for 50-60% of susceptibility [24-26]. The first genetic risk variant, 9p21, for CAD was discovered simultaneously by two independent groups [27,28]. This was followed by confirmation of 9p21 as a risk factor by several groups throughout the world [29]. This stimulated the formation of international consortiums with large sample sizes of over 80,000 and its amalgamation with CARDIoGRAM with The Coronary Artery Disease (C4D) Genetics Consortium into CARDIoGRAMPlusC4D has been a leading force. The public availability of the data collected by CARDIoGRAMPlusC4D and the recent availability of data from the UK Biobank has further enhanced the pursuit of genetic predisposition of CAD [30]. The collected efforts of these consortiums and other investigators have discovered 173 genetic risk variants predisposing to CAD and have been the subject of several recent reviews [29,31-33]. These variants are said to be genome-wide significant meaning they satisfied a Bonferroni correction having a P-value of 5x10-8 and have been replicated in an independent population. There were many more associated genetic risk variants predisposing to CAD but did not fulfill the stringent Bonferroni correction. The discovery of genetic variants predisposing to CAD has provided a unique insight into its pathogenesis. Over one-half of the risk variants do not mediate their risk through traditional risk factors [29,34]. This would indicate that some of the factors causing coronary atherosclerosis are yet to be discovered. One new pathway indicated by the genetic risk variants is that of inflammation. The Interleukins [31] and the MHC risk variant [32] for CAD were presumed to mediate their risk through inflammation. This was recently confirmed by the randomized placebo-controlled clinical trial CANTOS [35,36]. This study showed inhibition of interleukin (IL-1B) was associated with a 15% reduction in cardiac events. The pursuit of the mechanism mediating the risk of the remaining genetic risk variants will offer many more surprises, and more importantly, targets will unfold for the development of novel drugs [37].
The relative increased risk per genetic risk variants for CAD is less than 10 percent [29]. The overall genetic risk burden of CAD is proportional to the number of risk variants inherited rather than any single variant and can be summarized in a single number. Utilizing blood, saliva, or tissue, one can genotype the DNA for the number of genetic variants inherited by each individual. The number of copies for each single genetic risk variant can only vary from 0 to 2. It is 0 if neither of the parents has transmitted the genetic variant, 1 if only one of the parents transmits the genetic variant and 2 if both parents transmit the genetic variant. The risk of each variant is the odds ratio determined previously for each variant in a Genome-Wide Association Study (GWAS). The risk is usually weighted by the product of the number of copies of each genetic risk variant times the natural log of the odds ratio [38]. The number resulting from the summation of all of these products is the numerical Genetic Risk Score.
The interest to use genetic variants to risk stratify for CAD increased as the number of risk variants increased. These studies were accelerated by the availability of blood samples collected from patients enrolled in large clinical trials such as Jupiter which assessed the effect of statins on cardiac morbidity and mortality. The other available specimens and phenotype data were from recently developed biobanks such as UK Biobank. An initial attempt utilizing just 12 genetic risk variants showed the additional benefit over that of risk stratifying based on traditional risk factors was small [39]. Mega et al. [40], utilizing 27 genetic risk variants for CAD, retrospectively genotyped a population of 48,421 individuals who participated previously in primary and secondary prevention clinical trials assessing efficacy and safety of statin therapy. Individuals with the highest GRS (the high genetic risk group) received the most benefit from statin therapy. It offered increased discriminatory power of that of TRFs. The number to treat with a statin was only 25 to prevent one cardiac event in the high genetic risk group. Retrospective genotyping of the 4,910 individuals who participated in the West of Scotland Coronary Prevention Study (WOSCOP) provided similar encouraging results. The group with the high GRS score had a relative risk reduction of 44% compared to 24% in others [41]. In the GRS high-risk group, the number to treat to prevent one cardiac event was only 13, as opposed to 50 individuals based on stratification with TRFs, [42]. Abraham et al. [43] genotyped 16,082 individuals utilizing a microarray with 49,310 SNPs, which confirmed genetic risk stratification has greater discriminatory power than methods based on TRFs stratification. It was also relatively independent of TRS and complementary. Inouye et al. [44] using a microchip with 1.7 million risk variants for CAD, genotyped nearly 500,000 individuals obtained from the UK Biobank. They observed that in the top 20% risk group of the GRS, there was a fourfold increased risk for CAD. Khera et al. [30] utilized a computerized algorithm, LDpred, to predict genetic variants associated with a predisposition for CAD. This resulted in a microchip with 6.6 million variants which were used to genotype a sample size of 288,278, also from the UK Biobank. They observed that 8% of the population inherited a threefold increased risk for CAD, and 0.5% inherited a fivefold risk for CAD. Investigators from both studies indicate those at high risk for CAD would not have been identified using TRFs to risk stratification for CAD. Genetic risk stratification using either 1.7 million genetic variants, or 6.6 million increased the predictive power over that of previous efforts using either 27, 50, or 49,000 genetic risk variants [30,44].
The FOURIER trial, [45] used two microchips, one containing 27, the other 6.6 million variants predisposing to CAD to genotype 14,298 individuals. Patients with the highest genetic risk score had the greatest risk and the greater benefit from the lowering of plasma cholesterol. Individuals classified at high or intermediate genetic risk for CAD had 1.23 and 1.65 fold increased hazard for major coronary events, respectively. Patients receiving Evolocumab had a 13% relative risk reduction in the group with traditional risk factors, but without high genetic risk, and 31% relative risk reduction in patients with high genetic risk regardless of traditional clinical risk factors. Retrospective genotyping of the ODYSSEY trial [46], with a sample size of 11,953, used a microarray with over 6 million SNPs. The group with the highest GRS had the highest risk for CAD and the relative reduction of cardiac events by Alirocumab was 37% in the high GRS, versus a 13% reduction in the low GRS group.
The preceding studies consistently show the GRS is superior to risk stratifying programs based on TRFs, namely, Framingham Risk Score, Pooled Cohort equations, and Reynolds score. However, two studies have shown less favorable results, Elliot et al genotyped a UK Biobank population [47] of 352,660, and Mosley et al., a U.S. population [48] of 7,237. Both studies utilized a microarray having 6 million genetic risk variants for CAD. These studies concluded the GRS as a tool to risk stratify for CAD was statistically better than traditional risk factors, but the difference was small. Reconciliation of the results of these two studies with the previous studies appears difficult. Nevertheless, the investigators concluded the GRS would be more applicable than TRFs to risk stratify for CAD in the young.
A recent study by Aragam et al. [49] brings the application of genetic risk stratification for CAD a step closer to the bedside. The genetic risk score was used to risk-stratified for CAD in 47,108 individuals. They were selected from three independent healthcare systems, operating in three separate geographic locations in the US. The mean age of participants was 60 years. The genetic risk score, based on 6.6 million genetic variants, strongly associated with CAD. Based on the GRS, the group was divided into high, intermediate, and low risks. The high-risk group, which is approximately 20% of the population, had a 1.9 fold increased risk of CAD compared to the remaining 80%. The top 5% had a 2.3 fold increased risk of CAD. The important observation with respect to primary prevention of CAD is shown in Figure 1. Only 46% of the patients classified with high genetic risk would qualify for statin therapy based on the 10-year risk calculated by PCE of the current AHA/ACC guidelines and only 44 % according to the 2016 United States Preventive Services Task Force (USPSTF) recommendations. It is perhaps somewhat surprising that PCE stratification of the top genetic risk group and the bottom 80% into low, borderline, intermediate, and high risk gave no stratification differences.
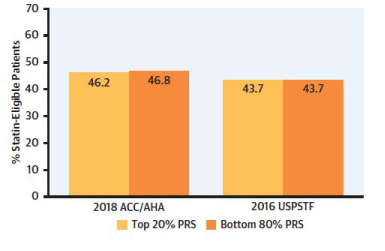
Figure 1. Shown here is a comparison of the percentage of patients that qualify for statin therapy based on the top 20% genetic risk score (PRS) versus that based on the Pooled Cohort Equation of the ACC/AHA 2018 guidelines. A similar comparison is shown between the genetic risk score and the U.S. Preventive Services Task Force (USPSTF). Among patients eligible by genetic stratification for statin therapy, whether in the top 20% or the bottom 80%, only 40% would qualify for statin therapy based on the ACC/AHA guidelines, or those by the USPSTF.
These results strongly suggest the PCE method is less-than-adequate in stratifying risk of CAD, even in those individuals of the age group of the 50s and 60s. This study confirmed the observed of multiple studies, namely, the GRS is relatively independent of the TRFs and complementary. This study showed utilizing the GRS would significantly increase the number of individuals qualifying for statin therapy as part of primary prevention in younger and older asymptomatic individuals. A particularly encouraging result of this study was showing genetic risk stratification was, as expected, effective in identifying the high-risk group in people of European descent, but also effective in Hispanics and African Americans. The odds ratio was greater for those of European descent, but still significant in Hispanic and African Americans.
It has long been considered, even among healthcare providers that what is in your genes is off-limits to treatment. This, of course, is a myth and decreasing genetic risk is the same as decreasing those resulting from lifestyle or environmental risk factors. Genes themselves live a very provincial life, is restricted to the nucleus. The plebiscites that carry out the functions of genes are the proteins. The mRNA template leaves the nucleus and attaches to the ribosomes to direct the synthesis of the proteins in the cytoplasm. Statin therapy, which inhibits the activity of the rate-limiting enzyme 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA), inhibits synthesis of cholesterol and indirectly blocks the function of the gene encoding for this enzyme. We have previously discussed several trials that show statin therapy significantly reduces genetic risk.
A randomized clinical trial performed by Khera et al. [50] consisted of 55,685 participants who genotyped prospectively with a microarray of 50 genetic risk variants predisposing to CAD. The endpoint was comparing a favorable lifestyle with that of an unfavorable lifestyle. A favorable lifestyle consisted of a healthy diet, frequent exercise, no current smoking, and a lack of obesity. An unfavorable lifestyle had at least two of these unfavorable components. Genetic risk stratification showed the top 20% of the GRS had a 91% higher risk of cardiac events than those with a low GRS. Individuals with a favorable lifestyle, in the top 20% of genetic risk, had a 40% lower risk for cardiac events than an unfavorable lifestyle.
To determine the effect of physical activity on genetic risk for CAD, Tikkanen et al. [51] genotyped 468,095 individuals. This population was provided by the UK Biobank. Physical activity was quantified on the basis of performing handgrip for three seconds, and a cardiorespiratory test of exercise on a stationary bicycle, during which oxygen consumption was quantified. Based on the genetic risk stratification, the population was dived into low, intermediate, or high. The individuals with the highest GRS had the most benefit from exercise, with a 49% lower risk for CAD.
Multiple studies indicate that risk stratification for CAD based on genetic risk variants predisposing to CAD is superior to those based on traditional risk factors. There are limitations to routine applications of the GRS. (1) It has been assessed primarily in individuals of European descent. (2) The test and validation population in most of these studies have been the same. (3) While more and more genetic risk variants are included to derive the GRS, it is likely that more are yet to be discovered. (4) The genetic risk variants are primarily tags, rather than the causal mutations.
Primary prevention currently depends on methods utilizing traditional risk factors, which are age-dependent and often not present until the 6th or 7th decade of life. Based on considerable data, including the recent Mendelian Randomization studies, early intervention such as lowering of plasma LDL-C is at least three times more effective than the same intervention later in life. Intervening in males should probably occur in the 20s or 30s, whereas females may get a similar benefit even if initiated in the 5th decade. Genetic risk stratification for CAD has now been evaluated in multiple studies involving over a million individuals. Genetic risk stratification is independent of age and provides the same risk at birth as anytime later in life since the risk is innate in one’s DNA and does not change in one’s lifetime. The risk determined by the genetic risk score has been shown to markedly decrease with statin therapy and lifestyle changes. The sensitivity of the GRS to differentiate between high and lower risk was shown to be equally applicable for primary and secondary prevention. In the recent genetic risk stratification study involving three clinical groups, over 50% of individuals in the genetic high-risk group were not recommended for statin therapy by the PCE method of the clinical guidelines of AHA/ACC. A potential solution is genetic screening followed by risk stratification based on the genetic risk score, as shown in Figure 2.
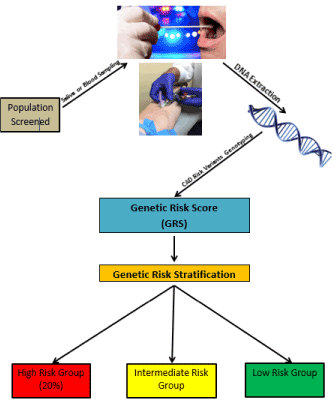
Figure 2. Shown here is a schema for genetic screening based on either blood or saliva. The DNA would be genotyped for genetic risk variants known to predispose to CAD. Based on the genetic risk score calculated from the genotyping, one can categorize the risk into high, intermediate, and low risk. Multiple studies indicate that the top 20% of genetic risk carries a one to three-fold increased risk for CAD. The percentage at high risk may vary with both geography and ethnicity.
The GRS is a simple test available throughout the world and can be performed on blood, saliva, or tissue sample. The test is relatively inexpensive, and its commercial cost is expected to be in the range of any simple blood test (100-200$). The sample can be transported across countries or continents, if necessary, to perform the genotyping from which the GRS is estimated. Therapy, such as off-patent statins, is inexpensive and available throughout the world. CAD is pandemic and continues to increase in low-income countries. In a high-income country, such as the US, one-third of all deaths are due to CAD and it is predicted that 50% of the population will have a cardiac event in their normal lifespan. The data available strongly indicates primary prevention for CAD is threefold more effective when initiated early in life. Genetic risk stratification for early primary prevention offers several advantages over screening based on TRFs. It is time for the Cardiology Clinical Guidelines to consider the incorporation of genetic risk screening for primary prevention so that those at risk can available therapy to halt the spread of CAD.
- World Health Organization (2017) Cardiovascular diseases (CVDs).
- Murray CJ, Lopez AD (2013) Measuring the global burden of disease. N Engl J Med 369: 448-457. [Crossref]
- Enos WF, Holmes RH, Beyer J (1953) Coronary disease among United States soldiers killed in action in Korea; preliminary report. J Am Med Assoc 152: 1090-1093. [Crossref]
- McNamara JJ, Molot MA, Stremple JF, Cutting RT (1971) Coronary Artery Disease in Combat Casualties in Vietnam. JAMA 216: 1185-1187. [Crossref]
- Strong JP, Malcom GT, McMahan CA, Tracy RE, Newman WP 3rd, et al. (1999) Prevalence and extent of atherosclerosis in adolescents and young adults: implications for prevention from the Pathobiological Determinants of Atherosclerosis in Youth Study. JAMA 281: 727-735. [Crossref]
- McGill Henry C, McMahan CA, Zieske AW, Tracy RE, Malcom GT, et al. (2000) Association of Coronary Heart Disease Risk Factors with Microscopic Qualities of Coronary Atherosclerosis in Youth. Circulation 102: 374-379. [Crossref]
- McGill Henry C, McMahan CA, Malcom GT, Oalmann MC, Strong JP (1997) Effects of serum lipoproteins and smoking on atherosclerosis in young men and women. The PDAY Research Group. Pathobiological Determinants of Atherosclerosis in Youth. Arterioscler Thromb Vasc Biol 17: 95-106. [Crossref]
- Pencina MJ, D’Agostino RB Sr, Larson MG, Massaro JM, Vasan RS (2009) Predicting the 30-year risk of cardiovascular disease: the framingham heart study. Circulation 119: 3078-3084. [Crossref]
- Navar-Boggan AM, Peterson ED, D’Agostino RBS, Neely B, Sniderman AD, et al. (2015) Hyperlipidemia in early adulthood increases long-term risk of coronary heart disease. Circulation 131: 451-458. [Crossref]
- Mozaffarian D, Benjamin EJ, Go Alan S, Arnett DK, Blaha MJ, et al. (2016) Executive Summary: Heart Disease and Stroke Statistics—2016 Update. Circulation 133: 447-454. [Crossref]
- Ference BA, Graham I, Tokgozoglu L, Catapano AL (2018) Impact of Lipids on Cardiovascular Health. J Am Coll Cardiol 72: 1141 -1156. [Crossref]
- Ference BA, Yoo W, Alesh I, Mahajan N, Mirowska KK, et al. (2012) Effect of Long-Term Exposure to Lower Low-Density Lipoprotein Cholesterol Beginning Early in Life on the Risk of Coronary Heart Disease. J Am Coll Cardiol 60: 2631-2639. [Crossref]
- Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, et al. (2012) Plasma HDL cholesterol and risk of myocardial infarction: a Mendelian randomisation study. Lancet 380: 572-580. [Crossref]
- Hopewell JC, Stari T, Parish S, Collins R, Clarke R (2012) Abstract 11959: The Impact of Genetic Variants Related to LDL-cholesterol on Risk of Ischemic Stroke and Coronary Heart Disease. Circulation 126: A11959-A11959.
- Cholesterol Treatment Trialists’ (CTT) Collaboration, Baigent C, Blackwell L, Emberson J, Holland LE, et al. (2010) Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 376: 1670-1681. [Crossref]
- Collins R, Reith C, Emberson J, Armitage J, Baigent C, et al. (2016) Interpretation of the evidence for the efficacy and safety of statin therapy. The Lancet 388: 2532-2561. [Crossref]
- Ference BA, Majeed F, Penumetcha R, Flack JM, Brook RD (2015) Effect of naturally random allocation to lower low-density lipoprotein cholesterol on the risk of coronary heart disease mediated by polymorphisms in npc1l1, hmgcr, or both: a 2 × 2 factorial mendelian randomization study. J Am Coll Cardiol 65: 1552-1561. [Crossref]
- Ference BA, Ginsberg HN, Graham I, Ray KK, Packard CJ, et al. (2017) Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J 38: 2459-2472. [Crossref]
- Pérez-López FR, Larrad-Mur L, Kallen A, Chedraui P, Taylor HS (2010) Gender differences in cardiovascular disease: hormonal and biochemical influences. Reprod Sci 17: 511-531. [Crossref]
- Grundy SM, Stone NJ, Bailey AL, Beam C, Birtcher KK, et al. (2018) 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 139: e1082-e1143. [Crossref]
- Lloyd-Jones DM, Larson MG, Beiser A, Levy D (1999) Lifetime risk of developing coronary heart disease. The Lancet 353: 89-92. [Crossref]
- American Heart Association (2000) Heart and Stroke Statistical Update.
- Swiger KJ, Martin SS, Blaha MJ, P. Toth P, Nasir K, et al. (2014) Narrowing sex differences in lipoprotein cholesterol subclasses following mid-life: the very large database of lipids (VLDL-10B). J Am Heart Assoc 3: e000851.
- Zdravkovic S, Wienke A, Pedersen NL, Marenberg ME, Yashin AI, et al. (2002) Heritability of death from coronary heart disease: a 36-year follow-up of 20966 Swedish twins. J Intern Med 252: 247-254. [Crossref]
- Wienke A, Holm NV, Skytthe A, Yashin AI (2001) The heritability of mortality due to heart diseases: a correlated frailty model applied to Danish twins. Twin Res 4: 266-274. [Crossref]
- Marenberg ME, Risch N, Berkman LF, Floderus b, U de Faire (1994) Genetic susceptibility to death from coronary heart disease in a study of twins. N Engl J Med 330: 1141-1146. [Crossref]
- Helgadottir A, Thorleifsson G, Manolescu A, Gretarsdottir S, Blondal T, et al. (2007) A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science 316: 1491-1493. [Crossref]
- McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, et al. (2007) A common allele on chromosome 9 associated with coronary heart disease. Science 316: 1488-1491. [Crossref]
- Assimes TL, Roberts R (2016) Genetics: implications for prevention and management of coronary artery disease. J Am Coll Cardiol 68: 2797-2818. [Crossref]
- Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, et al. (2018) Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet 50: 1219-1224.
- Erdmann J, Kessler T, Munoz Venegas L, Schunkert H (2018) A decade of genome-wide association studies for coronary artery disease: the challenges ahead. Cardiovasc Res 114: 1241-1257. [Crossref]
- Davies RW, Wells GA, Stewart AF, Erdmann J, Shah SH, et al. (2012) A genome-wide association study for coronary artery disease identifies a novel susceptibility locus in the major histocompatibility complex. Circ Cardiovasc Genet 5: 217-225. [Crossref]
- Roberts R, Chang CC (2020) A Journey through the Genetic Architecture and Predisposition of Coronary Artery Disease. Current Genomics 21: 382.
- Schunkert H, König IR, Kathiresan S, Reilly MP, Assimes TL, et al. (2011) Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet 43: 333-338. [Crossref]
- Libby P (2017) Interleukin-1 Beta as a Target for Atherosclerosis Therapy: Biological Basis of CANTOS and Beyond. J Am Coll Cardiol 70: 2278-2289. [Crossref]
- Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, et al. (2017) Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med 377: 1119-1131. [Crossref]
- Musunuru K, Kathiresan S (2019) Genetics of common, complex coronary artery disease. Cell 177: 132-145. [Crossref]
- Goldstein BA, Knowles JW, Salfati E, Ioannidis JP, Assimes TL (2014) Simple, standardized incorporation of genetic risk into non-genetic risk prediction tools for complex traits: coronary heart disease as an example. Front Genet 5: 254. [Crossref]
- Davies RW, Dandona S, Stewart AF, Chen Li, Ellis SG, et al. (2010) Improved prediction of cardiovascular disease based on a panel of single nucleotide polymorphisms identified through genome-wide association studies. Circ Cardiovasc Genet 3: 468-474. [Crossref]
- Mega JL, Stitziel NO, Smith JG, Chasman DI, Caulfield M, et al. (2015) Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. Lancet 385: 2264-2271. [Crossref]
- Natarajan P, Young R, Stitziel NO, Padmanabhan S, Baber U, et al. (2017) Polygenic risk score identifies subgroup with higher burden of atherosclerosis and greater relative benefit from statin therapy in the primary prevention setting. Circulation 135: 2091-2101. [Crossref]
- Torkamani A, Wineinger NE, Topol EJ (2018) The personal and clinical utility of polygenic risk scores. Nat Rev Gene 19: 581-590.
- Abraham G, Havulinna AS, Bhalala OG, Byars SG, De Livera AM, et al. (2016) Genomic prediction of coronary heart disease. Eur Heart J 37: 3267-3278. [Crossref]
- Inouye M, Abraham G, Nelson CP, Wood AM, Sweeting MJ, et al. (2018) Genomic risk prediction of coronary artery disease in 480,000 adults. J Am Coll Cardiol 72: 1883-1893. [Crossref]
- Marston NA, Kamanu FK, Nordio F, Gurmu Y, Roselli C, et al. Predicting benefit from evolocumab therapy in patients with atherosclerotic disease using a genetic risk score. Circulation 141: 616-623. [Crossref]
- Damask A, Steg PG, Schwartz GG, Szarek M, Hagström E, et al. (2020) Patients with high genome-wide polygenic risk scores for coronary artery disease may receive greater clinical benefit from alirocumab treatment in the odyssey outcomes trial. Circulation 141: 624-636. [Crossref]
- Elliott J, Bodinier B, Bond TA, Chadeau-Hyam M, Evangelou E, et al. (2020) Predictive accuracy of a polygenic risk score–enhanced prediction model vs a clinical risk score for coronary artery disease. JAMA 323: 636-645. [Crossref]
- Mosley JD, Gupta DK, Tan J, Yao J, Wells QS, et al. (2020) Predictive accuracy of a polygenic risk score compared with a clinical risk score for incident coronary heart disease. JAMA 323: 627-635. [Crossref]
- Aragam KG, Dobbyn A, Judy R, Chaffin M, Chaudhary K, et al. (2020) Limitations of Contemporary Guidelines for Managing Patients at High Genetic Risk of Coronary Artery Disease. J Am Coll Cardiol 75: 2769.
- Khera AV, Emdin CA, Drake I, Natarajan P, Bick AG, et al. (2016) Genetic risk, adherence to a healthy lifestyle, and coronary disease. N Engl J Med 375: 2349-2358.
- Tikkanen E, Gustafsson S, Ingelsson E (2018) Associations of fitness, physical activity, strength, and genetic risk with cardiovascular disease: longitudinal analyses in the UK Biobank study. Circulation 137: 2583-2591. [Crossref]