Objective: To determine if brain serotonergic activity increase induced by the treatment with fluoxetine plus metformin can decrease insulin resistance (IR) in adolescents with metabolic syndrome (MetS).
Methods: A quasi-experimental study was conducted in 40 adolescents with MetS and IR. IR was determined through homeostatic model assessment (HOMA). After IR was determined in MetS patients, treatment with fluoxetine and metformin was started and continued for 20 weeks. At the beginning and at the end of treatment, all patients had L-tryptophan free fraction (FFT), glucose and insulin plasma levels determined, as well as HOMA, lipid profile and intensity-dependent auditory-evoked potentials (IDAEPs) in order to measure brain 5-HT activity.
Results: At baseline, the adolescents had obesity, hyperglycemia, hyperinsulinemia, IR, dyslipidemia and decreased FFT, as well as a steeper AFS slope of the N1/P2 component of IDAEPs. The treatment with Fluoxetine and metformin reduced body weight, glucose, insulin, triglycerides and LDL-cholesterol and caused an increase in plasma FFT and decrease in the slope of the N1/P2 component of IDAEPs. Interestingly, the treatment also decreased IR at 20 weeks.
Conclusion: This work shows that the combined treatment with fluoxetine and metformin decreases insulin resistance concurrently with an increase in brain serotonergic metabolic and functional activity, expressed by an increase in plasma FFT and a decrease in the N1/P2 ASF slope of the IDAEPs in patients with MetS.
insulin resistance, metabolic syndrome, N1/P2 component of evoked auditory potential, brain serotonergic activity
Much of the research on metabolic syndrome (MetS) has focused on the relationships between its cardinal clinical features: insulin resistance (IR), excess abdominal adipose tissue, elevated blood pressure, lipid abnormalities and atherosclerosis [1,2]. Overeating and sedentary lifestyle is generally recognized to contribute to its development [3,4]. MetS research has also been focused on the different molecular and neurobiological mechanisms involved in its pathophysiology and etiology. The Central Nervous System, in addition to regulating health-related behavior, modulates the body’s metabolic processes through autonomic and neuroendocrine pathways. MetS has been associated with chronic activation of the hypothalamic-pituitary-adrenal axis [5,6]. Furthermore, there is evidence that has linked brain serotonergic activity with peripheral insulin sensitivity [7]. MetS, IR, type 1 and 2 diabetes are conditions that have been shown to exhibit a significant decrease in brain serotonergic neurotransmission [8-11]. Abdominal obesity and type 2 diabetes have also been associated with genetic variations of two serotonergic receptors (5-HT2A and 5-HT2C) [12,13]. The brain serotonergic system has several neuroanatomic and functional features that suggest its involvement in the pathophysiology of MetS. Serotonergic neurons are localized in the raphe nuclei of the brainstem and are connected to the cerebral cortex, hypothalamus, and major autonomic nuclei, where they exert broad regulatory control. Serotonergic activity has been shown to regulate several behaviors, including nutrition, locomotion, reproduction, sleep, pain, aggression and stress response [14], as well as other autonomic functions, such as thermoregulation, cardiovascular control, circadian rhythms and pancreatic function [15-17].
Recently, patients with MetS have been shown to exhibit a significant increase in the auditory cortical response recorded through an increase in the ASF slope of the N1/P2 component of IDAEPs as a result of low brain serotonergic activity [8]. More specifically, a strong increase in auditory-evoked cortical responses to increasing auditory stimuli intensities reflects low serotonergic activity, as it has been shown in patients with type 1 and 2 diabetes mellitus [9-11], whereas low-intensity dependence is assumed to result from an increased serotonergic function, as we have previously observed in intrauterine growth-restricted infants and rats [18,19]. There are data indicating that patients with MetS often have IR, a precursor of altered glucose tolerance that plays a key role in the pathophysiology of the syndrome as a result of complex molecular and cellular mechanisms, which, together with JNK (c-jun kinase activator) and several other mechanisms, leads to inflammation, which, in turn, damages adipose tissue by disturbing its function and its role in insulin signaling recognition [20-23]. IR in these patients with MetS has been treated with diet, sedentary lifestyle changes and exercise, and there is controversy regarding the use of metformin for their management [24,25]. Metformin is a complex drug with multiple sites of action and multiple molecular mechanisms. Physiologically, metformin acts directly or indirectly in the liver to lower glucose production, and also acts in the gut to increase glucose utilization, increase GLP-1 and alter the microbiome. At the molecular level, metformin inhibits the mitochondrial respiratory chain in the liver, leading to AMPK activation, enhancing insulin sensitivity (via effects on the fat metabolism) and lowering cAMP, thus reducing the expression of gluconeogenic enzymes. Metformin also has AMPK-independent effects in the liver that may include fructose-1,6-bisphosphatase inhibition by AMP [26]. On the other hand, fluoxetine is a selective serotonin reuptake inhibitor (SSRI), and it exerts its therapeutic effect by inhibiting the presynaptic reuptake of the neurotransmitter serotonin. As a result, 5-hydroxytryptamine (5-HT) levels are increased in different parts of the brain [27]. Bearing in mind all this information, and given that there is a close relationship between peripheral insulin sensitivity and brain serotonergic activity in patients with MetS [7,28,29], the purpose of this work was to determine whether the increase in brain serotonergic activity induced by fluoxetine together with glucose metabolism stimulation by adding metformin to the treatment would result in an improvement in IR decrease and normalize other anomalies associated with the syndrome in Mexican adolescents with MetS.
The study was approved by the research and ethics committees of the Health Research Coordination, Mexican Institute of Social Security, Mexico City. All parents of participating patients provided written informed consent after they were fully informed on the study procedures. A quasi-experimental study was carried out in 40 adolescents of both genders with MetS and IR. Average age was 14.64 ± 2.85 years. The MetS diagnosis was established according to the National Cholesterol Education Program (NCEP) and to the International Diabetes Federation (IDF) criteria, modified for children and adolescents [30]. IR was determined using the homeostatic model assessment (HOMA) [31]. The cutoff point established to consider IR was 3.59, according to Arellano-Ruiz et al [32]. All patients were treated with metformin 1,500 mg/day, divided into three doses administered with meals, and with fluoxetine at 20 mg/day, for 20 weeks. At the beginning and at the end of treatment, the body mass index (BMI) was determined in all patients. In addition, they had 5 mL of blood extracted from a peripheral vein, which was placed in borosilicate tubes with 450 µL of an ACD solution (3.6 mg sodium citrate, 9.9 mg citric acid, 11 mg dextrose, buffered with 50 mM Tris base, pH 7.40). The blood samples were obtained at between 07:00 and 08:00 h after a 12-h fast. The tubes with the blood samples were immediately cooled to 4 ºC and then centrifuged at 500 g for three minutes in an Avanti J-31 refrigerated centrifuge (Beckman Instruments, Fullerton, CA) to obtain the plasma. Plasma aliquots were taken for the different biochemical assays: FFT and total L-tryptophan (L-Trp), glucose, cholesterol, triglycerides, HDL-cholesterol, LDL-cholesterol and insulin, as well as the HOMA index. AEPs were also determined. No clinical signs or symptoms of other pathologies were observed in any of the patients. Exclusion criteria were the use of medications such as sedatives, antidepressants and neuroleptics. All patients included in the study had normal hearing (perception up to 20 dB confirmed by an audiologist), as well as cognitive functions.
Biochemical assays
Ultra-filtered plasma fractions were obtained (Nanosep 30 K Omega Pall Life Science, Ann Arbor, MI), from which the free fraction of L-Trp (FFT) was recovered; plasma pH changes were prevented by adding tris acetate buffer (50 mM, pH 7.40). High-performance liquid chromatography (HPLC) analyses were performed using Johansen et al. fluorescence method to quantify FFT and total L-Trp [33]. Plasma glucose, cholesterol, triglycerides and HDL- and LDL-cholesterol were quantified using a Flex® reagent cartridge (Dade Behring Inc, Newark, DE 19714, USA), which uses an enzymatic colorimetric method. Insulin was determined by chemiluminescence and HbA1C by reflectometry.
Auditory evoked potentials recording
Recording took place in an electrically shielded and sound-attenuated room adjacent to the recording apparatus Viking 4 (Nicolet Viking 4, Madison, WI). Adolescents were seated in a comfortable chair with a headrest. Evoked responses were recorded with two channels referred to vertex (Cz). AgCl electrodes were used (EEG disk electrode NE-101, 10-mm diameter). A total of 200 tones (1 kHz, 100 msec duration with 10 msec rise and 10 msec fall time, interstimulus interval between 1000 and 1500 msec) with three intensities (70, 90, or 103 dB) were used to assess intensity-dependence. Intensities were each separately binaurally presented in a sequential form using headphones. Data were collected with a sampling rate of 1000 Hz and an analogous bandpass filter (0.1-150 Hz). Pre-stimulus periods (200 msec) and post-stimulus periods (500 msec) were evaluated with 200 sweeps at each intensity. For the suppression of artifacts, all assays were automatically excluded from averaging if the voltage exceeded 50 µV in either of the two channels at any time during the averaging period. X-Y graphs of the AEPs were examined, and prominent peaks were identified and measured using a specific software (Viking 4, Nicolet). Plots shown in (Figure 1) are representative examples of AEPs long latency waves that were obtained with sequential stimulations of 70, 90 or 103 dB from adolescents with MetS during treatment with fluoxetine and metformin at (A) Start of treatment and, (B) 20 weeks of treatment. Latencies were also calculated in milliseconds and amplitudes in µV. The amplitude of the AEP N1/P2 component was considered as the sum of the crests of the N1 and P2 waves in µV. The N1 component of the individual dipole source is measured as the negative peak within 60-120 msec, and the P2 component is measured as the positive peak within 110-210 msec. These components are regarded as being representative of the auditory cortex integrative function, whose regulation is associated with cortical serotonergic innervation [34].
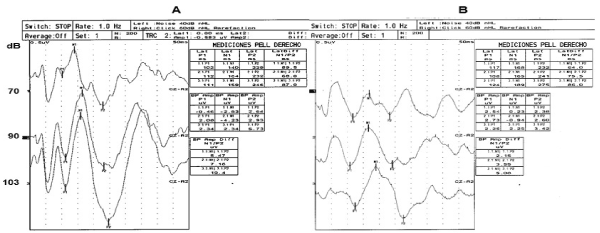
Figure 1. Illustrative examples of cortical auditory-evoked potentials (200 average responses) obtained at separate stimulation with 70, 90 or 103 dB sound pressure level. A) Treatment Start, (B) 20 weeks of treatment. Peak-to-Peak amplitude of the N1/P2 component was measured in this study. (Reproducibility tested by Levene and CV tests)
Average values and standard deviations were obtained for the results of clinical and biochemical data, the difference between the start and end of treatment with fluoxetine and metformin was established by Student’s t-test, with a P-value < 0.05 being considered significant. Peak-to-peak amplitude of the N1/P2 component was measured at 70-, 90- and 103-dB stimulus intensities, and the ASF slope was calculated at start and end of treatment with fluoxetine and metformin by linear regression analysis with a significance level of P < 0.05.
In general, the adolescents with MetS had obesity during the study period. Obesity was more evident at treatment initiation (P < 0.05) (Table 1). In table 1, body weight and body mass index are observed to have significantly decreased at end of metformin and fluoxetine treatment (P < 0.05). Blood glucose, as well as plasma insulin, HbA1c, triglycerides and LDL-cholesterol were significantly elevated in the adolescents at the beginning of treatment (Table 2). However, it is important to note that blood glucose, HbA1c, HDL-cholesterol and insulin resistance (HOMA) levels had significantly decreased at 20 weeks of treatment with metformin and fluoxetine in these adolescents (P < 0.05) (Table 2). Among the most important parameters measured in these adolescents were the different fractions of L-Trp in plasma. A significant increase in FFT was observed at 20 weeks of treatment with metformin and fluoxetine in comparison with the levels the adolescents had at the beginning of treatment (P < 0.5) (Figure 2). As for total L-Trp, there were no differences during the study period.
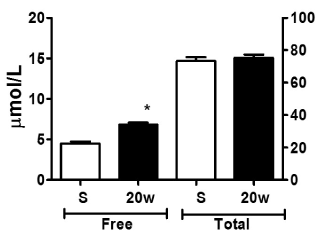
Figure 2. L-Tryptophan concentration in plasma. Each bar represents the mean value ± S.D. S) Treatment start, n = 40 and 20w) 20 week of treatment, n = 33. All determinations were made by duplicate. Differences were determined by Student-t test. * P < 0.05
Table 1. Clinical data of adolescents with metabolic syndrome and insulin resistance
Treatment time
(weeks) |
Start
n = 40 |
CV |
20 weeks
n = 33 |
CV |
Age (years) |
15.21 ± 2.5 |
0.16 |
15.71 ± 3.0 |
0.19 |
Sex
Male
Female |
22
18 |
|
18
15 |
|
Body weight (Kg) |
87.93 ± 8.9 |
0.10 |
80.62 ± 7.36* |
0.9 |
Length (m) |
1.64 ± 0.02 |
0.01 |
1.64 ± 0.17 |
0.1 |
Body mass index |
31.06 ± 0.84 |
0.02 |
28.64 ± 0.70* |
0.02 |
Waist circumference (cm) |
107.7 ± 14.0 |
0.13 |
98.90 ± 10.0 |
0.10 |
Blood pressure
Systolic (mm Hg)
Diastolic (mm Hg) |
115 .1 ± 12.1
71.6. ± 10.8 |
0.10
0.15 |
111.0 ± 8.0
69.60 ± 10.8 |
0.07
0.15 |
Each point represents the mean value ± S.D. Differences were determined by Student-t test. * P < 0.05, CV = Coefficient of variation.
Table 2. Biochemical data of adolescent with metabolic syndrome and insulin resistance
Treatment time (weeks) |
Start
n = 30 |
CV |
- weeks
n = 23 |
CV |
Glucose mg/dl |
106.10 ± 4.86 |
0.04 |
86.88 ± 1.74* |
0.02 |
HbA1c % |
5.36 ± 0.15 |
0.02 |
4.89 ± 0.90* |
0.18 |
Insulin µU/ml |
18.51 ± 1.06 |
0.05 |
10.95 ± 1.42** |
0.12 |
HOMA |
4.74 ± 0.35 |
0.07 |
2.76 ± 0.17** |
0.06 |
Cholesterol
mg/dl |
173.6 ± 5.32 |
0.03 |
170.4 ± 6.1 7 |
0.03 |
Triglycerides mg/dl |
198.0 ± 16.28 |
0.08 |
160.3 ± 14.80* |
0.09 |
HDL-cholesterol
mg/dl |
39.07 ± 1.86 |
0.04 |
44.73 ± 2.52* |
0.05 |
LDL-cholesterol
mg/dl |
100.5 ± 4.19 |
0.04 |
88.51 ± 4.76* |
0.05 |
Each point represents the mean value ± S.D. Difference were determined by Student-t test. * P < 0.05, CV = Coefficient of variation
Brain serotonergic activity was evaluated through the change in amplitude of the N1/P2 component of IDAEPs. The slopes calculated with these amplitudes provide a function that relates the amplitude in µV of the N1/P2 component to the stimulus intensity (ASF slope). Noteworthy, the ASF slope (-0.11 + 4.86 intensity, r2 =0.98) at start of treatment showed a significant decrease at 20 weeks of treatment with metformin and fluoxetine (-0.06 + 1.86, r2 = 0.97 (p < 0.001) (Figure 3). P1, N1 and P2 latencies (msec) showed a tendency to increase with auditory stimulus intensity in the patients, although it should be noted that latencies were similar at start and end of treatment (Table 3).
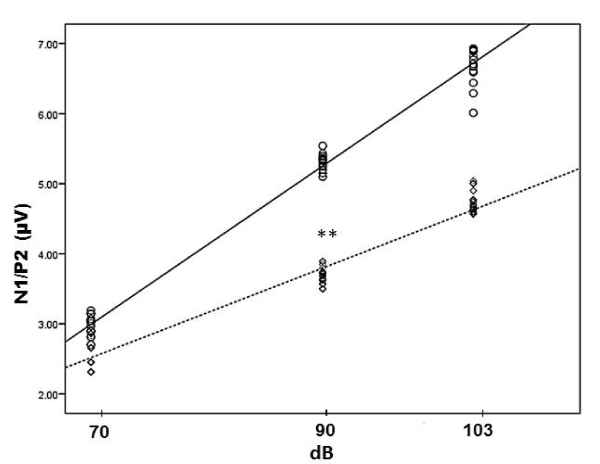
Figure 3. Linear regression analysis and scatter diagram. ○, ▬, treatment start (n = 40), ASF slope = -0.11 + 4.86 intensity, r2 = 0.98 and ◊, --- 20 weeks of treatment (n = 33), ASF slope = 1.86 + 0.06 intensity, r2 = 0.97. dB, sound pressure level. ** p < 0.001
Table 3. Latencies of P1, N1 y P2 components of auditory evoked potentials in adolescents with metabolic syndrome and insulin resistance
Treatment time |
Start
n = 30 |
20 weeks
n = 23 |
P1 |
90.8 ± 3.93 |
92.43 ± 3.25 |
97.25 ± 5.36 |
903.8 ± 7.54 |
905.5 ± 9.92 |
90.1 ± 9.42 |
N1 |
149.9 ± 7.52 |
158.5 ± 7.05 |
159.7 ± 8.50 |
153.6 ± 15.58 |
158.0 ± 8.49 |
163.6 ± 8.07 |
P2 |
220.5 ± 13.69 |
229.9 ± 11.54 |
233.7 ± 15.24 |
229.6 ± 18.20 |
225.6 ± 18.31 |
235.4 ± 21.52 |
Each point corresponds to the mean value ± S.D. The difference between groups was estimated using one-way ANOVA and Tukey’s multiple comparison. There was no statistical difference. dB = dB, (sound pressure level).
The biochemical analysis of this work confirms data observed in a former study in MetS patients [8] where FFT showed a significant decrease accompanied by an increase in the ASF slope of the IDAEPs, reflecting the activity of the primary auditory cortex, which may be interpreted as a consequence of a decreased neuronal firing rate by the brain serotonergic neurons. On the other hand, the decreased FFT levels in these patients cannot be explained by the increases in plasma glucose, HbA1c and lipids (triglycerides, HDL-cholesterol, LDL-cholesterol, and free fatty acids [FFA]),which would tend to favor an increase in FFT levels, since lipids are known to compete with L-Trp for binding to albumin [35]. The decrease in plasma FFT can rather be explained by a deviation of L-Trp to the major non-protein route of its metabolism, i.e., the synthesis of kynurenine (KYN) and KYN-nicotinamide adenine dinucleotide. The first, and rate-limiting step of the L-Trp-KYN pathway is regulated by inducible enzymes: indoleamine 2,3-dioxygenase (IDO) or L-Trp-2,3-dioxygenase (TDO) [22]. IDO is activated by pro-inflammatory mediators, (e.g., interferon-gamma (IFNG), tumor necrosis factor-alpha, IL-1 beta, and lipopolysaccharides), while TDO is induced by stress hormones, e.g., cortisol, estrogens, prolactin, and by the L-Trp substrate [36]. In addition, L-Trp can be degraded to alanine and acetoacetate through the reaction of 3-hydroxykynurenine cleavage to alanine and 3-hydroxyanthranilate by kynureninase, a pyridoxal 5’-phosphate-dependent enzyme. Alanine is glycogenic because its transamination product, pyruvate, which can be converted to glucose via hepatic gluconeogenesis, is frequently observed in patients with obesity, T2D and MetS as a compensatory mechanism during clinical evolution. IR plays also an important role in the pathophysiology of MetS [2,36]. The whole array of metabolic changes and the significant increase of neutral amino acids in plasma previously observed in adolescents with MetS [8] may cause an important decrease in FFT availability at the level of the blood-brain barrier and in its transport to the brain, so that the resulting balance would be a reduction of serotonin synthesis and function.
Various basic and clinical studies have proposed IDAEP (N1/P2 component) as an indicator of serotonergic neurons activity at the level of the primary auditory cortex [8-11,18,19,37-39] where low neuronal serotonergic activity leads to a higher intensity of the auditory cortical response [9-12, 45, 46, 8-11,38], with higher N1/P2 amplitude recorded through IDAEPs, as it was observed in the patients of this study at the beginning of the treatment with fluoxetine and metformin. The opposite effect is also observed when 5-HT neuronal activity increases in the auditory cortex, as we have previously observed in rats [18] and human infants with intrauterine growth restriction [19]. The increase in the ASF slope observed here in MetS patients, reflecting the activity of the primary auditory cortex, can be interpreted as a consequence of decreased firing of the serotonergic neurons in the dorsal nucleus at the brainstem. Therefore, this further supports that there is a functional relationship between the actual brain serotonin activity and alterations in the N1/P2 component of IDAEPs, which may reflect a cortical impaired functional activity associated with anomalies of brain serotonergic neurotransmission. We should underline several metabolic similarities between type 2 diabetes (T2D), MetS and early ontogenetic IUGR-induced undernutrition and its postnatal lasting consequences. The clinical evolution of MetS, T2D and early malnutrition, have similar metabolic changes, besides alterations of amino acids, particularly L-Trp and neutral amino acids, as well as lipid anomalies. Importantly, these metabolic and functional alterations can appear in any of these syndromes with apparently different presentations but comparable metabolic and functional changes. These pathophysiological common traits have been also observed in depressed and non-depressed diabetic patients and in post-myocardial infarction patients who develop depression [11,38]. It seems that their presence may precede depressive manifestations both in patients with diabetes and MetS patients. Therefore, these and previous results point at the existence of common pathophysiological traits in these metabolic disturbances that suggest a common early etiology with a very similar evolution, which also includes brain functional alterations of the neurotransmission system itself, i.e., brain serotonin [8-11,18,19,38]. Interestingly, the adolescents with MetS who were treated with fluoxetine and metformin in this study, were observed to have a significant decrease in body weight, as well as in plasma glucose, lipid profile, insulin concentrations and insulin resistance [39]. Noteworthy, there was also a significant increase in FFT and a decrease in the ASF slope at the end of treatment. Hence, these results support that fluoxetine, through its well-known effect of neuronal serotonin reuptake inhibition, and by increasing brain serotonergic activity, causes an effect that is recorded and expressed as a significant decrease in the slope of the N1/P2 component of the evoked hearing potential as a result of this treatment (Figure 3). In turn, metformin improves all peripheral metabolic parameters and reduces the levels of neutral amino acids, which regulates FFT uptake within the brain, thereby stimulating the synthesis of 5-HT by serotonergic neurons, as well as the activity of the latter [25,40].
Finally, the present results demonstrate that when the combined treatment of fluoxetine and metformin is administered to patients with metabolic syndrome and insulin resistance, it may represent a more effective approach to decrease insulin resistance in the short term. Therefore, our findings underline the relevance of the polypharmaceutical strategy as an effective intervention to increase the efficacy of the drugs that are used in the treatment of insulin resistance in patients with metabolic syndrome.
This work was carried out thanks to the financial support granted by the Mexican Institute of Social Security (FIS/IMSS/PROT/G16/1584) and to the donation of fluoxetine and metformin by Laboratorio Psicofarma, S.A de C.V. The authors acknowledge the editorial support of Victor Torrecillas.
The authors declare that they have no conflicts of interest concerning this article.
- Hansen BC (1999) The metabolic syndrome X: convergence of insulin resistance glucose intolerance, hypertension, obesity and dyslipidemias—searching for underlying defects. Ann NY Acad Sci 892: 1-24.
- Roberts Ch K, Hevener AL, Barnard RJ (2013) Metabolic syndrome and insulin resistance: Underlying causes and modification by exercise training. Compr Physiol 3: 1-58. [Crossref]
- Pereira MA, Jacobs DR Jr, Van Horn L, Slattery ML, Kartashov AI, et al. (2002) Dairy consumption, obesity, and the insulin resistance syndrome in Young adults: the CARDIA Study. JAMA 287: 2081-2089. [Crossref]
- Liu S, Manson JE (2001) Dietary carbohydrates, physical inactivity, obesity, and the ‘metabolic syndrome’ as predictors of coronary heart disease. Curr Opin Lipidol 12: 395-404. [Crossref]
- Björntorp P, Rosmond R (1999) Hypothalamic origin of the metabolic syndrome X. Ann NY Acad Sci 892: 297-307. [Crossref]
- Pasquali R, Gagliardi L, Vicennati V, Gambineri A, Colitta D, et al. (1999) ACTH and cortisol response to combined corticotropin releasing hormone-arginine vasopressin stimulation in obese males and its relationship to body weight, fat distribution and parameters of the metabolic syndrome. Int J Obes Relat Metab Disord 23: 419-424. [Crossref]
- Horacek J, Kuzmiakova M, Hoschl C, Andel M, Bahbonh R (1999) The relationship between central serotonergic activity and insulin sensitivity in healthy volunteers. Psychoneuroendocrinology 24: 785-797. [Crossref]
- Herrera-Marquez R, Hernández-Rodriguez J, Medina-Serrano J, Manjarrez-Gutierrez G (2011) Association of metabolic syndrome with reduced central serotonergic activity. Metab Brain Dis 26: 29-35. [Crossref]
- Manjarrez G, Herrera R, Leon M, Hernández R (2006) A low brain serotonergic neurotransmission in children with type 1 diabetes detected through the intensity dependence of auditory evoked potentials. Diabetes Care 29: 73-77. [Crossref]
- Manjarrez G, Vázquez F, Delgado M, Herrera R, Hernandez J (2007) A functional disturbance in the auditory cortex related to a low serotonergic neurotransmission in women with type 2 diabetes. Neuroendocrinology 86: 289-294. [Crossref]
- Manjarrez-Gutierrez G, Herrera-Marquez R, Mejenes-Alvarez SA, Godinez-Lopez T, Hernandez-RJ (2009) Functional change of auditory cortex related to brain serotonergic neurotransmission in type 1 diabetes adolescents with and without depression. World J Biol Psychiatry 10: 877-883. [Crossref]
- Yuan X, Yamada K, Ishiyama-Shigemoto S, Koyama W, Nonaka K (2000) Identification of polymorphic loci in the promoter region of the serotonin 5-HT2C receptor gene and their association with obesity and type II diabetes. Diabetologia 43: 373-376. [Crossref]
- Rosmond R, Bouchard C, Bjorntorp P (2002) Increased abdominal obesity in subjects with a mutation in the 5-HT(2A) receptor gene promoter. Ann NY Acad Sci 967: 571-575. [Crossref]
- Lucki I (1998) The spectrum of behaviors influenced by serotonin. Biol Psychiatry 44: 151-162. [Crossref]
- McCall RB, Clement ME (1994) Role of serotonin1A and serotonin2 receptors in the central regulation of the cardiovascular system. Pharmacol Rev 46: 231-243 [Crossref]
- Ramage AG (2001) Central cardiovascular regulation and 5-hydroxytryptamine receptors. Brain Res Bull 56: 425-439. [Crossref]
- Robinson R (2009) Serotonin’s Role in the Pancreas Revealed at Last. PLoS Biol 7: e1000227. [Crossref]
- Manjarrez G, Hernandez E, Robles A, Hernandez J (2005) N1/P2 component of auditory evoked potential reflects changes of the brain serotonin biosynthesis in rats. Nutr Neurosci 8: 213-218. [Crossref]
- Manjarrez G, Cisneros I, Herrera R, Vazquez F, Robles A, et al. (2005) Prenatal impairment of brain serotonergic transmission in infants. J Pediatr 147: 592-596. [Crossref]
- Shoelson SE, Lee J, Goldfine AB (2006) Inflammation and insulin resistance. J Clin Invest 116: 1793-1801. [Crossref]
- Samuel VT, Shulman GI (2012) Mechanisms for insulin resistance: common threads and missing links. Cell 148: 852-871. [Crossref]
- Oxenkrug G (2013) Insulin resistance and dysregulation of tryptophan-kynurenine and kynurenine-nicotinamide adenin dinucleotide metabolic pathways. Mol Neurobiol 48: 294-301. [Crossref]
- Lee JM (2006) Insulin resistance in children and adolescents. Rev Endocr Metab Disord 7: 141-147. [Crossref]
- Lim S, Ecklel RH (2014) Pharmacological treatment and therapeutic perspectives of metabolic syndrome. Rev Endocr Metab Disord 15: 329-341. [Crossref]
- Orchard TJ, Temprosa M, Goldberg R, Haffner S, Ratner R, et al. (2005) The effect of metformin and intensive lifestyle intervention on the metabolic syndrome: The Diabetes Prevention Program Randomized Trial. Ann Intern Med 142: 611-619. [Crossref]
- Rena G, Hardie DG, Pearson ER (2017) The mechanisms of action of metformin. Diabetologia 60: 1577-1585. [Crossref]
- Wenthur CJ, Bennett MR, Lindsley CW (2014) Classics in chemical neuroscience: Fluoxetine (Prozac). ACS Chem Neurosci 5: 14-23. [Crossref]
- Muldoon MF, Mackey RH, Korytkowski MT, Flory JD, Pollock BG, et al. (2006) The metabolic syndrome is associated with reduced central serotonergic responsivity in healthy community volunteers. J Clin Endocrinol Metab 91: 718-721. [Crossref]
- Muldoon MF, Mackey RH, Williams KV, Korytkowski MT, Flory JD, et al. (2004) Low central nervous system serotonergic responsivity is associated with the metabolic syndrome and physical inactivity. J Clin Endocrinol Metab 89: 266-671. [Crossref]
- Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (2001) Executive Summary of the Third Report of the national Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA 285: 2486-2497. [Crossref]
- Schwartz B, Jacobs DR Jr, Moran A, Steinberger J, Hong CP, et al. (2008) Measurement of insulin sensitivity in children: comparison between the euglycemic-hyperinsulinemic clamp and surrogate measures. Diabetes Care 31: 783-788. [Crossref]
- Arellano-Ruiz P, García-Hermoso A, Cavero-Redondo I, Pozuelo-Carrascosa D, Martínez-Viscaíno V, et al. (2019) Homeostasis Model Assessment-cut-off points related to metabolic syndrome in children and adolescents: a systematic review and meta-analysis. Eur J Pediatr 178: 1813-1822. [Crossref]
- Johansen PA, Jennings I, Cotton RG, Kuhn DM (1995) Tryptophan hydroxylase is phosphorylated by protein kinase A. J Neurochem 65: 882-888. [Crossref]
- Semple MN, Scott BH (2003) Cortical mechanism in hearing. Curr Opin Neurobiol 13: 167-173. [Crossref]
- Hansen KU (1990) Structure and ligand binding properties of human serum albumin. Dan Med Bull 37: 57-84. [Crossref]
- Nelson RA, Bremer AA (2010) Insulin resistance and metabolic syndrome in the pediatric population. Metb Syndr Relat Disor 8: 1-14. [Crossref]
- Hegerl U, Juckel G (1993) Intensity dependence of auditory evoked potentials as an indicator of central serotonergic neurotransmission: a new hypothesis. Biol Psychiatry 33: 173-187. [Crossref]
- Manjarrez GG, Ramírez CR, Borrayo SG, Hernández RJ (2013) Disturbance of serotonergic neurotransmission in patients with postmyocardial infarction and depression. Metab Brain Dis 28: 15-20. [Crossref]
- Hensch, T, Herold U, Diers K (2008) Reliability of intensity dependence of auditory-evoked potentials. Clin Neurophysiol 119: 224-236. [Crossref]
- Zemdegs J, Martin H, Pintana H, Bullich S, Manta S, Marqués MA, et al. (2019) Metformin Promotes Anxiolytic and Antidepressant-Like Responses in Insulin-Resistant Mice by Decreasing Circulating Branched-Chain Amino Acids. J Neurosci 39: 5935-5948. [Crossref]