Abstract
Moyamoya disease is rare and predominantly affects the Japanese population. Moyamoya disease is characterised by chronic idiopathic stenosis of the terminal internal carotid arteries with associated angiogenesis in the basal ganglia. Clinically, this results in strokes and cognitive impairment. To date there has not been a case of Moyamoya reported in the Australian Indigenous population. Here we present a case of Moyamoya disease masquerading as posttraumatic carotid dissection in an Indigenous patient for the purposes of stimulating scientific inquiry. This patient successfully underwent superficial temporal to MCA anastomosis and had a good result both radiologically and clinically, although long-term follow-up will be required.
Key words
moyamoya, indigenous Australian, stroke, carotid dissection, revascularisation
Case report
History and examination
A 19-year-old man presented to emergency with right facial droop, numbness and intermittent headache after being punched 1 week prior, leading to a brief loss of consciousness. He experienced intermittent paraesthesia and numbness in his upper and lower limbs bilaterally. After the assault he described generalised weakness and difficulty mobilising for one week.
At age 16 he had experienced a similar episode of aphasia and lower limb weakness.
MRI Brain revealed infarction in the watershed areas of the left cerebral hemisphere and recent dissection of the internal carotid artery from skull base through to the siphon (Figures 1 and 2). The provisional diagnosis was left internal carotid artery dissection, secondary to trauma. However a supplementary history of previous episodes along with angiographic abnormalities (Figures 3 and 4) revealed the true diagnosis of Moyamoya disease (MMD). He had no family history of MMD, but his mother had a history of sagittal sinus filling defect associated with papilloedema.
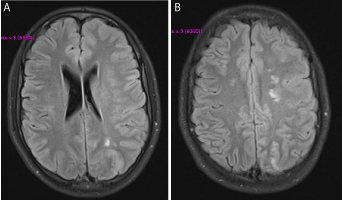
Figure 1. T1-weighted MR images through the infarcted (A) MCA-PCA and (B) ACA-MCA watershed areas.
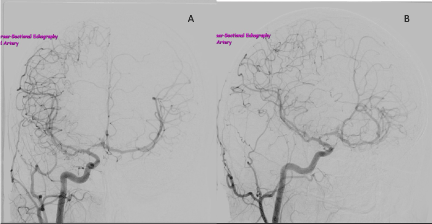
Figure 2. Right ICA angiogram, anteroposterior view (A) and lateral view (B), showing a patent right ICA that tapers within the carotid siphon, and marked stenosis of the A1 segment of the right ACA. The callosal branch of the right ACA is being drained through ACom to supplement the limited left circulation.
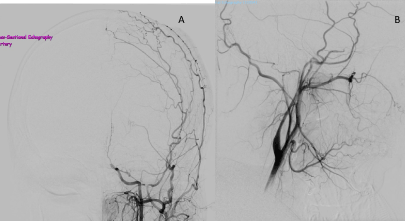
Figure 3. Left ICA aniogram, anteroposterior view (A) and lateral view (B).
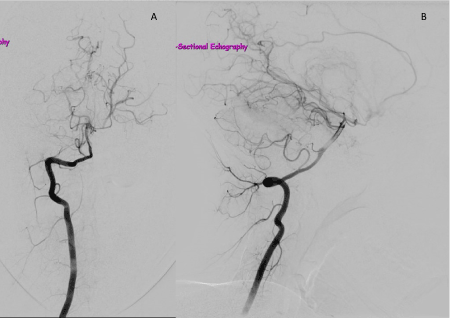
Figure 4. Right vertebral angiogram, anteroposterior view (A) and lateral view (B), showing dense networks of abnormal vessels characteristic of MMD.
MRA suggested trickle flow through the left internal carotid, and good collateral flow through the anterior communicating artery. The A1 segment of the right anterior cerebral artery was markedly stenosed. No significant posterior communicating artery was visualised: the left hemisphere was almost completely isolated from blood supply. He was given intravenous heparin infusion, before transfer to a neurosurgical unit for superficial temporal to middle cerebral bypass. Heparin was ceased and the patient was started on aspirin. Postoperatively, he initially showed word finding difficulties and slight decrease in power in his right hand, but this improved. Computed tomography angiography demonstrated graft patency.
Definition
In 1969, Suzuki and Takaku first described a chronic, progressive and idiopathic bilateral stenosis of the terminal internal carotid artery (ICA) followed by the development of “net like” formations of blood vessels at the base of the brain [1]. They assigned the term “moyamoya” to the typical angiographic appearance of these formations, resembling “puffs of smoke” (moyamoya in Japanese).
Unlike MMD, Moyamoya syndrome (MMS) is not congenital, but is instead characterised by the appearance of typical moyamoya on angiogram secondary to some other pathology. Causative conditions include atherosclerosis, autoimmune disease, trauma and congenital conditions such as Down’s syndrome and neurofibromatosis type 1.
Angiographic MMD diagnosis requires 1) stenosis/occlusion at the terminal ICA and/or proximal ACA and/or MCA, and 2) abnormal vascular networks (“moyamoya”) in the basal ganglia in arterial phase, and 3) bilateral findings (if findings are only unilateral, then it is a probable case) [2].
Epidemiology and genetics
MMD carries a prevalence of 0.086 per 100,000, with a bimodal age distribution peaking at age five and in the mid-forties [3,4]. It is twice as common in females as males. The Japanese have a particularly high incidence with 10 times as many people affected by the disease than in the Caucasian population [3,4]. The incidence of MMD in the Australian Indigenous population is unknown. Contributing significantly to discrepancies of MMD incidence in various ethnic groups is the carrier frequency of the RNF213 gene, which is by far the highest in the Japanese population: 1/72 people carry the gene [4]. The presence of seven single nucleotide polymorphisms in RNF213 carries an odds ratio >190, however the mechanism of this is unknown4. Other genes have been sought and found (e.g. HLA-D6S441) in the hope of better elucidating the pathophysiology [5].
The inheritance mode of MMD is multifactorial and is thought to be autosomal dominant with incomplete penetrance and imprinting [6]. As a result, MMD cases aggregate in families: ~10% of patients have a positive family history for Moyamoya [6].
Acknowledgements
We thank Prof. David B. Williams, Dr Jeffrey Blackie and Ms Amber Newphrey for their assistance.
Funding
2021 Copyright OAT. All rights reserv
This work did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
References
- Suzuki J, Takaku A (1969) Cerebrovascular ‘moyamoya’ disease: Disease showing abnormal net-like vessels in base of brain. Arch Neurol 20: 288–299. [Crossref]
- 2 Fukui M (1997) Members of the Research Committee on Spontaneous Occlusion of the Circle of Willis (Moyamoya Disease) of the Ministry of Health and Welfare J. Guidelines for the diagnosis and treatment of spontaneous occlusion of the circle of Willis (`Moyamoya’ disease)1. Clinical Neurology and Neurosurgery 99: S238–S240.
- 3 Scott RM, Smith ER (2009) Moyamoya Disease and Moyamoya Syndrome. New England Journal of Medicine 360: 1226–1237.
- 4 Kamada F, Aoki Y, Narisawa A, Abe Y, Komatsuzaki S, et al. (2011) A genome-wide association study identifies RNF213 as the first Moyamoya disease gene. J Hum Genet 56: 34–40. [Crossref]
- 5 Roder C, Nayak NR, Khan N, Tatagiba M, Inoue I, et al. (2010) Genetics of Moyamoya disease. J Hum Genet 55: 711–716.
- 6 Mineharu Y, Takenaka K, Yamakawa H, Inoue K, Ikeda H, et al. (2006) Inheritance pattern of familial moyamoya disease: autosomal dominant mode and genomic imprinting. J Neurol Neurosurg Psychiatry 77: 1025–1029. [Crossref]