The objective of this work is to evaluate the implication of mutations in exon 4 of the TP53 gene in OCCs. Analysis was performed by PCR-sequencing for 24 tissues (cancerous and healthy adjacent) using Mutation Surveyor, DnaSP, MEGA, SIFT, Polyphen2, mutationTaster2 and ClinVar. A low variability of exon 4 was noted, and non-synonymous mutations were exclusively similar in the two groups of tissues. 70.83% of the mutations led to the replacement of proline by arginine, and 16.6% led to the change of glutamic acid in stop codon, which is the only pathogenic mutation. The analysis in a combined manner of mutations of this gene in cancerous and adjacent normal tissues seems to be useful for predicting the risk of recurrence.
cancer, oral cavity, TP53
Globally, oral cavity cancers (OCC) constitute the most common malignant tumors in the subcategory of head and neck cancers, but especially in developing countries [1]. They represent the 6th most common cancer in the world, with a high prevalence particularly in South Asia [2,3].
Despite current advances in surgery and other treatment options, survival rates have not improved in recent decades due to heterogeneous etiology, genetic aberrations, and poor treatment outcomes [4]. Moreover, these cancers, which used to affect only elderly men, have now become more frequent among women, but especially among young people who have no history of tobacco or alcohol consumption, which are the major risk factors for these pathologies. Therefore, this has imposed the search for other factors such as the human papillomavirus (HPV) [5-6]. HPV contributes to oral carcinogenesis through the synthesis of two proteins: E7 and E6. The latter binds and promotes the degradation of the TP53 tumor suppressor gene product [7]. This gene is widely used in the study of cancers as it is commonly mutated in human cancers and in about 50% of malignant tumors [1,8]. Several mechanisms lead to the alteration of the P53 protein response, but somatic mutations in the gene are one of the most universal mechanisms during oral carcinogenesis. Its prognostic value in OCC has long been debated. Conflicting results have been published on the presence or absence of mutations in this gene, with variations in their frequencies across OCCs [8].
In the majority of cases, oral cavity cancer is detected at an advanced stage. At this stage, they are disfiguring and painful, treatment options are reduced, and survival rates are low [9]. The key to reducing patient suffering and increasing their survival rate is early detection of pathology and, therefore, the search for alternative approaches to biopsy [10]. Molecular biology is very promising in this regard. This is a procedure that detects molecular changes well before histological ones [11]. It is in this context that this study was conducted, with the aim of determining the implication of mutations in exon 4 of the TP53 gene in OCC in Senegal.
Population study
This study was approved by the Research Ethics Committee (CER) of the Cheikh Anta Diop University (Reference: Protocol 0272/2018/CER/UCAD). The study involved 13 cancerous tissues and 11 healthy tissues collected from the Department of Stomatology and Maxillofacial Surgery of the Hospital Center University Aristide Le Dantec.
DNA extraction, polymerase chain reaction and sequencing
DNA from each tissue sample was extracted using the Zymo research kit, following the manufacturer's protocol. Exon 4 of the TP53 gene was amplified with a reaction volume of 25 μl containing: 4 μl of DNA extract; 1 μl of MgCl2; 14.4 μl of ultrapure water; 2.5 μl of 10X buffer; 0.5 μl of dNTP; 0.1 μl of Taq polymerase and 1.25 μl of each primer, which are: F 5'-TCCCCCTTGCCGTTCCAA-3’ and R 5'-CGTGCAAGTCACAGACTT-3’. PCR was performed under the following conditions: initial denaturation at 94° (5 min); repeat of 35 cycles of denaturation at 94° (30 s), hybridization at 58° (45 s) and elongation at 72° (40 s); final elongation at 72° (5 min). Sequencing reactions were performed with ABI 3730XL DNA Analyzer.
Molecular analyses
The sequences of exon 4 of the TP53 gene were thoroughly verified and corrected with BioEdit software version 7.0.5.3 using the Clustal W multiple alignment algorithm [12,13]. Chromatograms were analyzed using Mutation Surveyor software (https://softgenetics.com/mutationSurveyor.php) version 5.1.2 (SoftGenetics, 2020). The mutations found were then searched in the IARC TP53 database (https://p53.iarc.fr/TP53GeneVariations.aspx), in order to distinguish between those already described in the literature and those that are new.
Genetic diversity parameters were determined in order to evaluate the degree of gene variability in our populations. Indeed, two groups were formed and compared with each other: healthy tissues (TS) and cancerous tissues (TC). These parameters, including the number of polymorphic sites, the total number of mutations, the number of haplotypes, the average number of nucleotide differences, as well as the genetic diversity indices (haplotypic and nucleotide diversity) are evidenced by the DnaSP software version 5.10 [14]. On the other hand, the nature (transition or transversion) and rate of mutations were determined using MEGA software version 7.0.14 [15]. Amino acid changes were analyzed with MEGA software version 7.0.14 by transforming nucleotide sequences into amino acids with the choice of the best reading frame [15].
Prediction of mutational consequences was performed with the SIFT tool (https://sift.bii.a-star.edu.sg/) and the IARC TP53 database to see if an amino acid substitution would affect protein function. Similarly, the tools of MutationTaster2 (http://www.mutationtaster.org/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and the ClinVar database (https: // www.ncbi.nlm.nih.gov/clinvar/) were used to assess the pathogenicity of mutations.
Statistical tests
The association between the mutational status of TP53 exon 4 and some clinico-pathological features was analyzed by logistic regression using R software version 4.0.0 (R Core Team, 2020). These parameters are: advanced age (greater than 50 years), male gender, maturity of tumor cells, presence of lymphadenopathy, advanced stage of the disease, and tobacco or alcohol consumption. The significance of the odds ratio (OR) was assessed by Pearson's chi-square test. For all statistical tests, a p value <0.05 is considered significant.
Mutations of interest
After correction and alignment, the sequences obtained lie between codon 46 and codon 125 of the TP53 gene. Comparison of the chromatograms with the reference indicates a total of 16 heterozygous mutations, grouped into 5 types, and presented in Table 1. There is a recurrent mutation (c.215 C > G), where proline changes to arginine, present on both TS and TC with a frequency of 75% (12/16); whereas the other 4 mutations occur only once (only on TS). All 5 types of mutations were found in the IARC TP53 database, three (3) of them are non-synonymous (missense), and only the c.215 C> G mutation was recognized as a single nucleotide polymorphism (validated SNP). Chromatograms showing the nature of the mutations are shown in Figure 1.
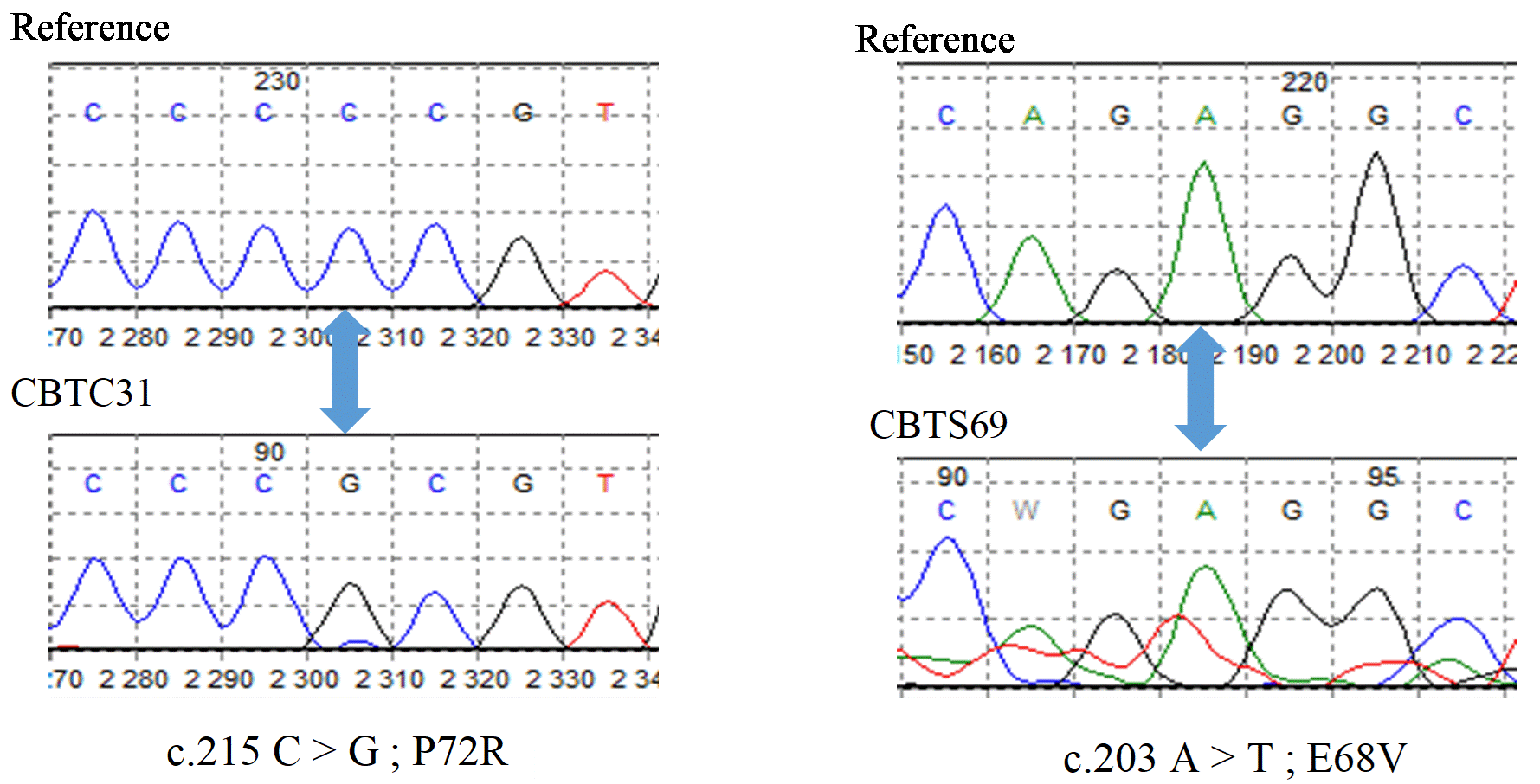
Figure 1. Nature of mutations in exon 4 of the TP53 gene. The position of the mutations is indicated by the blue arrows
Table 1. Summary of the different mutations found with Mutation Surveyor
Pos. Genome
(GRCh38.p12) |
Mutations
Pos. CDS |
Amino acids |
Nature |
Tissues Affected |
SNP |
ID
dbSNP |
chr17: 7676154 |
c.215 C > G |
P72R |
False sense |
5 TC / 7 TS |
Validated |
rs1042522 |
chr17: 7676166 |
c.203 A > T |
E68V |
False sense |
1 TS |
- |
- |
chr17: 7676164 |
c.205 G > T |
A69S |
False sense |
1 TS |
- |
- |
chr17: 7676162 |
c.207 T > G |
A69A |
Silent |
1 TS |
- |
- |
chr17: 7676135 |
c.234 A > G |
A78A |
Silent |
1 TS |
- |
rs375099397 |
Pos.: Position; GRCh38.p12: latest version of the human reference genome; chr17: chromosome 17; CDS: Coding Sequence; ID: Identifier; dbSNP: database of single nucleotide polymorphism
Nucleotide diversity
The parameters indicate little variability and very few differences between healthy and cancerous tissues (Table 2). Indeed, for both TS and TC, only 4 sites show variability out of 238 in total, and each of these sites presents a single type of substitution with the number of Eta mutations, which is equal to the number of variable sites. Among the TCs, three sites are informative. The average number of nucleotide differences is slightly higher in TS (k = 1.7) than in TC (k = 1.3). The mutation rate is zero (R = 0). This reflects the fact that all substitutions are of the transversion type (v = 100%). Genetic diversity indices of cancerous tissues show high haplotypic diversity (hd = 0.821) and very low nucleotide diversity (Pi = 0.005).
Table 2. Genetic diversity parameters for each tissue population
Parameters |
TS |
TC |
Population size (n) |
11 |
13 |
Total number of sites (N) |
238 |
238 |
Variable sites |
4 |
4 |
Informative sites |
4 |
3 |
Total number of mutations (Eta) |
4 |
4 |
Average number of nucleotide differences (k) |
1,709 |
1,333 |
Number of haplotypes (h) |
7 |
6 |
% Transition (s)/% Transversion (v) |
0%/100% |
0%/100% |
Mutation rate (R) |
0 |
0 |
Haplotypic diversity (hd) |
0.909 |
0.821 |
Nucleotide diversity (Pi) |
0.007 |
0.005 |
Diversity of amino acids
Figure 2 shows the amino acid sequence of a reference sequence (NM_000546.6) and each tissue (TS and TC). The four variable sites at the nucleotide level show an amino acid change (highlighted in yellow), with a total of 24 changes. These amino acid changes are: Proline to Arginine (P47R) at the beginning of the sequence, present in 4 TS and 3 TC (29.16%); Aspartic Acid to Valine (D48V) in 2 TS and 1 TC (12.5%); Glutamic Acid to stop codon (E68*) in 2 TC and 2 TS (16.6%); and a Proline to Arginine at codon 72 already described with the analysis of chromatograms. Thus, a substitution of proline by arginine, in duplicate for 4 tissues, is mostly found with a frequency of 70.83%. All these variants are listed in the IARC TP53 database. Their characteristics are established in Table 3.

Figure 2. Substitutions on aligned amino acid sequences
Table 3. Characteristics of substitutions on amino acid sequences
Pos. Genome
(GRCh38.p12) |
Mutations
Pos. CDS |
Amino Acids |
Nature |
Tissues
Affected |
SNP |
ID
dbSNP |
chr17: 7676167 |
c.202 G > T |
E68* |
Nonsense |
2 TC / 2 TS |
- |
rs869312782 |
chr17: 7676226 |
c.143 A > T |
D48V |
False sense |
1 TC / 2 TS |
- |
- |
chr17: 7676229 |
c.140 C > G |
P47R |
False sense |
3TC / 4 TS |
- |
rs1597375038 |
(*) stop codon
Predicting the impact of non-synonymous mutations
The transactivation function seems to be unaffected except for the P47R mutation, which could induce an increase in this function (a supertrans protein). Most of the mutations (P72R, E68V, A69S and D48V) appear to be harmless; on the contrary, the mutation inducing a stop codon (E68*) is damaging (induces the disease). The P47R mutation appears suspicious because it was predicted as possibly damaging with PolyPhen-2, but considered harmless by other software.
For the P72R and E68* variants, the impact of the mutation on protein function is not available in the IARC TP53 database. The SIFT and PolyPhen-2 tools do not predict a stop codon change, and variants without dbSNP identifiers (E68V, A69S, and D48V) are absent from the ClinVar database. All of these results are presented in Table 4.
Table 4. Effect of non-synonymous mutations on protein function and their pathogenicity
Effect on the protein |
Pathogenicity |
Mutations |
CIRC/TP53 |
SIFT
(score) |
MutationTaster 2
(probability) |
PolyPhen-2
(score) |
ClinVar |
P72R |
- |
Tolerated (0.5) |
Polymorphism (0.9) |
Benign (0.083) |
Benign |
E68V |
Functional |
Tolerated (0.25) |
Polymorphism (0.9) |
Benign (0.004) |
- |
A69S |
Functional |
Tolerated (0.73) |
Polymorphism (0.9) |
Benign (0.000) |
- |
E68* |
- |
~ |
Causative disease (1) |
- |
Pathogenic |
D48V |
Functional |
Tolerated (0.24) |
Polymorphism (0.9) |
Benign (0.322) |
- |
P47R |
Supertrans |
Tolerated (0.62) |
Polymorphism (0.9) |
Possibly damaging (0.921) |
Probably benign
(uncertain) |
(~) Automatically damaging because of the stop codon
Relationship between mutations and epidemiological characteristics
The analysis of the association between the presence of at least one mutation and the clinico-pathological parameters shows that there would be no significant association between them, with P values greater than 0.05. The same is true for the potentially damaging mutations (P47R and E68*) and the P72R polymorphism, taken separately. These results are reported in Table 5.
Table 5. Association between mutation status and some clinico-pathological parameters
Parameters |
Mutants
OR (P-value) |
P72R
OR (P-value) |
P47R
OR (P-value) |
E68*
OR (P-value) |
Gender (M) |
3.93e+8 (0.99) |
4.94e-9 (0.99) |
6.60e+7 (0.99) |
3.6 (0.32) |
Age (> 50 years) |
0.37 (0.43) |
1.25 (0.83) |
2.5 (0.48) |
0.8 (0.83) |
Maturity (mature) |
4.70e-8 (0.99) |
7.05e-8 (0.99) |
7.83e-9 (0.99) |
1.41e-8 (0.99) |
ADP (presence) |
0.61 (0.72) |
0.86 (0.92) |
1.94e-8 (0.99) |
6.80e+7 (0.99) |
Stage (advanced) |
0.43 (0.53) |
3.75e-1 (0.39) |
6 (0.18) |
0.37 (0.39) |
Tobacco (positive) |
3.85e+7 (0.99) |
6 (0.29) |
3.85e+7 (0.99) |
6 (0.29) |
Alcohol (positive) |
1.42e+7 (0.99) |
8.50e+7 (0.99) |
1.16e+8 (0.99) |
8.51e+7 (0.99) |
OR: Odds Ratio; ADP: Lymphadenopathy
Gene variability was studied by comparing via parameters of genetic diversity the nucleotide sequences of cancerous tissues to adjacent tissues assumed to be healthy. The results revealed very few differences between the two tissue groups. The search for mutations by chromatogram analysis showed that 4/5 of the mutations are found only in the TS; similarly, the average number of nucleotide differences is slightly higher at the TS level (1.7 against 1.3 for the TC). All of these results could raise doubts about the implication of the gene in OCCs, or suggest that healthy tissue samples would not really be involved. However, Singh, et al. [4] also found a higher percentage (52.2%) of cases with mutations in adjacent normal tissues when working on the TP53 gene including exon 4. Mutations in histologically normal adjacent mucosa are believed to increase the risk of local recurrence [16]. Indeed, Braakhuis, et al. [17] demonstrated this by attempting to genetically explain the concept of the "carcinogenesis field", a process described in several organ systems including the head and neck (oral cavity, oropharynx and larynx). This is a histologically normal epithelial area around the tumor site, but with genetic alterations of which those of TP53 constitute an early marker in the process. Moreover, even after removal of the primary tumor, this area could evolve into a cancer later on. However, this step would require alteration of other additional genes such as CCND1, CDKN2A, and EGFR [17-18]. Thus, the development of a field of carcinogenesis is a possible cause of the presence of mutations in adjacent healthy tissue, which could therefore be considered pre-neoplastic.
Genetic diversity indices, showing high haplotypic diversity and very low nucleotide diversity, indicate rapid evolution of neoplastic and pre-neoplastic cells from a small ancestral population. For a fairly high proportion of cases in the oral cavity and other cancers, it has been conclusively demonstrated that there is a common clonal origin of the cancer cells [17]. Evidence for this monoclonal origin is based on the similarity of genetic changes [18]. Indeed, the exact same types of mutations were found in both groups of tissues when looking at the amino acid changes. This further proves the possibility of adjacent healthy tissue evolving into cancer.
Exon 4 is the only one in TP53 that partially encodes 3 functionally important domains of the protein [19]. With a low total number of mutations (Eta = 4), this study reveals that exon 4 is very little variable. It has been shown that almost all TP53 mutations observed in head and neck cancers (HNCs) occur primarily at the DNA binding domain in exons 5 to 9 [20]. Based on these preliminary data, most researchers have limited their analyses to these exons. However, in the study by Singh, et al. [4] on oral cancer where exons 4 to 9 were analyzed, the detected mutations were predominantly grouped in exon 4 (39.1%). Possible explanations for the discrepancy in mutation rate between the Singh, et al. [4] study carried out in India and this one could be ethnic and geographic factors, etiological factors (more smokers), but also the difference in sample size.
All nucleotide substitutions found were of the transversion type. They cause greater changes in DNA structure, and are more likely to result in amino acid substitution [21]. This could explain the fact that almost all of these transversions lead to missense (non-synonymous) mutations, which is in line with previous studies, since it is well known that the majority of TP53 mutations in cancers are missense mutations [22-24]. The superiority of non-synonymous mutations demonstrates that exon 4 of the TP53 gene is under positive selection. Thus, a proliferative advantage is conferred on cancer cells with mutations in this gene, which further implicates it in the evolution of oral cancer.
The C > G transversion is mostly found among the substitutions (70.83%), and leads to the replacement of proline by arginine at codons 47 and 72. 9/24 tissues (37.5%) present this change only once and 4/24 (16.6%) present it twice. Thus, the increase of Arginine at the expense of Proline level could be significant regarding the early detection of the disease. The mutation at codon 72, which is located in the proline-rich region involved in apoptosis, was the most frequent of all mutations found in this study, as in that of Batta & Pandey for the TP53 gene [25,26]. This variant (P72R) had already been classified as a polymorphism of the gene, and the mutant R72 allele does not have a significant role in the susceptibility of several cancers, including those of the mouth as shown by mutation prediction results [27].
Regarding the other C > G transversion at codon 47 (P47R), based on functional assays performed by Kato, et al. [22], the IARC TP53 database predicts that this mutation would have the ability to increase the transactivation activity of P53 (supertrans protein). Theoretically, this phenotype could be due to subtle variations in DNA binding affinity/specificity, higher tetramer stability, or weaker interactions with negative regulators [28]. Similarly, Shi, et al. [29] found at the same codon, the P47S mutation which also induces a supertrans protein. This codon is located at the second transactivation domain of the TP53 protein, but other supertrans mutants were found at the DNA binding domain, and were more efficient at inducing growth inhibition and apoptosis compared to the normal protein [25,30]. Thus, these supertrans alleles may prove useful in cancer gene therapy protocols aimed at restoring the function of TP53 to tumor cells.
Another transversion (G > T) present in 2 TCs and 2 TSs where a stop codon replaces the glutamic acid at position 68 (E68*), leading to a truncated protein, again underlines the essential role of TP53 in carcinogenesis. Mutations leading to a truncated protein have already been found on TP53 in several HNC studies, with either nonsense or reading frame shift mutations being the source [17,23,29]. Shi, et al. [29] found 5 mutations giving a stop codon and, in 3 cases, it was glutamic acid that was replaced as in the present study. The presence of a stop codon makes this mutation automatically detrimental to the functioning of the protein due to the truncation of the latter, and consequently would induce the pathology, as shown by the prediction results. Furthermore in HNCs, a TP53 truncating mutation is associated with a poor prognosis for patient survival [23].
In addition, for certain types of cancers, evidence has been obtained that TP53 mutations are associated with worse clinical outcomes. However, in this case, no significant association was found between the mutations and the clinico-pathological parameters. Likewise, in the study by Singh, et al. [4], no significant association was found between mutations and these parameters, except for small mutant tumors (less than 4 cm), where there was a significant risk of recurrence. However, the number of patients in the present study is small to draw conclusions about any association between these two parts. It should also be noted that there was generally low exposure to tobacco smoke or alcohol regarding etiology.
This study showed that mutations in exon 4 of the TP53 gene generally demonstrate incomplete penetrance. Indeed, of the 13 TCs, 8 carry the mutation (61.54%), a frequency not far from what has been previously described: 75.6% [24]. Moreover, of the 6 non-synonymous mutations, the one giving a truncated protein is the only one that is sure to inactivate the protein and therefore to be pathogenic; and only 15.38% of CTs (2/13) present this mutation. However, it has been shown that multiple mutations are required to inactivate TP53 transactivity and that most are in the DNA binding domain [22]. Also, the implication of other genes seems obvious, since the alterations in TP53 concern a priori only in the early stages of the carcinogenesis process [17-18].
Genetic analyses revealed low variability in exon 4 of the TP53 gene, with the presence of similar mutations in healthy and cancerous tissues. Despite the recurrence of the replacement of proline by arginine, the penetrance of mutations in this gene is incomplete. The change of the glutamic acid in the stop codon, resulting in a truncated protein, is the only one that is pathogenic despite its low frequency. These two mutations could constitute molecular markers in OCCs, and possibly contribute to early diagnosis.
- Ragos V, Mastronikolis NS, Tsiambas E, Baliou E, Mastronikolis SN, et al. (2018) P53 mutations in oral cavity carcinoma. J BUON 23: 1569-1572. [Crossref]
- Ghanghoria S, Ghanghoria A, Shukla A (2015) P53 Expression in oral cancer: A study of 50 cases. J pathol Nepal 5: 747-751.
- Su SC, Lin CW, Liu YF, Fan WL, Chen MK, et al. (2017) Exome sequencing of oral squamous cell carcinoma reveals molecular subgroups and novel therapeutic opportunities. Theranostics 7: 1088-1099. [Crossref]
- Singh RD, Patel KR, Patel PS (2016) P53 mutation spectrum and its role in prognosis of oral cancer patients: a study from Gujarat, west India. Mutat Res 783: 15-26. [Crossref]
- Dao A, Benchakroun N, Bouchbika Z, Tawfiq N, Jouhadi H, et al. (2012) Epidemiopathological profile of squamous cell carcinomas of the ENT sphere in young subjects. Cancer/Radiother 16: 546-547.
- Dieng MM, Dem A, Gaye PM, Diouf D, Touré S, et al. (2012) Cancers of the oral cavity: about 145 cases at the Joliot-Curie Institute in Dakar. Cancer/Radiother 16: 547.
- Rivera C (2015) Essentials of oral cancer. Int J Clin Exp Pathol 8: 11884-11894. [Crossref]
- Patel KR, Vajaria BN, Singh RD, Begum R, Patel PS (2018) Clinical implications of P53 alterations in oral cancer progression: A review from India. Exp Oncol 40: 10-18. [Crossref]
- Dzebo S, Mahmutovic J, Erkocevic H (2017) Quality of life of patients with oral cavity cancer. Mater Sociomed 29: 30-34. [Crossref]
- Bagan J, Sarrion G, Jimenez Y (2010) Oral Cancer: Clinical Features. Oral Oncol 46: 414-417. [Crossref]
- Jurel SK, Gupta DS, Singh RD, Singh M, Srivastava S (2014) Genes and oral cancer. Indian J Hum Genet 20: 4-9. [Crossref]
- Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser 41: 95-98.
- Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22: 4673-4680. [Crossref]
- Librado P, Rozas J (2009) DnaSP version 5: a software for comprehensive analysis of DNA polymorphism data. Bioinformation 25: 1451-1452.
- Tamura K, Stecher G, Kumar S (2016) MEGA7: Molecular Evolutionary Genetics Analysis version 7.0.14 for bigger datasets. Mol Biol Evol 33: 1870-1874. [Crossref]
- Thode C, Bilde A, Von Buchwald C, Dabelsteen E (2010) TP53 mutations in clinically normal mucosa adjacent to oral carcinomas. J Oral Pathol Med 39: 662-666. [Crossref]
- Braakhuis BJ, Tabor MP, Kummer JA, Leemans CR, Brakenhoff RH (2003) A genetic explanation of Slaughter’s concept of field cancerization: evidence and clinical implications. Cancer Res 63: 1727-1730. [Crossref]
- Leemans CR, Braakhuis BJM, Brakenhoff RH (2011) The molecular biology of head and neck cancer. Nat Rev Cancer 11: 9-22. [Crossref]
- Bukovac A, Kafka A, Hrašcan R, Vladušic T, Pecina-Šlaus N (2019) Nucleotide variations of TP53 exon 4 found in intracranial meningioma and in silico prediction of their significance. Mol Clin Oncol 11: 563-572. [Crossref]
- Bahethi RR, Stepan KO, Pinotti R, Li R, Agrawal N, et al. (2020) Genetic mutations in young nonsmoking patients with oral cavity cancer: A systematic review. OTO Open 4: 2473974X20970181. [Crossref]
- Guo C, McDowell IC, Nodzenski M, Scholtens DM, Allen AS, et al. (2017) Transversions have larger regulatory effects than transitions. BMC Genom 18: 394. [Crossref]
- Kato S, Han SY, Liu W, Otsuka K, Shibata H, et al. (2003) Understanding the function–structure and function–mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci USA 100: 8424-8429.
- Lindenbergh-van der Plas M, Brakenhoff RH, Kuik DJ, Buijze M, Bloemena E, et al. (2011) Prognostic significance of truncating TP53 mutations in head and neck squamous cell carcinoma. Clin Cancer Res 17: 3733-3741. [Crossref]
- Zhou Ge, Liu Z, Myers JN (2016) TP53 mutations in head and neck squamous cell carcinoma and their impact on disease progression and treatment response. J Cell Biochem 117: 2682-2692. [Crossref]
- He F, Borcherds W, Song T, Wei X, Das M, et al. (2019) Interaction between P53 N terminus and core domain regulates specific and nonspecific DNA binding. PNAS 16: 8859-8868.
- Batta N, Pandey M (2019) Mutational spectrum of tobacco associated oral squamous carcinoma and its therapeutic significance. World J Surg Oncol 17:198. [Crossref]
- Olivier M, Hollstein M, Hainaut P (2010) TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol 2: a001008. [Crossref]
- Inga A, Monti P, Fronza G, Darden T, Resnick MA (2001) P53 mutants exhibiting enhanced transcriptional activation and altered promoter selectivity are revealed using a sensitive, yeast-based functional assay. Oncogene 20: 501-513. [Crossref]
- Shi Q, Xiao K, Wei W, Zhang BY, Chen C, et al. (2013) Associations of TP53 mutations, codon 72 polymorphism and human papillomavirus in head and neck squamous cell carcinoma patients. Oncol Rep 30: 2811-2819. [Crossref]
- Menendez D, Inga A, Resnick MA (2006) The biological impact of the human master regulator p53 can be altered by mutations that change the spectrum and expression of its target genes. Mol Cell Biol 26: 2297-2308. [Crossref]
Editorial Information
Editor-in-Chief
Terai Masanori
Tokyo Ariake University of Medical and Health Sciences
Article Type
Research Article
Publication history
Received: October 05, 2021
Accepted: October 25, 2021
Published: November 05, 2021
Copyright
©2021 Diatta H, Mbaye F, Gueye MD, Ndiaye MM, Samb MD. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation
Diatta H (2021) Genetic mutations of exon 4 at TP53 gene in oral cavity cancers. Oral Health Care 6: DOI: 10.15761/OHC.1000201.