Background: Report on the changes of diagnostic criteria since 1970 and correspondence due to the development of new therapeutic drugs during long term clinical period.
Patients and method: The subjects were Fifty-five ET patients who were 21-95years old who newly diagnosed Sendai Blood Disease Center (renaming Sendai Tomita Hospital from March 2013) between 1991 year and 2010 year. As a substitute for bone biopsy for convenient diagnostic tool, we diagnosed essential thrombocythemia (ET) by the smear sample of bone marrow aspiration focusing on platelet production of megakaryocyte cytoplasm.
Result: We classified megakaryocytes into 7 types (0 to 5 and U) with platelet counts in the cytoplasm. We named Type U which was filled and spilling out platelets from megakaryocyte. Type U was not observed in our experienced 25 chronic myeloid leukemia cases, 10 polycythemia vera cases and another hematologic malignancies. The proportion of seven types was Type 0 was 35.9%, type I 4.7%, type II 6.0%, type III 3.0%, type IV 0.8%, type NK 4.4% and type U was 45.2% respectively.
30 males and 25 females, the age at the time of diagnosis was the median 64 years and range 21 to 95 with 61.8% above the age 60 years. The observation periods was from 17 months to 303 months and the median was 94 months.
The main goal in ET is to prevent thrombohemorrhagic complications and the same for older patients. The frailty of elderly people, 15 of the 19 patients who stopped receiving medical examination was brought about by falls and aspiration pneumonia which caused the physical condition to decrease, not accompanied by ET treatment. The cause of frailty in older ET patients was irrespective ET treatment, and so on based on risk-adapted therapy was necessary for older ET patients.
Conclusion: Megakaryocytes of ET patients showed platelet-free production and overproduction of platelets in cytoplasm. Based on risk-adapted therapy was necessary for frail older ET patients.
essential thrombocythemia, outcome, diagnosis, megakaryocyte, bone marrow aspiration smear
Essential thrombocythemia (ET) is one of a group of related myeloproliferative neoplasms (WHO 2008). The earliest description of thrombocythemia by di Guglielma in 1920 and by Epstein and Goedel in 1934. In 1960, thrombocythemia was established as a separate disease entity on a clinic pathologic basis. Strict diagnostic criteria for ET were first established in the 1970s by the polycythemia vera study group (PVSG) [1,2]. And the approach to the diagnosis and management of ET is steadily changing and strengthening to lower platelet count [3-5], influenced by advances in molecular biology.
Even in an era when ET was little known about the molecular pathogenesis, and showed a clonal disorder, the majority of patients with ET can expect a normal life expectancy in the first 15 years of disease [6].
Patients who were diagnosed in the age when molecular/pathologically defined entities are not established, have reached the stage of disease progression while sending a long term almost normal life and necessary to review the treatment policy [6,7].
Regarding the diagnosis of ET, before 2001 World Health Organization criteria for thrombocythemia [8] were disseminated, we are based on PVSG criteria and we focused on the platelet count and the megakaryocyte cytoplasm and hiatus of megakaryocyte proliferation by bone marrow aspiration. Unlike the bone marrow biopsy method, the bone marrow aspiration method is immediately after obtaining the specimen and has the advantage of rapid diagnosis.
Thrombosis and hemorrhagic complications are the most frequent clinical complication observed in ET patients [9-12]. Vascular complications [13-17] had been studied as the predictive factor.
Normalization of platelet counts was recommended [10,18,19] but hydroxycarbamide (HC) was reported mutagenesis or leukemogenesis [20].
Tefferi proposed a risk stratification schema for patients with ET based on epidemiological evidence [21]. Consensus-based practice guidelines for the therapy of ET developed in Italy [22] and an algorithm was recommended in ET patients [7,23].
In 2009, European Leukemia Net proposed response criteria for ET [24,25]. ET patients expected to have long-normal life should be treated with drugs without adverse effects and without vascular complications [14,26].
Therapy goals are individualized and driven by the patient’s stratification and symptomatology [27-29]. We report the clinical course of ET patients during a quarter century in single institution in Japan as it responded to the change of the diagnostic criteria and the development of therapeutic drugs and reported on the outcome for frail older patients.
Patients
The subjects were Fifty-five ET patients excluded by careful review of medical history and current and previous laboratory data who were 21-95years old who newly diagnosed Sendai Blood Disease Center (renaming Sendai Tomita Hospital from March 2013) between 1991 year and 2010 year.
Diagnosis of ET was an unexplained and persistent thrombocytosis and reactive thrombocytosis was excluded by careful review of the medical history and current and previous laboratory data. The number of platelets which is the diagnostic criterion was 600X109/L [8,15] until 2008 and thereafter 450X109/L [30,31]. Diagnosis of ET was strengthened platelet count accordance to guidelines.
In Japan, since bone biopsy had not been widely done till recently, bone marrow aspirate which could be carried out on a daily popular, and megakaryocytes in bone marrow blood smear were carefully assessed with the platelet counts within the megakaryocyte cytoplasm for evaluating feasibility of the diagnosis of the ET.
In the bone marrow aspirated smear sample, we classified megakaryocytes with platelet counts seven types, especially filled and spilling out with adjacent shedding platelets as type U (Figure 1), because, megakaryocyte with filled and spilling out with adjacent shedding platelets was not observed in 25 Philadelphia positive myeloid leukemia cases ,10 polycythemia vera cases and another hematologic malignancy cases. type 0; Megakaryocytes without platelet production, type I, Platelet production; less than 10, type II; Platelet production more than 10, type III, Prominent, type IV; Extremely high platelet production and type V; naked megakaryocytic nuclei.
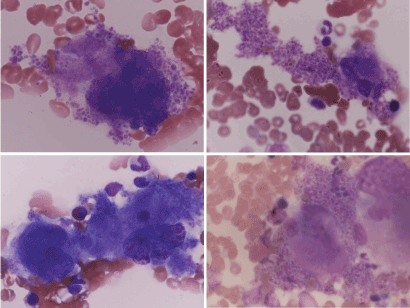
Figure 1. Type U of megakaryocyte
Gene mutation
Unable to obtain patient consent because of high cost.
Therapy
After diagnosis with ET, according to diagnostic criteria [8,15,30] treatments were started immediately or sometimes after observation.
Antiplatelet agent
We recommended the use of low dose aspirin (81 mg/day, range 40 mg/day – 100 mg/day) in all patients. The use of aspirin with anagrelide deserved specific consideration given [14].
Cytoreductive therapy
From 1991 to 2002, 27 ET patients were started the cytoreductive therapy with Esquinone (CQ) [ 32] 0.5 mg as a platelet-lowering oral agent. From 2002 to 2010, 28 ET patients were started with HC 1000 mg orally. Until the number of platelets was normalized, CQ was adjusted to 0.25 mg each and that the normalized amount dose was continuously administered for maintenance. CQ was prescribed until June 2005, when production and sales were canceled, after which it switched to Hydroxycarbamide (HC).
Anagrelide
After commencing sales anagrelide on Nov. 2014 in Japan, as a general rule, we changed prescriptions with patient consent from hydroxyurea to anagrelide while prescribing data on the literature of both drugs because anagrelide is more expensive than hydroxyurea.
Treatment goal
Once cytoreductive therapy was instituted, we adjusted to maintain the normalizing platelet count without myelosuppression and adverse effects.
Statistical analysis
The visit duration curves were plotted by the Kaplan-Meier method and compared by the log rank test.
The main clinical and laboratory features of the 55 patients at diagnosis are presented in table 1. 30 patients were male and 25 were female. The median age, at diagnosis was 64 years, range 21 to 95, with 61.8% above the age 60 years. The median platelets count was 871×/109/L, range 510-2839. The median leukocyte count was 10.1×109/L, range 4.3- 23.0. The median hemoglobin was 13.6g/dL, range7.3-18.3. No patient had an abnormality in chromosome examination. In Japan, the JAK2V617F mutation was not covered by public health insurance, and so, no one wanted the examination.
Table 1. Clinical and laboratory characteristics before treatment
Male/Female, n (male %) |
30/25 |
Median age (range), years |
64 (21-95) |
Age>60 years n (%) |
34(61.8) |
Median platelet count (range), X109/L |
871 (510-2839) |
Median leukocyte count (range), X109/L |
10.1 (4.3-23.0) |
Median hemoglobin (range), g/dL |
13.6 (7.3-18.3) |
Elevated lactate dehydrogenase, n (%) |
11 (20) |
Chromosome abnormaliteis, n (%) |
0 |
Palpable splenomegaly, n (%) |
29 (52.7) |
Previous hemorrages, n (%) |
0 |
Previous thrombosis, n (%) |
9 (16.4) |
Thrombosis after diagnosis, n (%) |
1((1.8) |
Hypertension, n (%) |
29 (52.7) |
Median follow up (range), months |
94 (17-303) |
In the bone marrow aspirated smear sample, we classified megakaryocytes with platelet counts of megakaryocyte cytoplasm type 0; Megakaryocytes without platelet production, type I, Platelet production; less than 10, type II; Platelet production more than 10, type III, Prominent, type IV; Extremely high platelet production and naked megakaryocytic nuclei and type U (Figure 1): filled and spilling out platelets from megakaryocyte. The average percentage was that type 0 was 35.9%, type I 4.7%, type II 6.0%, type III 3.0%, type IV 0.8%, type NK 4.4% and type U was 45.2% respectively (Table 2). Type 0 and type U were a large proportion.
Table 2. The proportion of the typing according to platelet counts in megakaryocyte cytoplasm
Megakaryocyte |
Number |
Type 0 |
Type I |
Type II |
Type III |
Type IV |
Type NK |
Type U |
|
X109/L |
% |
% |
% |
% |
% |
% |
% |
average |
0.51 |
36 |
4.6 |
6 |
3 |
0.8 |
4.4 |
45.2 |
range |
0.22 - 2.22 |
0-88 |
0-27 |
0-35 |
0-30 |
0-10 |
0-40 |
10 - 100 |
Type 0: Meg akaryocytes without platelet production, Type I: Platelet production less than 10, Type II: Platelet production more than 10, Type III: Prominent, Type IV: Extremely high platelet production and Type V: naked megakaryocytic nuclei
29 patients (52.7%) showed splenomegaly. Before diagnosis, 15 patients were suffered cardiac or cerebral vascular complications (Table 1).
From 1991 to 2002, 27 patients started with CQ 1.0 mg per days orally as the first-line treatment and who had 11 to 140 days until the platelet count reached 500 ×109/L or less, and the median was 60 days. Maintenance therapy was switched equivalent dose from CQ 0.5 mg to HC 500mg after June 2005.
28 patients who administered HC starting dose 1000 mg reached platelet count 500 ×109/L or less for 5 to 85 days, and the median 17days. Maintenance therapy was making fine adjustments while checking the platelet count.
After November 2014 which became indication of public health insurance in Japan, for 16 applicants, maintenance therapy was switched to anagrelide from HC. Five out of the 16 patients were discontinued between dosage 3 mg and less and administration period 2 and 3 months as insufficient platelet-lowering effect despite a high cost drug.
5 patients receiving anagrelide 1 to 2mg were achieving better likely long-term platelet count than HC. Six of 16 patients examined leukocytosis after
Only one patient got a transient ischemic attack. Two patients observed transition to polycythemia and one was leukocytosis under cytoreductive treatment. Outcome summary showed in table 3. Fifteen patients between 72 to 98 years old did not visited for frailty. The frailty of elderly people was brought about by falls and miscarriage pneumonia which caused the physical condition to decrease, not accompanied by ET treatment. Four patients between the ages 71 and 84 years old passed out of receiving visit in less than 1 year. Two patients died with leukemic transformation after 8years and 5 years of diagnosis and four people died after repeating infection caused by leukopenia by myelofibrotic change. Figure 2 showed the visit duration curves, and there was a significant difference under 60 years old and above (p < 0.0028)
Table 3. Outcome during follow up
|
Number (%) |
|
Number (%) |
Years follow-up |
|
Deaths |
|
less than 5years |
22 (40) |
Thrombotic cause |
0 |
5 - <10 years |
17 (30.9) |
Hemorrhagic cause |
0 |
10 - <15 years |
8 (14.5) |
Post-ET MF |
4 |
15 - <20 years |
6 (10.9) |
Post-ET AML |
2 |
more than 20years |
2 (3.6) |
Solid tumors |
2 |
|
|
Other causes |
2 |
Thrombosis |
|
|
|
AM |
1 |
Fade-out |
|
TIA |
1 |
asymptome |
4 |
|
|
frailty |
15 |
Hematologic transformation |
|
|
|
MF |
6 |
|
|
AML |
2 |
|
|
Abbreviation: AM: Acute Myocardial Infarction, TIA: Transient Ischemic Attack MF: Myelofibrosis, AML: Acute Myelogenous Leukemia, ET: Essential Thrombocythemia
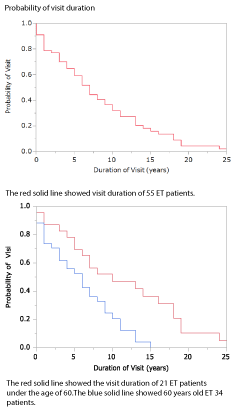
Figure 2. Probability of the visit duration in 55 ET patients.
Even after the diagnostic criteria of ET was proposed in the 1975 by the polycythemia vera study group (PVSG) [1,2], the platelet count was above 1,000 ×109/L, there was much discussion the platelet count for diagnostic criteria for ET. In 1986 PVSG modified the minimum platelet count for diagnosis of ET to 600 ×109/L15. ET is both long-term life expectancy in asymptomatic condition, and thrombotic and hemorrhagic complication associated [32,33]. Hydroxyurea (HU), one of cytoreductive agents remains about leukemogenicity [20]. It is important that a diagnostic approach to ET distinguish from reactive thrombocytosis. After the criteria for ET include a persistent platelet count of greater than 600 ×109/L, it has been suggested that the platelet count criterion should be reduced to greater than 400 ×109/L since, in long-term follow-up studies the clinical course of patients with platelets count between 400 and 600 ×109/L was found to be distinguishable from that of patients with a clear of ET. In 2008, the World Health Organization (WHO) classification system has strengthened the diagnostic criteria for ET by lowering the threshold platelet count.
2021 Copyright OAT. All rights reserv
It has been suggested that ET can be positively diagnosed by careful quantitative examination of the bone marrow biopsy [34-37]. But, in Japan bone marrow biopsy did not feel necessity and was not performed easily until the beginning of 2000. The bone marrow aspirate which could be carried out on a daily popular, and megakaryocytes in bone marrow blood smear were carefully assessed type U with filled and spilling out platelets from megakaryocyte cytoplasm for evaluating feasibility of the diagnosis of the ET.
Diagnosis of ET is currently based on WHO which does require bone marrow biopsy in particular to differentiate “true” ET from prefibrotic/early primary myelofibrosis (prePMF) [31]. Since the bone marrow aspiration examination is obtained results easily and faster, we would like to investigate the relationship between Type U megakaryocyte and biopsy findings and molecular finding.
Now, an acquired mutation of JAK2 was detected as approximately half of ET patients, but before the JAK2V617F mutation examination was not easy popular and covered by public health insurance in Japan so diagnosis by simple, routine and convenient examination was important.
Thrombosis and hemorrhagic complications are the most frequent clinical complications observed in ET patients, and so the ET patients were classified based on thrombotic and hemorrhagic risk [7,9]. We continued prescription of cytoreductive drugs for asymptomatic patients with a clinical dilemma of mutagenicity. After the diagnosis, in order to avoid the occurrence of thrombohemorrhagic complications, the treatment goal was set to 400 ×109/L platelets, and red blood cells and white blood cells were within the normal range. No patients suffered from thrombosis.
ET patients with other hematological transition or resistant or refractory to HC [29] are good candidates for novel agents, such as JAK2 inhibitor [28]. Reduced platelet counts with therapies in ET patients affected vascular modulator changes [38]. The main goal in ET is to prevent thrombohemorrhagic complications and the same for older patients. The cause of frailty in older ET patients was irrespective ET treatment, and so on based on risk-adapted therapy was necessary for older ET patients.
ET patients expect normal long life. Treatment is required not only to control the number of platelets to prevent complications including cardiovascular symptoms but also to respond to changes in disease state while confirming red blood cells and white blood cells.
We thanks to all the members of our hospital for their careful and enthusiastic medical records in and out.
This study was supported by the Sendai Blood Disease Center (SBDC) and no specific funding was disclosed.
The authors made no disclosures.
- Berlin NI (1975) Diagnosis and classification of the polycythemias. Semin Hematol 12: 339-351. [Crossref]
- Laszlo J (1975) Myeloproliferative disorders (MPD): myelofibrosis, myelosclerosis, extramedullary hematopoiesis, undifferentiated MPD, and hemorrhagic thrombocythemia. Semin Hematol 12: 409-432. [Crossref]
- Sacchi S, Vinci G, Gugliotta L, Rupoli S, Gargantini L, et al. (2000) Diagnosis of essential thrombocythemia at platelet counts between 400 and 600 x10(9)/L. Gruppo Italiano Malattie Mieloproliferative Croniche(GIMMC). Haematologica 85: 492-495. [Crossref]
- Tefferi A, Hoagland HC (1994) Issues in the diagnosis and management of essential thrombocythemia. Mayo Clin Proc 69: 651-655. [Crossref]
- Tefferi A, Silverstein MN, Hoagland HC (1995) Primary thrombocythemia. Semin Oncol 22: 334-340. [Crossref]
- Murphy S, Peterson P, Iland H, Laszlo J (1997) Experience of the Polycythemia Vera Study Group with essential thrombocythemia: a final report on diagnostic criteria, survival, and leukemic transition by treatment. Semin Hematol 34: 29-39. [Crossref]
- Finazzi G, Harrison C (2005) Essential thrombocythemia. Semin Hematol 42: 230-238. [Crossref]
- Imbert M, vardiman JW, Pierre R, Brunning RD, Thiele J, et al. (2000) Esssential thrombocythemia. Pathology & Genetics Tumours of haematopoietic and lymphoid tissues. 3.
- Cortelazzo S, Viero P, Finazzi G, D'Emilio A, Rodeghiero F, et al. (1990) Incidence and risk factors for thrombotic complications in a historical cohort of 100 patients with essential thrombocythemia. J Clin Oncol 8: 556-562. [Crossref]
- Regev A, Stark P, Blickstein D, Lahav M (1997) Thrombotic complications in essential thrombocythemia with relatively low platelet counts. Am J Hematol 56: 168-172. [Crossref]
- Colombi M, Radaelli F, Zocchi L, Maiolo AT (1991) Thrombotic and hemorrhagic complications in essential thrombocythemia. A retrospective study of 103 patients. Cancer 67: 2926-2930. [Crossref]
- Lahuerta-Palacios JJ1, Bornstein R, Fernández-Debora FJ, Gutiérrez-Rivas E, Ortiz MC, et al. (1988) Controlled and uncontrolled thrombocytosis. Its clinical role in essential thrombocythemia. Cancer 61: 1207-1212. [Crossref]
- Besses C, Cervantes F, Pereira A, Florensa L, Sole F, et al. (1999) Major vascular complications in essential thrombocythemia: a study of the predictive factors in a series of 148 patients. Leukemia 13: 150-154. [Crossref]
- Campbell PJ, MacLean C, Beer PA, Buck G, Wheatley K, et al. (2012) Correlation of blood counts with vascular complications in essential thrombocythemia: analysis of the prospective PT1 cohort. Blood 120: 1409-1411. [Crossref]
- Murphy S, Iland H, Rosenthal D, Laszlo J (1986) Essential thrombocythemia: an interim report from the Polycythemia Vera Study Group. Semin Hematol 23: 177-182. [Crossref]
- Haider M, Gangat N, Lasho T, Abou Hussein AK, Elala YC, et al. (2016) Validation of the revised International Prognostic Score of Thrombosis for Essential Thrombocythemia (IPSET-thrombosis) in 585 Mayo Clinic patients. Am J Hematol 91: 390-394. [Crossref]
- Carobbio A, Thiele J, Passamonti F, Rumi E, Ruggeri M, et al. (2011) Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: an international study of 891 patients. Blood 117: 5857-5859. [Crossref]
- Barbui T (2003) Indications for lowering platelet numbers in essential thrombocythemia. Semin Hematol 40: 22-25. [Crossref]
- Tefferi A (1998) Is hydroxyurea leukemogenic in essential thrombocythemia? Blood 92: 1459-1460. [Crossref]
- Weinfeld A, Swolin B, Westin J (1994). Acute leukaemia after hydroxyurea therapy in polycythaemia vera and allied disorders: prospective study of efficacy and leukaemogenicity with therapeutic implications. Eur J Haematol 52: 134-139. [Crossref]
- (1999) Risk-based management in essential thrombocythemia. The American Society of Hematology Education Program Book: 172-177.
- Barbui T, Barosi G, Grossi A, Gugliotta L, Liberato LN, et al. (2004) Practice guidelines for the therapy of essential thrombocythemia. A statement from the Italian Society of Hematology, the Italian Society of Experimental Hematology and the Italian Group for Bone Marrow Transplantation. Haematologica 89: 215-232. [Crossref]
- Finazzi G, Barbui T (2005) Risk-adapted therapy in essential thrombocythemia and polycythemia vera. Blood Rev 19: 243-252. [Crossref]
- Barosi G, Birgegard G, Finazzi G, Griesshammer M, Harrison C, et al. (2009) Response criteria for essential thrombocythemia and polycythemia vera: result of a European LeukemiaNet consensus conference. Blood 113: 4829-4833. [Crossref]
- Carobbio A, Finazzi G, Antonioli E, Vannucchi AM, Barosi G, et al. (2010) Hydroxyurea in essential thrombocythemia: rate and clinical relevance of responses by European LeukemiaNet criteria. Blood 116: 1051-1055. [Crossref]
- Falchi L, Bose P, Newberry KJ, Verstovsek S (2017) Approach to patients with essential thrombocythaemia and very high platelet counts: what is the evidence for treatment? Br J Haematol 176: 352-364. [Crossref]
- Tefferi A, Barbui T (2013) Personalized management of essential thrombocythemia-application of recent evidence to clinical practice. Leukemia 27: 1617-1620. [Crossref]
- Alimam S, Wilkins BS, Harrison CN (2015) How we diagnose and treat essential thrombocythaemia. Br J Haematol 171: 306-321. [Crossref]
- Hernandez-Boluda JC, Alvarez-Larran A, Gomez M, Angona A, Amat P, et al. (2011) Clinical evaluation of the European LeukaemiaNet criteria for clinicohaematological response and resistance/intolerance to hydroxycarbamide in essential thrombocythaemia. Br J Haematol 152: 81-88. [Crossref]
- Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, et al. (2009) The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 114: 937-951. [Crossref]
- Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, et al. (2016) The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127: 2391-2405. [Crossref]
- Uzuka Y, Saito Y, Takahashi H, Komatsu M (1982) Carboquone therapy for hematologic neoplasms. Tohoku J Exp Med 138: 151-160. [Crossref]
- Randi ML, Stocco F, Rossi C, Tison T, Girolami A (1991) Thrombosis and hemorrhage in thrombocytosis: evaluation of a large cohort of patients (357 cases). J Med22: 213-223. [Crossref]
- Thiele J, Kvasnicka HM (2003) Diagnostic differentiation of essential thrombocythaemia from thrombocythaemias associated with chronic idiopathic myelofibrosis by discriminate analysis of bone marrow features--a clinicopathological study on 272 patients. Histol Histopathol 18: 93-102. [Crossref]
- Florena AM, Tripodo C, Iannitto E, Porcasi R, Ingrao S, et al. (2004) Value of bone marrow biopsy in the diagnosis of essential thrombocythemia. Haematologica 89: 911-919. [Crossref]
- Brousseau M, Parot-Schinkel E, Moles MP, Boyer F, Hunault M, et al. (2010) Practical application and clinical impact of the WHO histopathological criteria on bone marrow biopsy for the diagnosis of essential thrombocythemia versus prefibrotic primary myelofibrosis. Histopathology 56: 758-767. [Crossref]
- Thiele J, Kvasnicka HM, Zankovich R, Diehl V (2000) Relevance of bone marrow features in the differential diagnosis between essential thrombocythemia and early stage idiopathic myelofibrosis. Haematologica 85: 1126-1134. [Crossref]
- Piccin A, Steurer M, Feistritzer C, Murphy C, Eakins E, et al. (2017) Observational retrospective study of vascular modulator changes during treatment in essential thrombocytemia. Transl Res 184: 21-34. [Crossref]