The role of genetic mutation in the pathogenesis of cardiomyopathies (CM) has been long-established. In contrast, the involvement of familial (hereditary) neuromuscular diseases (NDs) in the development of CM remains under-researched. Little attention has been paid to better characterize and understand myocardial involvement in NDs. There is evidence that early diagnosis may improve therapeutic response in ND patients with CM. However, diagnosis can be easily missed because signs and symptoms of cardiac dysfunction are non-specific or can overlap with that of muscular wasting (reduced physical activity) and respiratory failure due to the underlying ND. Diagnosis can also be delayed because of the late manifestations of the traditional non-invasive imaging markers of cardiac dysfunction particularly left ventricular (LV) systolic function and LV volumes or dimensions on echocardiography and late gadolinium enhancement (LGE) on cardiac magnetic resonance imaging (CMRI). For these reasons, a better understanding of longitudinal progress, cardiac complications and long-term cardiac outcomes is warranted. The clinician should also be familiar with the therapeutic options in CM associated with NDs. Thus, this review discusses important clinical and diagnostic features of CM in patients with NDs as well as emerging diagnostic and therapeutic options.
familial cardiomyopathy, dilated cardiomyopathy (DCM), neuromuscular diseases (ND), muscular dystrophies, fredrick ataxia (FA), neurofibromatosis (NF)
The genetic basis of cardiac diseases came to the fore after large population-based studies associated parental heart failure (HF) of non-ischemic origin with increased prevalence of left ventricular (LV) systolic dysfunction and elevated risk of HF in offspring [1]. Similarly, classification systems of cardiomyopathies (CMs) recognize the involvement of genetics in the pathogenesis of myocardial dysfunction by grouping CMs into specific morphofunctional phenotypes and sub-classifying each phenotype into familial (hereditary) and non-familial forms [2-6]. In the familial form, more than one family member has the same CM phenotype caused by the same genetic mutation while in the non-familial form, systemic diseases and/or exposure environmental toxins are the primary aetiologic agents [5].
Unfortunately, classifying CMs based on the underlying genetic mutation is not clinically feasible since the pathway from diagnosis to treatment rarely begins with the identification of the causative genetic defect, rather patients usually present with symptoms or incidentally discovered to have clinical signs or abnormal screening tests [5]. On the other hand, grouping CMs according to ventricular morphology and function is clinically oriented and remains the most useful method for diagnosing and managing patients with CM [4]. Nevertheless, the recognition of the genetic basis of CMs is important in the management of CMs as well as in identifying at risk offspring. While a direct effect of genetic mutation on the pathogenesis of CMs is well recognized, the evidence of CMs due to familial neuromuscular diseases remains sporadic and the understanding their diagnosis and management remains tenuous. This paper presents an overview of past and recent research on familial neuromuscular CMs and their role in the diagnosis and management of CMs in general. The review also identify critical gaps in knowledge to move forward with research to fill them.
The involvement of familial neuromuscular diseases in the pathogenesis of CMs has been described but mostly within the context of the five major morphofunctional phenotypes – dilated (DCM), hypertrophic (HCM), restrictive (RCM), arrhythmogenic right ventricular (ARVC) and left ventricular non-compaction (LVNC).
Duchenne muscular dystrophy
Overview: Muscular dystrophies encompasses a heterogeneous group of disorders that share clinical characteristics of progressive muscular weakness. In the strictest pathologic sense, the term “dystrophy” refers to chronic and severe myopathic changes within the muscle. Most muscular dystrophies share the pathologic features of fibrosis and fatty replacement particularly late in the course of the disease [7]. Duchenne muscular dystrophy (DMD) is the most prevalent neuromuscular disorder affecting up to 1 in 3,600 male births worldwide [8]. It is an X-linked recessive disorder caused by mutation in the dystrophin gene on the X chromosome leading to the absence of functional dystrophin – a sarcolemmal protein linking the cytoskeleton to the extracellular matrix by interacting with a large number of protein [9]. The clinical signs are absent at birth and the average age of diagnosis is at four years at onset of symptoms [10]. The disease progresses rapidly usually necessitating a wheelchair at the age of 10 years [11].
Dilated CM is a common clinical manifestation in DMD patients and contributes to significant morbidity and mortality. The use of corticosteroid therapy and artificial respirators have significantly extended life expectancy of DMD patients [11]. The reduction of respiratory related deaths in turn has contributed to increased cases of DMD-CM due to improved survival and advanced age of DMD patients [12,13] since the incidence of DMD-CM increases with age [14-16]. An estimated 25% of DMD patients have CM at 6 years of age and 59% by 10 years of age although cardiac dysfunction occurs in more than 90% of men ≥ 18 years of age dysfunction [17]. A severe CM has an onset at about 10 years and is prevalent in most patients by 20 years of age [17,18]. Death can occur due to HF, heart block or ventricular arrhythmias [18]. Distinct dystrophin mutation correlate to an increased incidence of CM and response to treatment [20].
Pathogenesis: Dystrophin is a protein found in the inner side of the skeletal and cardiac muscle cells with a major structural role: links the cytoskeleton to the extracellular matrix [21]. Dystrophin interacts with several membrane proteins and is involved in the regulation of signal transduction [22]. Dystrophin comprises of four distinct structural domains – (i) N-terminal actin-binding domain; (ii) a middle-rod domain with spectrin-like repeats; (iii) a cysterin-rich domain, and (iv) a carboxyl-terminal domain [23]. The amino terminus of dystrophin binds to actin and the carboxyl terminus binds to the dystrophin-associated protein complex (DAPC) at the sarcolemma. The DAPC comprises of transmembrane, cytoplasmic and extracellular proteins as well as include dystrophin, sarcoglycans, dystroglycan, dystrobrevins, syntrophins, sarcospan and nitric oxide (NO) synthase [23]. The absence of dystrophin in DMD patients affects the link between the cytoskeleton and the extracellular matrix leading to progressive muscle deterioration and weakness [21].
Cardiomyocyte degeneration in the heart results in fibrosis, which initially affects the posterobasal segment of the LV [24]. The loss of cardiomyocyte leads to an increase in wall stress and afterload [25]. The absence of dystrophin in the myocardium renders it vulnerable to pressure overload than in the normal myocardium [26]. Overtime, pressure overload leads to a progressive decrease in systolic function and an increase in myocardial oxygen consumption, ultimately leading to LV dilatation and dysfunction. In mdx mouse model, the lack of dystrophin is associated with a decrease in the myocardial neural NO and in dystrophic hearts an increase in the production of NO. Nitric oxide is associated with the development of cardiac fibrosis [27]. Abnormalities in calcium homeostasis may also be involved in cardiac muscular deterioration [28]. Dystrophin-deficient myocardium is mechanically weak and contraction of the cardiomyocyte leads to membrane damage [29-31]. The loss of membrane integrity results in a cascade of increased calcium influx into the cell and eventual cell death [14]. Increased intracellular calcium may activate proteases and the production of reactive oxygen species (ROS) resulting in alterations of protein and cellular membrane [32].
Diagnosis: The recognition of HF signs and symptoms in DMD patients is challenging because they are physically inactive and often presents with other respiratory complaints that can obscure diagnosis [33]. Current clinical guidelines recommend performing the initial cardiac screening at the time of DMD diagnosis, every two years until the age of 10 years and then yearly thereafter [34]. A majority of DMD patients often have abnormal ECG tracing – tall R waves, increased R/S amplitude in lead V1, Q waves in the left precordial leads, right axis deviation or complete right bundle branch block (RBBB) [35-37]. ECG findings precede echocardiographic findings in CM although no correlation has been established between ECG findings and the presence of CM [35,38]. Arrhythmias are also a common finding in DMD patients due to fibrosis of the conduction system in addition to the myocardium. Sinus tachycardia is common although atrial arrhythmias (atrial fibrillation, atrial flutter and atrial tachycardia) can also occur. Ventricular tachycardia (VT), premature ventricular complexes and other conduction abnormalities have been noted with a higher burden in patients with significantly depressed systolic function (LVEF < 35%) [14]. Although useful in the diagnosis of cardiac dysfunction, the diagnostic role of serum biomarkers such as cardiac troponin or B-type natriuretic peptides [BNPs] has not been established for DMD screening and further examination are required to clarify their diagnostic value [38,39].
Common cardiac imaging modalities used in evaluating DMD patients are echocardiography and cardiac magnetic resonance imaging (CMRI) although their use is technically challenging in patients with end-stage DMD because of scoliosis, ventilation abnormalities and contractures, which limits diagnostic yield [14]. Echocardiography demonstrates abnormalities in regional wall motion in the posterior basal wall, LV dilatation and depressed systolic function [40]. Current guidelines recommend echocardiography at diagnosis or at six years of age with repeat screening every 1-2 years until 10 years of age and thereafter, annual screening for LV function [41]. Cardiac MRI is used to image DMD patients and can provide an accurate assessment of LV size and function [42-44]. Late gadolinium enhancement (LGE) by CMRI has been demonstrated in the dystrophic heart, and LGE by echocardiography was present even with preserved systolic function [42]. LGE by CMRI has also been demonstrated in the posterobasal region of the LV in sub-epicardial distribution [45] and LGE in basal inferior and inferolateral walls [39,46]. Myocardial fibrosis by LGE in DMD patients increase with age and correlates with a decline in LVEF [47,48]. Thus, LGE-CMRI may provide earlier detection of cardiac involvement in DMD patients, allow for accurate and reproducible quantification of the LV function and size and promote the initiation of early cardioprotective treatment [14]. The use of CMRI has been proposed at the time of diagnosis and annually after 10 years of age unless contraindicated [14].
Management: Currently, DMD has no curative treatment although corticosteroids is the gold standard therapy that can delay DMD progression but associated with several adverse effects (weight gain, nervous system disturbance, gastrointestinal symptoms, metabolic disorders or osteoporosis), which can be reduced by finding an optimal standard regimen [49,50]. Since DMD can lead to multisystem complications, a multidisciplinary approach is essential for effective treatment. In addition to corticosteroid therapy, adjunctive treatment is crucial and involves physiotherapy, nutritional advice and respiratory support [16]. Many drugs have been assessed for the treatment of DMD – glucocorticoids, myostatin, premature termination codon, proteasome inhibitors, NO-releasing anti-inflammatory agents and histone deacetylase inhibitors but their efficacy remains unclear [51-55]. In patients with signs and symptoms of HF, management rests on conventional HF therapy including angiotensin-converting enzymes – inhibitors (ACE-I), beta-blockers and diuretics. Many experimental therapies (exon skipping, gene therapy or stem cells therapy) targeting skeletal muscle in order to restore dystrophin expression in myofibres have failed to improve cardiac function [14,16,21].
Limb-girdle muscular dystrophy
Overview: Limb-girdle muscular dystrophy (LGMD) is an umbrella term encompassing childhood or adult-onset of genetically determined progressive muscular disorders (dystrophies) that are distinct from the much more common X-linked dystrophinopathies such as DMD and Becker muscular dystrophy. It may be inherited in an autosomal dominant or recessive fashion. As such, may affect both genders rather than only males as in the case in X-linked dystrophinopathies. Typically, LGMDs are non-syndromic, with clinical involvement limited to skeletal muscle. LGMD patients generally show weakness and wasting restricted to the limb musculature, proximal weakness (muscles closer to the centre of the body – shoulder, pelvic girdle or upper thighs/arms) greater than distal weakness (muscles farther from the centre of the body – lower legs/feet or lower arms/hands) and muscle degeneration or regeneration on muscle biopsy [56]. Autosomal dominant inherited LGMDs are termed LGMD1 divided into 8 sub-groups (LGMD 1A-1H) and autosomal recessive inherited are termed LGMD2, divided into 16 sub-groups (LGMD2A-2O and 2Q) based on the affected gene locus [57,58]. LGMD has a predominantly proximal distribution of weakness that may spare the distal, facial and extraocular muscles early in the course of the disease.
Pathogenesis: The involvement of myocardium may manifest as HCM or DCM accompanied by cardiac arrhythmias potentially leading to premature deaths. Although its pathogenesis has not been elucidated, available evidence suggests similarity to that of DMD-CM. Studies have frequently documented severe cardiac dysfunction in LGMD2C, 2D, 2E, 2F and LGMD1B and rarely in LGMD1C, 2A and 2B, and severe cardiac involvement manifesting as either HCM or DCM. LGMD1B patients often exhibit findings of both CM and dysrhythmias [59]. Same to the skeletal muscles of DMD patients, the pathoanatomical evidence of cardiac involvement is the replacement of myocardium by connective tissue. LGMD patients also have an increased risk of LV thrombus formation in HF with reduced ejection fraction [59].
Several studies and case reports have reported cardiac involvement in FSHD patients [60-63]. Of 100 LGMD patients, 24%, 29% and 67% of LMGD2A, LMGD2I and LMGD2E respectively had evidence of cardiac involvement [60]. In a related study, three sisters with genetically demonstrated FSHD at the third decade of life had echocardiographic evidence of DCM and evidence of LV dysfunction by cardiac catheterization, and had the same paternal allele for three potential markers pf LMGD but different maternal alleles [61]. In a case of 57-year old female with LGMD presenting with progressive shoulder and pelvic girdle muscle weakness, exertional dyspnoea and trace bipedal oedema has LV thrombus in the setting of severe CM [62]. In a CMRI study of 16 LMGD 2I and 2B, all but one had normal LV size and systolic function, one LMDG2I had severe DCM. Of 15 patients with preserved systolic function LGE- CMRI revealed focal myocardial fibrosis in 7 (47%) of the patients. Up to 30% of patients with HF may have evidence of intracardiac thrombi on echocardiography [63]. These findings suggest in LMDG patients, the severity of cardiac dysfunction and CM varies depending on the affected gene (LMDG type).
Diagnosis: Diagnosis rests on demonstrating the evidence of LGMD mutation and cardiac dysfunction. Establishing the type of LGMD can be useful in determining the clinical course of the disease and for genetic counselling purposes, which involves obtaining medical and family history, physical examination and laboratory testing including serum CK concentration and muscle biopsy for histologic examination and protein testing [57]. Only dysferlin immunoblotting of muscle is currently thought to be specific and sensitive and findings of immunostaining of muscle should be confirmed with molecular genetic testing when it is available. However, molecular genetic testing to establish specific LGMD is problematic because of involvement of many genes, the lack of common pathogenic variants and about 50% of currently identified genes have no molecular diagnosis [57].
In the diagnosis of cardiac dysfunction, ECG that is characteristically abnormal in muscular dystrophy does not necessarily mirror the presence of significant myocardial involvement [61]. However, ECG may reveal sinus rhythm, left anterior fascicular block and non-specific T-wave abnormalities [63]. Thus, in patients with LGMD, screening with non-invasive cardiac imaging techniques is recommended since early diagnosis and therapy for CM in LGMD patients may improve long-term prognosis [61]. Transthoracic echocardiography (TTE) may reveal dilated and diffusely hypokinetic LV, a large LV apical thrombus and a severely reduced global RV and LV function (ejection fraction of 20-35%) and chest radiography may be consistent with pulmonary congestion [61,63]. In a CMRI study of 16 patients with LGMD 2B and 2I, and 8 controls, LGMD patients had normal LV size, global systolic function and peak circumferential strain but with LGE-CMRI evidence of subclinical myocardial fibrosis in 47% of LGMD patients accompanied by diastolic dysfunction in 31% of LGMD patients with myocardial fibrosis [62]. The findings demonstrates a link between myocardial fibrosis (sometimes in addition to fatty replacement) and diastolic dysfunction irrespective of aetiology [62].
Management: Treatment of LGMD-CM is not straightforward. Cardiac transplantation has been effective in some patients with LGMD1B and other subtypes who succumb to end-stage HF. Pacemaker or ICD-implantation should be considered to reduce the risk of sudden cardiac death due to bradycardia or ventricular arrhythmia. Otherwise, supportive management has been advocated for LGMD-CM patients since there is no specific therapy to modify the underlying LGMD disorder. Cardiologists plays an important responsibility in the multidisciplinary approach to the disease in order to screen and manage significant cardiovascular comorbidities and possible complications [63].
Fascioscapulohumeral dystrophy
Overview: Fascioscapulohumeral dystrophy (FSHD) is the third most common inherited muscular dystrophy after DMD and myotonic dystrophy [64] with an estimated prevalence of 1 in 14,000 – 20,000 individuals [65-68]. The term “facioscapulohumeral” is derived from the muscle groups that are initially affected by weakness and atrophy, which are the facial, scapular, and humeral muscles [64,69]. FSHD is inherited in autosomal dominant pattern associated with subtelomeric deletion of chromosome 4q with loss of a subset of repeat units in the D4Z4 macrosatellite repeat array [65]. The loss of ten or fewer repeats causes FSHD and in general the lower the number of repeats the more clinically serious the disorder. However, the function of the particular gene or genes that causes FSHD is not clear [69,70]. The initial common clinical presentation for FSHD patients include shoulder girdle weakness, facial muscle weakness, ankle dorsiflexor-muscle weakness or pelvic girdle weakness [64]. The availability of molecular diagnostic tests has broadened the spectrum of clinical presentation – calf muscle weakness and quadriceps-femoris and hamstring muscle weakness. Even with presentation of scapular muscle, shoulder girdle, and lower-extremity muscle weakness in the presence or absence of family history of similar problems requires a thorough medical examination to rule out FSHD [64].
Pathogenesis: Cardiac involvement is well known in a number of skeletomuscular dystrophies but not in FSHD, which has undermined the understanding of its specific role in the pathogenesis of CM. However, the evidence of immune cell infiltrate by FSHD histology suggests FSHD gene mutation that cause muscle weakness and deterioration may trigger a primary immune response in the pathophysiology of FSHD-CM [70]. Imaging studies in FSHD patients also support the role of immune process in the pathophysiology of FSHD-CM showing an association between CMRI characteristics consistent with an inflammatory process and the presence of infiltrating T-cells in FSHD muscle [71]. Another study shows DUX4 regulated genes as mis-expressed in some FSHD muscles with inflammatory MRI characteristics [72]. Other two related studies indicate edematous/inflammatory MRI characteristics progress to fatty-infiltration [73-75]. These studies suggest the possibility that DUX4 target gene expression and T-cell infiltrates may correlate with inflammatory characteristics on CMRI. Further studies correlating molecular characteristics of FSHD muscle with early CMRI changes may clarify the relationship between FSHD gene and inflammatory characteristics on CMRI.
Diagnosis: Diagnosis of FSHD rests on medical and familial history taking, and genic testing to identify the D4Z4 pathogenic contraction (D4Z4 repeat number less than 10) on a 4qA chromosome 4 [70]. There is no specific diagnosis for subsequent myocardial involvement and there is limited data to confirm diagnosis of CM in FSHD patients, which is often incidental or at autopsy. Only three cases of FSHD patients have been reported to have CMs providing evidence of pathological pathologic myocardial involvement [76-78].
The first was a 71-year old female with progressive cardiac insufficiency in FSHD confirmed on two other affected daughters by molecular analysis. The patient die due to cardia disease. Autopsy analysis revealed focal inflammatory infiltrates in the diaphragm and cardiac muscle involvement and histological changes resembled those observed in primary CM despite normal muscle mass volume [76]. The second patient was a 50-year old male. ECG revealed incomplete RBBB, ST elevation in leads V2-V4, tall T waves in leads V3-V5, and signs of hypertrophy. Echocardiography revealed LV myocardial thickening of the posterior wall and septum [77]. The third patient was a 38-year old male incidentally found with ECG abnormalities. Echocardiogram demonstrated mild dilatation of the LV and poor contractility. Cardiac histopathology indicated hypertrophic CM. later developed muscle weakness in the right arm and scapular winging and asymmetrical facial weakness. Muscle biopsy indicated FSH genetically confirmed in daughter with infantile symptoms [78].
The above three case reports [76-78] suggest that CM in genetically confirmed FSHD patients may manifest with non-specific ECG abnormalities, and LV myocardial thickening and poor contractility on echocardiography. With insufficient data on FSHD-CM, cardiologists should contribute to the literature on diagnosis of FSHD-CM by providing case reports and cohort studies to document the diagnostic role of ECG, non-invasive cardiac imaging and biopsy in FSHD patients with signs and symptoms of cardiac dysfunction.
Management: Current management of HSFD is supportive. There are no curative genetic or pharmaceutical treatment for FSHD although there is emerging consensus on approach to new therapeutic interventions targeting: (i) enhancing epigenetic repression of the D4Z4; (ii) the DUX4 mRNA; (iii) blocking the activity of the DUX4 protein; or (iv) inhibiting the DUX4-induced process that leads to pathology. Already, drugs that decrease epigenetic expression are now in wide clinical use such as decitabine, an inhibitor of DNA methylation, or SAHA, an inhibitor of histone de-acetylases [70]. Despite these novel drugs, the mainstay of FSHD management targets symptomatic impairment aimed to maximize functional abilities and improve quality of life of patients [64]. Physical therapy in treatment and management of pain, and improving strength, aerobic fitness, functional abilities and quality of life [70,71]. Patients with signs and symptoms of HF may require conventional HF therapies as recommended by the current expert consensus guidelines.
Myotonia atrophica (dystrophy)
Overview: Myotonia atrophica (or dystrophy: DM) is a progressive familial degenerative multisystem disease characterized by atrophy and myotonia of skeletal muscle groups but may also affect endocrine, ocular, gastrointestinal and nervous systems. Myotonia, or delay in relaxation of muscle contraction, is a hallmark feature of DM [79]. Myotonic atrophica can be divided into two clinical and genetic sub-types: (i) type 1 (DM1) or Steinert’s disease’ and (ii) type 2 (DM2) proximal myotonic myopathy. DM1 is the most common form of adult onset whereas DM2 tends to exhibit a milder phenotype with later onset of symptoms [80]. The prevalence of DM1 range from 1 per 10,000 in Iceland to 1 per 100,000 in Japan, and European prevalence of 3-15 per 100,000 [80]. Both DM1 and DM2 are inherited in autosomal dominant pattern. DM1 develops from the expansion of trinucleotide sequence on chromosome 19 and DM2 from expansion of a tetranucleotide repeat sequence on chromosome 3 [81]. DM1 is further divided into three subtypes based on the age of onset and severity; (i) congenital subtype, the most severe with onset at birth or the first year of life; (ii) child form; and (iii) adult onset [81].
Pathogenesis: Pathological features underlying myocardial findings in CM patients appear to involve cardiomyocyte hypertrophy, interstitial fibrosis, lymphocytes and/or fatty infiltration of the myocardium and the condition system [82,83]. Imaging modalities often identify evidence of structural heart disease before the onset of symptoms. A large registry of over 400 DM patients found some form of structural disease in about 20% of the patients but only 2% has overt symptoms of HF [83]. Radionuclide imaging and Doppler echocardiography may reveal diastolic dysfunction early in the disease course and later show systolic dysfunction in LV or RV. LV hypertrophy, LV dilatation and LA dilatation may also occur [80]. Regional wall motion may also occur due to non-ischemic fibrosis. In some DM patients with normal resting ejection fraction, stress testing may reveal failure of EF to augment with exercise [80]. Tanaka et al. [84] described a case of CM in a 31-year old DM female who presented with slight cardiac symptoms. Light microscopy revealed considerable degree of fatty infiltration in the myocardium, slight interstitial fibrosis and degenerated myocardial cells. Electron microscopy revealed marked mitochondriosis and small number of vacuoles consisting of single limiting membrane. Endomyocardial biopsy (EMB) was useful to detect myocardial changes even in the early stages of the disease and electron microscopy examination of the myocardium may provide finding that is pathognomonic for the development of CM in DM patients [84].
Conduction disturbances prevalent in DM patients affecting between 30% and 75% of DM1 patients [85]. Relative to DM1, DM2 patients are less studied but also have a high incidence of prolonged PR interval and evidence of heart block [82]. About two-thirds of DM1 patients have abnormal ECG findings. Conduction abnormalities are a consequence of myocyte hypertrophy, fibrosis, focal fatty infiltration and lymphocytic infiltration, which can occur anywhere along the conduction system including the HIS-Purkinje system [86]. Prolonged PR segment affect about 20 to 40% and QRS widening 5 to 25% of DM patients. These depolarization abnormalities may lead to the presence of Q waves on ECG and late potential, considered predictors of ventricular arrhythmias, may be evident. These abnormalities result from delayed myocardial activation of the His-Purkinje system rather than propagation of action potentials via focal islands of fibrosis [80]. In addition to conduction abnormalities, both atrial and ventricular arrhythmias can occur in DM patients. Atrial fibrillation, flutter and tachycardia are the most prevalent occurring in 25% of DM patients [85]. Ventricular arrhythmias (monomorphic VT, polymorphic VT and ventricular fibrillation) are less common but their presence raise more concern because of their life-threatening potential [80]. Several mechanisms may underlie ventricular arrhythmias including fibro-fatty degeneration of myocardium or the fascicles serving as a catalyst for re-entry within the ventricular wall and fascicular re-entry. DM patients are prone to bundle branch re-entry VT (BBRVT) due to a diseases conduction system and its diagnosis is vital since it is curable by radiofrequency ablation [87].
Diagnosis: The gold standard for the diagnosis of DM is genetic testing performed on blood leukocytes. Initially DM1 testing is performed followed by DM2 if clinical suspicion warrants [80]. DM is associated with a wide range of cardiovascular aberrations. The most frequently observed are conduction disturbances, arrhythmias and CM. DM1 patients may develop age-related cardiovascular complications including coronary artery disease and valvulopathies. Detection of these cardiac defects rests on a combination of laboratory, ECG and non-invasive imaging tests [84,88,89]. In a study examining the incidence and progression of ECG abnormalities in 45 DM patients, 58% on entry had at least one ECG abnormality and 38% had conduction abnormalities. New abnormalities developed in 47% during a mean follow up of 4.6 years. The overall incidence increased to 78% and conduction abnormalities to 62%. However, there was no correlation between ECG abnormalities and disease severity [88]. Muscle biopsy may also reveal variation in the size of muscle fibres as well as atrophy and fibrosis. It may demonstrate ring fibres in which myofibril bundles appear perpendicular to each other rather than in parallel [81].
Ventricular dysfunction and structural changes have been documented by non-invasive imaging (echocardiography and CMRI) [83,90].echocardiography has been show to detect diastolic dysfunction in the early phase of the disease and LV/LA dilatation and LV hypertrophy later in the course of the disease [83,84]. Cardiac MRI has greater sensitivity and reproducibility to demonstrate early abnormalities when standard cardiac evaluation is unremarkable [91]. Cardiac MRI can detect functional and structural abnormalities in 44% of DM patients: LV systolic dysfunction (57%), LV dilatation (20%) and LV hypertrophy (17%) and myocardial fibrosis in 13% strongly correlating with ECG [92]. In comparison to controls, CMRI reveals DM patients have lower end-diastolic volume index, cardiac index, and shorter myocardial T1 time, higher T1 time had a positive association with LV mass index, LV end-diastolic volume index, filtered QRS duration and low-amplitude [93]. CMRI can detect subtle changes in serial assessment and offers the promise of better defining the natural course of DM and opportunity to develop novel therapeutic approaches [91]. Finally, strain analysis promises to be a more sensitive modality to detect serial impairment in LV function compared to LVEF and thus may be of great value to assess treatment efficacy in DM patients [43].
Management: Clinical management of CM in DM patients is primarily about cardioprotective therapies to slow adverse cardiac remodelling and attenuate HF symptoms. Medical therapy for DM patients should include standard CM treatment, ACE-I or angiotensin receptor blockade (ARBs) and beta-blockers. The aim of these drugs is to reverse remodelling. ACE-I has been shown to slow-down the onset of CM in DMD patients and mortality benefits but these benefits have not been confirmed in DM patients [94]. Medical therapy for atrial arrhythmias should be administered with close monitoring since DM patients are prone to bradyarrhythmias and conduction system block at the atrioventricular node. Anticoagulation therapy in DM patients with atrial arrhythmias should consider skeletal muscle symptoms and gait instability that renders prone to falling [94]. More recent trials have evaluated carbamazepine or mexiletine for myotonia [95]. Larger longitudinal studies of DMI patients have calcified Indications for device therapies including pacemakers and defibrillators. Surface ECG sis useful in determining patients who may benefit from device implantation since patients with prolonged PR (> 240msec) and widened QRS (>120) are at an increased risk [96]. For DM patients and prolonged HV intervals, pacemakers protect from bradyarrhythmias and heart block but also allow for the diagnosis of potentially life-threatening tachyarrhythmias through device interrogation [81].
Myasthenia gravis
Overview: Myasthenia gravis (MG) is a potentially serious but treatable organ specific autoimmune disease in which antibodies bind to acetylcholine receptors (AChR) or to functionally related molecules in the postsynaptic membrane at the neuromuscular junction [97,98]. The antibodies induce weakness and fatigability of the skeletal muscles, which are the hallmarks of the disease [98-100]. The prevalence of MG ranges between 2 and 7 per 10,000 persons in the U.K. [101] and 1.5 per 10,000 in central Virginia and Western Virginia [102]. The disease can present at any age but a bimodal peak of incidence – the first peak in the third decade (predominant in women) and the second peak in the sixth and seventh decades ([predominantly in men) and the incidence falls after 70 years of age [97]. The disease has a slight female preponderance, with a sex ratio of 3 to 2 [103]. Cardiac involvement in MG takes several forms ranging from asymptomatic ECG changes to VT, myocarditis, conduction disorders, HF and sudden death. MG patients have a higher prevalence (10-15%) of cardiac manifestation in the present of thymoma [104]
Pathogenesis: A review of literature on cardiac manifestations in MG associated thymoma, anti-striational (especially anti-Kv 1.4) antibodies and advancing age with cardiac involvement. In addition to AChR antibodies specific to skeletal muscles but do not bind the heart, nearly half (48%) of all MG cases and 97% of all thymoma-associated MG patients have antibodies towards the heart muscle [104]. Although support for the pathogenic role these antibodies are lacking, they may influence cardiac function by complement activation and T-cell proliferation [104]. Cardiac involvement in MG patients has also been associated with giant cell myocarditis (GCM) and Takotsubo CM as common comorbidities [104]. GCM is a severe form of myocarditis with myonecrosis and serpiginous infiltrate of chronic inflammatory cells including giant cells [104]. Twenty-two cases of GCM with thymoma and 12 autopsy cases of MG with thymoma, polymyositis and myocarditis have been reported in which giant cells were found in half of the cases in both myocardium and skeletal muscles [105,106]. Case reports describe giant lymphocytes in the myocardium and skeletal muscles of a 68-year old female with MG after she developed myocarditis [107] and GCM in a 72-year old male with MG both cases having thymoma [105]. Stress-induced CM is a reversible form of LV dysfunction precipitated by emotional or physical stress causing a surge in catecholamine resulting in suppressed myocardial function [108]. MG can precipitate severe stress during crisis episode [104]. Stress-induced CM has been observed in patients on plasmapheresis and intravenous immunoglobulin during myasthenia crisis [109-113]. MG and CM developed concurrently in 68-year old female after mitral valve replacement associated with myasthenia crisis [114]. Older patients with severe MG appear to be at an increased risk of stress-induced CM while GCM occurs more often in elderly cases with thymoma [104].
Diagnosis: Diagnosis of cardiac dysfunction in MG patients remains a clinical challenge. Overlap of symptoms such as fatigue, dyspnoea and poor exercise tolerance between MG and cardiac disease may result in under-recognition of cardiac manifestations [104]. Most published data based on case reports [103,115] and case-controlled studies indicate the presence of ECG abnormalities in MG patients [116]. In a case report of two MG patients, one had symptomatic conduction disturbances with frequent episodes of syncope requiring pacing and the second had giant diffuse T waves [103]. In another case of a 65-year old MG male, he developed worsening chest pain and his ECG revealed ST-segment elevation in multiple leads who subsequently went into cardiac arrest due to monomorphic VT [115]. In a retrospective analysis of 58 MG patients who underwent ECG and echocardiography, 56.8% had ECG abnormality: atrial fibrillation (10.3%), AV block (6.8%), non-specific ST depression (29.3%) and negative T-wave (29.3%). On echocardiography, there was no significant difference in E/E’ between patients with and without ECG abnormality (11.2±3.2 vs. 8.7±2.2, p = 0.03). Fourteen (14) out of 15 patients with ECG abnormality had evidence of cardiac damage compared to 6 out of 33 without ECG abnormality. Reduced LVEF was observed in 8.6% of patients with ECG abnormalities and none without ECG abnormality. Diagnostic sensitivity, specificity, positive predictive value and negative predictive value of ECG was 70%, 71%, 56% and 82% respectively [116]. These findings suggest ECG may aid as the first step for further examination of cardiac damage in MG patients. Prospective studies will be needed to make recommendation for differential diagnosis by non-invasive cardiac screening in MG patients since it often occurs in the setting of other cardiac diseases particularly GCM and stress-induced CM.
Management: There is no curative therapy for MG but medical treatment with immunosuppressants (azathioprine) or cholinesterase inhibitors can be useful in symptom control [117]. Immunosuppressants alter the body’s immune system to produce fewer antibodies that cause MG while cholinesterase inhibitors improve communication between nerves and muscles effective in MG patients with mild symptoms [115]. In the case of a tumour, the thymus gland may be surgically removed. Plasmapheresis (depleting the body of blood plasma without depleting blood cells may remove unwanted antibodies [117]. These treatments are only effective in the short-term and their effect on cardiac structure and function have not been demonstrated [104].
Juvenile spinal muscular atrophy
Overview: Spinal muscular atrophy (SMA) refers to a group of genetic neuromuscular disorders characterized by the degeneration of alpha motor neurons (anterior horn cells) of the spinal cord (considered the most salient feature) leading to weakness of the lower motor neurons and progressive muscular atrophy [118-120]. SMA affects 1 in 6,000 to 10,000 infants with a carrier frequency of 1 in 40 [118]. The most common form of SMA accounting for 95% of the cases is autosomal recessive proximal SMA caused by mutations in the survival of motor neurons (SMN1) gene localized to 5q11.2-q13.3 [118,119,121]. SMA includes a wide range of phenotypes classified based on age of onset: infantile (Type 1); Intermediate (Type 2), Juvenile (Type III) and adult onset (Type IV) [120,121]. Mortality and morbidity of SMA inversely correlate with age at onset [122]. Although cardiac manifestation is prevalent in type I/II, only type III, a juvenile form of slowly progressive spiral amyotrophy (Kugelberg-Welander disease [KWD]) has been associated with the development of CM but the incidence of CM is difficult to appreciate, as cases are sporadic.
Pathogenesis: The pathogenesis of CM in SMA patients remain incompletely understood possibly attributed to a very low prevalence of the disease and a paucity of studies. Cardiac involvement in general has been described in SMA Type 1 patients who present at birth with a high degree of pulmonary involvement in which SMA may be secondary to respiratory insufficiency [122]. Cardiovascular and autonomic nervous systems have also been involved in SMA but direct evidence of myocardial involvement is lacking in these patients [123,124]. A retrospective study of SMA Type 1 found 24% experienced symptomatic bradycardia with no clinical nor instrumental signs of CM. However, ECG findings revealed signs of RV overload in 37% of the patients possibly provoked by pulmonary hypertension due to respiratory anomalies [123]. A systematic review of 72 studies enrolling 264 patients reported various cardiac dysfunctions in SMA Type I and II including abnormalities in the position and connection of the heart, atrial and atrial septum, AV valves and AV septal defect, ventricles and ventricular septum, coronary arteries, arterial duct and pericardium. The study did not find any reported case of CM in these patients [124].
Although the juvenile form of SMA is rare, cases of CM in the progressive form of this disease have been reported [125-129]. Tanaka et al. [125] reported two cases of KWD-associated CM. The first patient, a 24-year old man had atrial flutter with complete AV block and on echocardiography has increased LA and LV dimensions. The second patient, a 26-year old man, had AV junctional rhythm, Deep Q waves, RS pattern in lead V1 and slight myocardial interstitial fibrosis on histologic examination. These findings suggest the involvement of the atrium, ventricular myocardium and AV conducting tissue and atrial arrhythmias, AV conduction disturbances and HF may occur in KWD [125]. Kimura et al. [126] reported a case of a 21-year old KWD woman with conduction disturbance (atrial standstill and AV junctional rhythm) and cardiomegaly on chest x-ray. In a prospective study of 8 KWD patients, Elkohen et al. [127] reported classical features of atrial hyperexcitability with variable degrees of atrioventricular block and one patients was hospitalized with DCM with a fatal outcome. Roos et al, [128] described a case of a 43-year old male with AV block and implanted with dual chamber pacemaker, who later developed non-sustained VT progressing in duration and frequency and on electrophysiology study had prolonged His-ventricular interval duration and induction of sustained VT. Takahashi et al. [129] reported complete RBBB and transient complete AV block with escape rhythm in a 51-year old KWD male. The presence of CM has been described in these patients although cardiac involvement has been suggested to be secondary to chronic respiratory insufficiency typical of the disease.
Studies on mouse model of SMA examined arrhythmias and cardiac defects as a feature of SMA [130-132]. These studies reported a severe model of SMA mice suffered from severe bradyarrhythmias characterized by progressive heart block and impaired ventricular depolarization. Further investigations revealed evidence of both sympathetic innervation defects and DCM ay the later stages of the disease [130-134]. Myocardial fibrosis is frequent in both severe and intermediate SMA believed to contribute to arrhythmias in SMA with arrhythmias. Bradycardia is common due to delays in the cardiac electrical conduction system causing various types of AV blocks and BBB. However, since myocardial fibrosis is a hallmark of normal aging, and limited number of patients precludes a definitive conclusion whether impulse conduction disorders in SMA are due to pre-senile cardiac fibrosis secondary to SMN deficiency [122].
Diagnosis: Patients with KWD should be evaluated regularly for cardiac disease. Common features of cardiac dysfunction in SMA includes conduction disturbances, and atrium and ventricular dilatation and LV dysfunction. ECG and non-invasive cardiac imaging should be performed to assess conduction disturbance and atrial and ventricular structure and function [127,135]. There is need for prospective studies to evaluate pathological cardiac features associated with SMA to support diagnosis by non-invasive cardiac imaging.
Management: No medical treatment for juvenile progressive SMA is available although several candidate treatment agents have been identified and are in various stages of development [119]. Current treatment strategies are largely supportive, targeting the management of arrhythmias, and signs and symptoms of HF. Arrhythmias are common and portend an ominous prognosis as well as are associated with increased mortality and morbidity in these patients [126-128]. Management of arrhythmias is a common therapeutic target. Successful use of implantable cardiac pacemaker [126], a dual chamber pacemaker upgraded to a prophylactic dual chamber cardioverter defibrillator [128], emergency temporary pacing followed by permanent pacemaker implantation [129] have been reported. Further studies are warranted to shed light on the efficacy of standard HF medication on SAM patients with severe cardiac dysfunction.
Friedreich's ataxia
Overview: The term “ataxia” refers to impaired coordination of voluntary muscle movement [136]. Friedreich ataxia (FA) is an autosomal recessive inherited neurodegenerative disease that most often presents in childhood or in young adulthood with progressive ataxia leading to loss of ambulation after 10-15 years [137,138]. It is the most common inherited ataxia in Europe with a prevalence of 1 in 20,000 in south-west Europe and 1 in 250,000 in north and east Europe [139]. In a majority of cases, FA results from a homozygous guanine-adenine-adenine (GAA) triplet repeat expansion in frataxin gene and the shorter repeat expansion length correlated with age at onset and disease severity. Besides the characteristics features of spinocerebellar ataxia, the heart may also be affected, and patients may experience HCM eventually progressing towards HF and death [140]. A substantial proportion of FA patients also develop CM usually presenting as progressive LV hypertrophy, thickening of ventricular walls (FA-CM), which significantly reduces the mean life expectancy to approximately 40 years, and about 60% of FA patients die from arrhythmia or cardiac failure [141,142]. Cardiac morphology varies and cardiac symptoms may develop early in life but the extent and timing of cardiac involvement correlate poorly with the level of neurological disability. Histological changes in LV consists of cellular hypertrophy, diffuse fibrosis and focal myocardial necrosis [143]. Patients with FA-CM develop heart involvement early during their lifetime (< 40 years) [144].
Pathogenesis: The most common pathology in FA-CM heart is concentric hypertrophy with reduced ventricular sizes and thickened walls, and extensive endomysial fibrosis [145]. Although this results from FA, the exact association between neurological and cardiac involvement in patients with FA remains incompletely understood. Nevertheless, the major pathophysiological feature of FA is neurodegeneration of axons in the spinal cords and spinal roots resulting in loss of myelinated axons in peripheral nerves increasing with age and disease duration [144,145]. Unmyelinated fibres in sensory roots and peripheral sensory nerves are spared. Myocardial muscle fibres also show degeneration and replaced by macrophages and fibroblasts. Chronic interstitial myocarditis develops with hypertrophy of cardiac muscle fibres and the loss of fibre striations. This is followed by swelling and vacuolation, and finally interstitial fibrosis [145]. In a study of 205 FA patients, Weidemann et al. [144] found pathological features in CM include progression towards LV hypertrophy and a decrease in global LV function. In mild FA-CM, the thickness of the LV walls is homogenous and LV is not dilated while in severe FA-CM, the septum is significantly thicker than the posterior wall [144].
Further, autopsy and biopsy studies suggest that FA patients can develop myocardial fibrosis, which might be responsible for the observed shrinking of the myocardium in severe FA-CM. The thinned posterior wall compared to the septum in FA suggests the progression towards fibrosis is more advanced in the posterior cardiac segments, which is consistent with findings of posterior wall showing lowest values for regional longitudinal function [146]. The phenomenon of developing fibrosis particularly in the posterior wall is well known in other genetic CM such as DMD and Fabry disease [147,148]. Thus, FA-CM progress is characterized from initial concentric remodelling in early stages followed by concentric hypertrophy in advanced stages. At this time, myocardial fibrosis gradually develops influencing heavily myocardial morphology and function. Fibrosis leads to LV wall thinning and LV dilation while ejection fraction remains stable for a long time and decreases only in end-stage hearts. Therefore, FA-CM patients in advanced stages present with a slightly thicker septum compared to the posterior wall but this difference is not easily recognizable [140,144].
Diagnosis: The demonstration of myocardial involvement in FA-CM patients rests on non-invasive cardiac imaging by echocardiography and CMRI. Echocardiographic hallmark of FA-CM is LV hypertrophy. The typical pattern is concentric LVH with an end-diastolic wall thickness < 15 mm in the absence of outflow tract obstruction [144,149]. Cross-section analyses in large FA cohorts reveal altered LV geometry based on LV mass index and diastolic relative wall thickness in 80% of all FA patients and 40% of all FA patients show concentric remodelling, 35% concentric hypertrophy and only 5% have eccentric hypertrophy [150]. Global systolic function is normal in many patients and only end-stage FA-CM patients develop reduced ejection fraction with global hypokinesia and slightly dilated LV [144,150]. The diagnostic use of CMRI in FA-CM has been recent. In CMRI, LV mas positively correlates with the GAA repeat number (GAA1 repeats > 6000) and with the age of disease onset. LV mass also decreases with longer disease duration (> 15 years) suggesting cardiac thinning with prolonged disease [144].
On ECG, QRS duration is normal in most FA-CM patients even with significant LVH, patients do not show prolonged QRS duration indicating LBBB is an uncommon finding, which is in contrast with other HCM [144]. In the advanced stages of FA-CM, patients may present with high S-wave in leads V1 and V2, and high R-wave in V5 and V6 indicating ECG signs of LVH [151,152]. T-wave abnormalities (flattening or inversion) is common in almost all FA-CM patients in the left chest leads [153]. Although it has been suggested that some patients with advanced FA-CM may suffer from supraventricular tachycardia like atrial fibrillation and flutter and AV re-entry tachycardia [154], QTc interval are normal in most patients suggesting they may not be particularly susceptible to malignant ventricular arrhythmias [152]. Thus, it remains unclear whether a specific arrhythmic event is responsible for many deaths of FA-CM patients [154].
Clinical signs of cardiac develop are not straightforward and usually develop later in the course of the disease. In addition, neurological involvement determines limitation in physical activity rather than the stage of CM, and it is difficult to quantify the progression of HF using the New York Heart Association functional classification [153]. Thus, in patients with advanced neurological disease, subclinical CM may be present despite the absence of exertional symptoms [144].
Management: Specific tailored treatment regimens for FA-CM are lacking although treatment options such as antioxidants (idebenone) and iron chelation (deferiprone) are in use but do not lead to sustained clinical improvement [155]. With no effective specific treatment, conventional treatment including HF medical therapy, anti-arrhythmic drugs, and device implantation are used in FA-CM patients for management of HF symptoms, LV systolic dysfunction or arrhythmia. Management of HF symptoms often requires salt restriction and diuretic therapy and the use of afterload reduction agents such as ACE-I/ARBs may be beneficial in long-term treatment of the heart in FA-CM patients but beta-blockers that slow heart rate may not be well-tolerated [149]. Cardiac transplantation may be offered as a therapy for patients with end-stage HF in FA [144].
Neurofibromatosis
Overview: Neurofibromatosis (NF; or von Recklinghausen disease) is a hereditary condition due to mesodermal and neuroectodermal dysplasia transmitted as an autosomal dominant condition although 50% of the cases may arise as spontaneous mutations [156,157]. The incidence ranges between 1 in 2,000 and 1 in 4,000 with no gender bias [156]. It has two genetic distinct sub-types: NF1 affects 1 in 35,000 and NF2, which is much rarer, affects 1 in 25,000 persons [158]. NF1 is genetically inherited disease with formation of nerve tissue tumours (neurofibromas), which may be asymptomatic or can cause symptoms by local tissue compression. The disease affects all neural crest cells (Schwan cells, melanocytes and endoneural fibroblasts). NF1 present with variable array of clinical expression due to dysplasia of mesodermal and neuroectodermal tissues [156]. Cardiovascular manifestations of NF include hypertension (the most common affecting 6% of NF1 patients) as a result of renal artery stenosis, congenital heart disease, HCM and less frequently Pheochromocytoma [156-158].
Pathogenesis: Initial studies suggested that congenital cardiac defects were more common in NF1 population but a subsequent review found no clear evidence of any link [157]. The evidence of CM in NF1 patients has also been inconsistent or circumstantial. In a retrospective study that analysed medical records of 65 NF1 patients (60% with sporadic form while 40% with the familial form), none had evidence of renal arterial stenosis, HCM, mitral valve prolapse or coarctation of the aorta [160]. However, other cardiac abnormalities (mitral valve regurgitation without mitral valve prolapse, tricuspid valve regurgitation and mild aortic valve regurgitation was reported [160]. On the other hand, two case reports specifically reported HCM on NF patients [161,162]. Jurko et al. [161] reported a case of 18-year old boy with NF1 and HCM with systolic anteward movement of the anterior leaflet of the mitral valve, and LV outflow gradient (85 mmHg) secondary to subvalvular aortic stenosis with LV diastolic dysfunction. Fitzpatrick et al. [162 also reported two siblings from a family with NF inherited as an autosomal dominant had clinical evidence of HCM. ECG showed considerable LVH. Cross-sectional and M-mode echocardiography showed HCM with asymmetric septal hypertrophy with preserved systolic function. However, there are doubts of whether NF1 and HCM is a coincidental finding in two conditions that have similar incidence. Fitzpatrick et al. [161] suggests that the LVH may be secondary to NF and because of the abnormal metabolism of catecholamines or nerve growth factor, or that both disease represent defects of neural crest tissue. Moreover, neurofibromas may involve the heart causing both hypertrophy and outflow tract obstruction [157].
Diagnosis: NF1 patients can develop CM with potentially progressive course. Based on case reports, and bibliographic data, NF1 patients should be followed up in the cardiology clinic and echocardiography should be carried out early, with any changes on ECG or thoracic x-ray. At present, there are no validated diagnostic methods for CM in NF1 patients, with case reports identifying HCM in NF1 patients. Additional studies are warranted to develop differential diagnosis for CM in NF1 and HCM (Table 1).
Table 1. Summary of the included studies
1st Author [Ref#] |
Year |
Sample (Trtnt/ Ctrl) |
Mean Age |
Patients |
The Aim of the Study |
Summary of Major Findings |
Silva [147] |
2007 |
7/3 |
13.1±3.6 |
D/BMD + MF |
To analyze whether CMRI can detect and quantify myocardial damage in early stages of CM in DMD |
CMRI can identify MF and may be useful for detecting early stages of CM in DMD |
Turkbey [93] |
2012 |
33/13 |
46.3±14.7 |
DM |
To define functional and post-contrast myocardial T1 time CMR in DM patients |
CMRI associate DM with structural alterations - shorter post-contrast myocardial T1 time compared to controls reflecting the presence of diffuse MF |
Hor [47] |
2013 |
113/201 |
15.2±5.1 |
DMD - LGE positive |
To establish the prevalence and distribution f LGE in DMD population and relationship with age and LEVF |
LGE occurs early, is progressive and increases with both age and decreasing LVEF. Time course and distribution of LGE promises to be clinical biomarker to aid management of DMD-CM patients |
Gaur [166] |
2016 |
12/20 |
8.0±2.75 |
DMD |
To assess CMRI biomarkers of muscle fat percentage and cardiac T1 and T2 relaxation times |
CMRI imaging of upper arm skeletal muscle and heart provides clinically useful tests for diagnosing, monitoring disease progression and therapeutic response. |
Mavrogeni [167] |
2017 |
12/34 |
10.5±1.3 |
DMD |
To evaluate cardiovascular function in DMD/BMD patients using CMRI |
DMD/BMD patients under deflazacort and perindopril treatment have preserved LV function and lack of LGE. |
West [168] |
2018 |
27/34 |
14(12,16) |
DMD - LGE positive |
To describe the utilization of CMRI in DMD patients for identification of LV fibrosis |
CMRI is an accurate screening tool that can detect fibrosis without volume changes and help with treatment intensification |
Iwase [169] |
2010 |
7 |
57.0±6.0 |
DMD carriers |
To assess the utility of CMRI for the detection of myocardial damage in female carriers of DMD |
CMRI is a useful modality for detecting cardiac involvement in DMD carriers – 57% had LGE-CMR findings characterized by subepicardial LGE localized at inferolateral segments |
Giglio [170] |
2014 |
30 |
36.0±5.0 |
DMD carriers |
To assess whether CMRI in DMD carriers may index any cell milieu of LV dysfunction |
A typical myocardial LGE pattern location was a common finding in DMD carriers frequently subepicardial and mid myocardial with preserved LV systolic/diastolic function 47% LGE-CMRI |
Lang [171] |
2015 |
22 |
NR |
DMD carriers |
To characterize the degree of myocardial fibrosis and LV dysfunction in DMD carriers |
18% LV dysfunction, 35% had LGE-CMRI. A high rate of LGE and LV dysfunction in DMD carriers |
Schelhorn [172] |
2015 |
15 |
32.3±10.2 |
DMD carriers |
To investigate asymptomatic DMD carriers for cardiac abnormalities using CMRI |
High frequency of cardiac pathologies: increased normalize LVEDV (7%), LVESV (20%), reduced EF (33%) LGE 60% predominantly in midmyocardial and subepicardial |
Mah [173] |
2019 |
57/20 |
NR |
DMD carriers vs. non-carriers |
To evaluated diagnostic value of graded exercise and CMRI to determine prevalence of cardiac disease in DMD carriers |
Exercise test and CMRI in genetic carries of DMD/BMD shows prevalence of disease (44% carriers vs.5%1 non-carrier had fibrosis |
Raman [174] |
2010 |
26/8 |
35.8±11.7 |
FA |
To test the hypothesis that abnormal myocardial perfusion and fibrosis represent early manifestation of CM |
FA patients have abnormal perfusion reserved and fibrosis in the absent of significant hypertrophy. |
Weidemann [144] |
2012 |
25/65 |
28.8±11.1 |
Severe FA |
To assess myocardial hypertrophy in FA-CM |
Echocardiography-based IVSTd and ejection fraction provides a readily applicable clinical grouping of CM associated with FA and neurological status. |
Weidemann [175] |
2015 |
13/2 |
32.0±11.0 |
Severe FA |
To provide a detailed description of various disease stages in FA-CM and described cardiac progression of cardiac involvement overtime. |
ECG signs may be early signs to unravel established CM in FA patients |
BMD: Becker Muscular Dystrophy; DM: Myotonia Dystrophy; DMD: Duchenne Muscular Dystrophy; FA: Fredrick Ataxia; LGE: Late Gadolinium Enhancement; IVSTd: End-diastolic Wall Thickness of the Interventricular Septum; M F: Myocardial Fibrosis;
Management: Disease modifying therapy in NF patients is neuromuscular blocking agents. However, NF1 patients with renal impairment or concurrent medication (anticoagulant) should have neuromuscular transmission monitored because of potential interference with the normal pharmacokinetics or pharmacodynamics of neuromuscular blocking drugs [157]. However, the effect of these drugs on cardiac function has not been demonstrated. Further studies on conventional HF medication in these patients is needed to develop a more effective clinical management approach. In the case of HCM, it is necessary to follow-up these patients as they may need surgical intervention [161].
Neuromuscular diseases (ND) particularly muscular dystrophies and FA are fatal familial disorders that can lead to progressive muscular weakness, respiratory failure, CM and ultimately HF. This particular form of CM has an ominous prognosis; thus, early diagnosis and monitoring disease progression is vital to improve therapeutic response. However, definitive diagnosis of CM in ND patients by echocardiography, the widely available non-invasive cardiac imaging modality, is often delayed or even missed because findings are usually inconsistent or become clinically relevant during the advanced stages of CM [163,165]. Although not in widespread use, CMRI is the clinical gold standard non-invasive imaging for high quality anatomical, structural and functional cardiac imaging. Cardiac MRI typically incorporate high-resolution, static and black-blood imaging to evaluate cardiovascular anatomy, cine white-blood imaging to assess regional and global cardiac function and post contrast static LGE for evaluating the presence or absence of myocardial fibrosis [163]. Cardiac MRI promises to improve diagnosis of CM associated with familial NDs by detecting subclinical myocardial changes even before the onset of myocardial dysfunction. The present meta-analysis pools published data on the use of CMRI in clinical evaluation of cardiac function in patients with muscular dystrophies including female carriers of X-linked inherited muscular dystrophies.
The search for relevant studies was performed on PubMed and Google Scholar using the following search terms: cardiac magnetic resonance imaging (CMRI) and neuromuscular diseases and with each of the following individual diseases – Duchenne muscular dystrophy, limb-girdle muscular dystrophy, facioscapulohumeral dystrophy, myotonia dystrophy, myasthenia gravis, juvenile spinal muscular atrophy and Fredrick ataxia. Studies were included if they included patients with genetically confirmed neuromuscular diseases and evaluated them using CMRI modalities. Studies were excluded if they did not compare ND patients with CM to age-matched healthy controls (except studies that enrolled DMD carriers) or did not provide raw quantitative data on diagnostic findings. Categorical data was expressed as frequency and percentage, continuous data as mean and standard deviation and dichotomous data as weighted mean difference (WMD) and 95% confidence interval. Heterogeneity across studies was estimated by the inconsistency index I2, which also formed the basis of selecting either fixed or random effect model for pooled analysis. Differences were considered statistical significant at p < 0.05.
Study/Patient characteristics
The systematic electronic search yielded 112 publications of which only 14 that met the inclusion criteria were included in the final dataset for analysis [47,93,144,147,166-175]. The 14 studies were recent, published between 2007 and 2019. Only three muscular dystrophies (DMD, BMD and DM) with myocardial fibrosis or with the presence of LGE were evaluated in eleven studies [47,93,144,147,166-173]. Of the 11 studies, six [47,93,144,147,166-168] evaluated cardiac changes in DMD male patients while the remaining five [169-173] evaluated cardiac changes in DMD female carriers. The other three studies assessed myocardial changes in FA patients [144,174,175]. In total, the 14 included studies enrolled 799 patients consisting of 399 with CM due to NDs and 400 age-matched healthy controls. The mean age of the patients was 27.4±15.4 years. All DMD patients were male, all DMD carriers were female whereas FA patients had an almost equal gender representation (male = 49%; female = 51%).
Diagnostic findings
Common conventional CMRI features evaluated in the 14 studies were LV systolic function, cardiac index and LV structure and dimensions. Pooled analysis of weighted mean difference differences between CM patients and healthy controls revealed significantly reduced LVEF in CM patients (WMD: -8.39%; 95% CI: -12.6% to -4.17%; p = 0.000) [93,144,147,166,167,174,175]. Significantly increased LV mass index (LVMI - WMD: 8.38; 95% CI: 1.39 to 15.37; p = 0.019) [93,47,144,166,174,175] and slightly but significantly increased LV end systolic volume index (LVESVI – WMD: 11.73; 95% CI: 1.30 to 22.16; p = 0.028) [93,144,147,166,167,174]. Despite the differences, LVEF, LVMI and LVESVI were all within normal ranges in CM patients suggesting the absence of myocardial pathologic changes. There were no significant differences in LV end diastolic volume and cardiac index between CM patients and healthy controls (p > 0.05) (Table 2).
Table 2. Weighted mean difference for CMRI indices between treatment and control
MRI Feature |
Studies |
WMD |
95% CI |
P-value |
LVEF |
[93,144,147,166,167,174,175] |
-8.39 |
-12.61 to -4.17 |
0.000 |
LVMI |
[93,47,144,166,174,175] |
8.38 |
1.39 to 15.37 |
0.019 |
LVEDVI |
[147,93,47,166,167,174] |
1.55 |
-5.84 to 8.94 |
0.681 |
LVESVI |
[93,144,147,166,167,174] |
11.73 |
1.30 to 22.16 |
0.028 |
Cardiac Index |
[47,147] |
0.056 |
-0.575 to 0.688 |
0.861 |
WMD: Weighted Mean Difference
Further analysis of newer CMRI cardiac markers suggested subclinical myocardial changes. Pooled analysis in 11 studies [47,93,147,168-173] evaluating the presence of LGE in CMRI revealed it was present in 252 out of 588 patients, translating into an event rate of 52.6% (95% CI: 39.8% to 65.0%: Figure 1). The presence of LGE was also prevalent in female DMD carriers occurring in 77 out of 129 carriers (Event rate: 55.9%; 95% CI: 38.8% to 71.8%) [169-173] (Figure 2). The mean myocardial T1 time of CM patients was significantly shorter compared to healthy controls (WMD: -1.754; 95% CI: -3.5 to -0.003; p= 0.05: Figure 3) [93,166]. Individual studies also reported evidence of subclinical myocardial changes. Myocardial to skeletal muscle T1 ratio was significantly reduced in CM patients compared to healthy controls (0.67±0.08 versus 0.76±0.08) suggesting greater accumulation of gadolinium in the myocardium relative to skeletal muscle [93]. Impaired myocardial perfusion reserve index (MPRI) in CM patients. The ratio of endocardial to epicardial MPRI revealed significant impairment in CM patients compared to controls (0.80±0.18 versus 1.22±0.36; p = 0.01) [174].
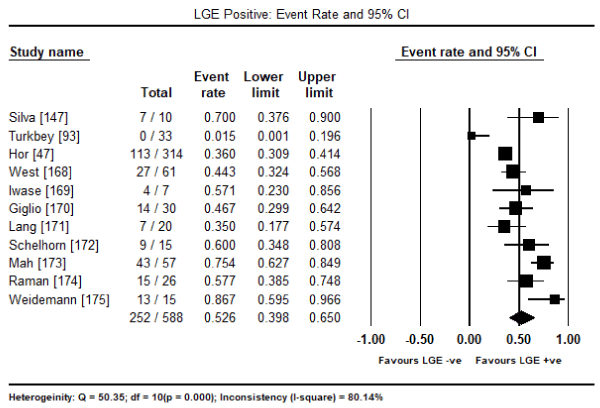
Figure 1. LGE event rate and 95% confidence interval in DMD patients
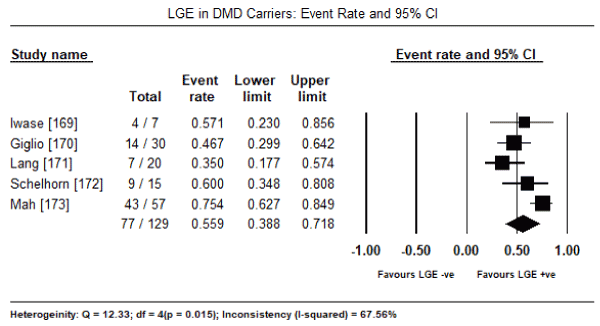
Figure 2. LGE event rate and 95% confidence interval in DMD carriers
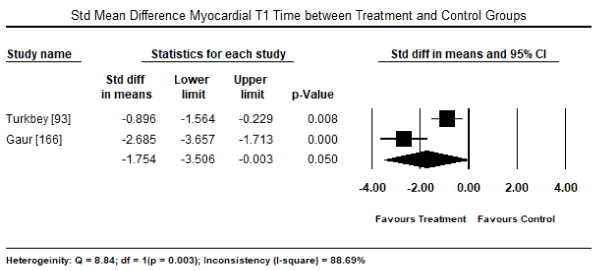
Figure 3. Weighted mean difference - myocardial T1 time between patients and controls
Key findings
Early diagnosis of CM in patients with NDs remains a clinical challenge because signs and symptoms of cardiac dysfunction are non-specific, and physical inactivity in these patients in the presence of other respiratory complaints can complicate diagnosis [33]. Late manifestations of echocardiographic evidence of cardiac dysfunction in ND patients often delays diagnosis. Cardiac MRI is emerging as a promising non-invasive modality for the detection of subclinical myocardial changes as well as monitoring disease progression to improve therapeutic response. In the present meta-analysis, although conventional CMRI markers such as LVEF, and increased LV systolic and diastolic volumes are reduced and elevated respectively in CM patients relative to healthy controls, they are within normal ranges. On the other hand, the presence of LGE-CMRI in 53% of ND patients and 56% of asymptomatic female DMD carriers is a marker for myocardial fibrosis, which is believed to be a key feature in the pathophysiology of CM. In addition, significantly shortened mean myocardial T1 time and reduced myocardial to skeletal muscle T1 ratio and impaired myocardial perfusion reserve index all suggest subclinical myocardial changes before the onset of CM.
Conventional CMRI markers
The role of CMRI in the clinical evaluation of cardiac structure and function in patients with NDs is consistent with current evidence on the value of CMRI in evaluating cardiac health in HF patients as well as in consensus reports on the diagnosis, treatment and monitoring of HF patients [176,177]. In recent decades, cardiac MRI has emerged as the clinical gold standard for high quality anatomical, structural and functional imaging of the heart. CINE and LGE- CMRI are common modalities used in clinical evaluation of ND patients. CINE CMRI permits the evaluation of LV remodelling and wall thickening as well as quantitative LV global function.
On CINE CMRI, the quantitative LV remodelling index (LVRI) defined as the ratio between LV mass and volume is useful in distinguishing HCM from DCM, which is relevant in DMD patients who nearly always present with the DCM phenotype. However, although LVRI is significantly lower in DMD patients compared with normal controls, the median LVRI in DMD patients falls within normal published LVRI limits and thus is not effective in identifying detrimental LV remodelling in DMD patients [178]. Quantitative analysis of CINE images yields useful markers of cardiac function – LVEDV, LVESV, LVEF, LV stroke volume, LV cardiac output and LV mass as well as RV indices [179]. LVEF from CINE images is the most widely relied on marker for a variety of cardiac diseases and is an indicator of cardiac dysfunction in DMD boys. A cut-off of <50% clinically defines abnormal cardiac function and <45% is a good predictor of fatal cardiac outcomes [180,181]. However, only DMD patients with significant cardiac involvement present with reduced LVEF and in subclinical disease, LVEF remains preserved as observed in the present meta-analysis.
CMRI tissue characterization by LGE permits evaluation of changes in the myocardial extracellular matrix remodelling during disease [182]. In regions of focal or dense fibrosis, gadolinium extravagates, which enhances its appearance on CMR images while normal myocardium appears dark. In the present meta-analysis, LGE-CMRI was present in more than half of DMD boys including asymptomatic female carriers and has been shown to correlate with reduced LVEF in these patients [42]. In LGE negative DMD males (myocardial fibrosis absent) LVEF declined 0.58±0.1% per year while in LGE present DMD males mean annual decline is 2.2±0.31% [48]. Overall, LGE presence has a clinical value for identifying the presence of advanced cardiac involvement in DMD but does not appear to be useful indication early stages of the disease [42,163].
Emerging CMRI markers
Myocardial perfusion imaging with CMRI permits differentiation of ischemic and non-ischemic myocardium but its use has not been widely reported in DMD males. The present findings indicate impaired myocardial perfusion in DMD males compared to healthy controls. Cardiac perfusion deficits in DMD males have been measured using positron-emission tomography (PET) imaging [183,184] although further investigation by CMRI is warranted to confirm injection of contrast and to define the time of injection prior to LGE imaging [163]. Emerging CMRI markers of cardiac microstructure rely on tissue specific contrast mechanisms of CMRI governed by T1 and T2 relaxation time constants. Normal and pathologic (fibrotic or fatty) tissue types exhibit different relaxation times and these difference can be useful in diagnosing myocardial disease, determine severity of involvement and monitor response to therapy [163]. In the present findings, ND patients had significantly shorter myocardial T1 relaxation time and significantly reduced myocardium to skeletal muscle T1 ratio. However, additional studies are warranted to confirm reduced T1 relaxation times in ND patients. Other newer CMRI markers such as native T1 maps, post-contrast maps and extracellular volume (ECV) maps have gained increased attention because they permit capturing earlier myocardial changes such as diffuse fibrosis resulting from increased collagen deposition due to cellular damage in CM compared to LGE-CMRI findings that demonstrate overt and focal fibrosis [185].
Clinical implications
Current evidence suggests that LGE and CINE CMRI imaging modalities are widely available with demonstrated accuracy for diagnosing cardiac dysfunction and fibrosis that occur during the intermediate and late stages of the disease process but neither modality is effective in identifying occult cardiac dysfunction prior to overt evidence of cardiac disease or the onset of patient symptoms. Emerging CMRI markers such as myocardial perfusion imaging, myocardial T1 relaxation time and T1 mapping promise to detect subclinical myocardial changes with benefits of improved diagnosis, disease monitoring and evaluation of novel therapeutics. There is a clear need for more sensitive CMRI markers that can provide insights into the progression of cardiac dysfunction and for earlier and more effective treatment. However, the use of CMRI in clinical settings suffers from several drawbacks that affects its diagnostic utility in patients with ND. An important drawback to current CMRI examination is the requirement for repeated breath holding for periods of 5 to 20 seconds. This requirement can prove quite difficult especially for boys with neuromuscular diseases with compromised respiratory function and/or muscular contractures or in younger less complaint patients, total exam duration and the lack of widespread availability of validated CMRI marker sequences and analysis tools [163].
Familial neuromuscular diseases (NDs) are inherited diseases characterized by progressive muscular degeneration and weakness, which can potentially lead to cardiomyopathy (CM). Common NDs that can cause CM include muscular dystrophies (DMD, LGMD, FSHD, DM, MG and SMA) and neurological diseases (FA and NF). Muscular dystrophies often always result in the DCM phenotype. Its pathophysiology involves cardiomyocyte degeneration leading to myocardial fibrosis and subsequently wall stress and pressure overload, which in turn leads to a progressive decrease in systolic function, increased myocardial oxygen consumption and ultimately LV dilatation and dysfunction. Clinical presentation and diagnosis is challenging because of non-specific signs and symptoms, and an overlap between signs and symptoms of physical inactivity and respiratory failure with that of cardiac dysfunction. Definitive diagnosis rests on non-invasive cardiac imaging after the confirmation of the underlying neuromuscular disease by genetic and laboratory tests. Although conventional quantitative echocardiographic and CMRI markers such as LV systolic function and LV volumes may detect cardiac dysfunction in ND patients, these are often late manifestations in the course of the disease, which often lead to delayed diagnosis and an ominous prognosis. Emerging CMRI markers of myocardial microstructural remodelling such as myocardial perfusion, myocardial T1 relaxation time and T1 mapping can provide earlier insight into cardiac involvement in ND patients enabling improved cardiac care and evaluation of the efficacy of new and emerging therapies. Therapies for ND patients is largely based on disease modifying treatment or supportive treatment targeting symptom management.
- Lee DS, Pencina MJ, Benjamin EJ, Wang TJ, Levy D et al. (2006) Association of parental heart failure with risk of heart failure in offspring. N Engl J Med 355: 138-147. [Crossref]
- Richardson P, McKenna W, Bristow M, Maisch B, Mautner B et al. (1996) Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation 93: 841-842. [Crossref]
- Bozkurt B, Colvin M, Cook J, Cooper LT, Deswal A et al. (2016) Current diagnostic and treatment strategies for specific dilated cardiomyopathies: a scientific statement from the American Heart Association. Circulation 134: e579-e646. [Crossref]
- Pinto YM, Elliott PM, Arbustini E, Adler Y, Anastasakis A et al. (2016) Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J 37: 1850-1858. [Crossref]
- Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F et al. (2007) Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 29: 270-276. [Crossref]
- Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D et al. (2006) Contemporary definitions and classification of the cardiomyopathies: an American Heart Association scientific statement from the council on clinical cardiology, heart failure and transplantation committee; quality of care and outcomes research and functional genomics and translational biology interdisciplinary working groups; and council on epidemiology and prevention. Circulation 113: 1807-1816. [Crossref]
- Flanigan KM (2012) The Muscular Dystrophies. Semin Neurol 32: 255-263. [Crossref]
- Chung J, Smith AL, Hughes SC, Niizawa G, Abdel-Hamid HZ et al. (2016) Twenty-year follow-up of newborn screening for patients with muscular dystrophy. Muscle Nerve 53: 570-578. [Crossref]
- Hoffman EP, Brown Jr RH, Kunkel LM (1987) Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51: 919-928. [Crossref]
- Pane M, Scalise R, Berardinelli A, D’Angelo G, Ricotti V et al. (2013) Early neurodevelopmental assessment in Duchenne muscular dystrophy. Neuromuscul Disord 23: 451-455. [Crossref]
- Wu B, Cloer C, Lu P, Milazi S, Shaban M et al. (2014) Exon skipping restores dystrophin expression, but fails to prevent disease progression in later stage dystrophic dko mice. Gene Therapy 21: 785-793. [Crossref]
- American Academy of Paediatrics Section on Cardiology and Cardiac Surgery (2005) Cardiovascular health supervision for individuals affected by Duchenne or Becker muscular dystrophy. Paediatrics 116: 1569-1573. [Crossref]
- Eagle M, Baudouin SV, Chandler C, Giddings DR, Bullock R et al. (2002) Survival in Duchenne muscular dystrophy: improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul Disord 12: 926-929. [Crossref]
- Kamdar F, Garry DJ (2016) Dystrophin-deficient cardiomyopathy. J Am Coll Cardiol 67: 2533-2546. [Crossref]
- Van Westering T, Betts C, Wood M (2015) Current understanding of molecular pathology and treatment of cardiomyopathy in Duchenne muscular dystrophy. Molecules 20: 8823-8855. [Crossref]
- Falzarano M, Scotton C, Passarelli C, Ferlini A (2015) Duchenne muscular dystrophy: from diagnosis to therapy. Molecules 20: 18168-18184. [Crossref]
- Nigro G, Comi LI, Politano L, Bain RJ (1990) The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int J Cardiol 26: 271-277. [Crossref]
- Markham LW, Spicer RL, Khoury PR, Wong BL, Mathews KD et al. (2005) Steroid therapy and cardiac function in Duchenne muscular dystrophy. Pediatr Cardiol 26: 768-771. [Crossref]
- Undorkar R, Al-Yafi M, Romano S, Levin BR, Farzaneh-Far A (2018) Cardiomyopathy in muscular dystrophy. QJM 111: 267-268. [Crossref]
- Jefferies JL, Eidem BW, Belmont JW, Craigen WJ, Ware SM et al. (2005) Genetic predictors and remodelling of dilated cardiomyopathy in muscular dystrophy. Circulation 112: 2799-2804. [Crossref]
- Fayssoil A, Nardi O, Orlikowski D, Annane D (2010) Cardiomyopathy in Duchenne muscular dystrophy: pathogenesis and therapeutics. Heart Fail Rev 15:103. [Crossref]
- Lapidos KA, Kakkar R, McNally EM (2004) The dystrophin glycoprotein complex: signalling strength and integrity for the sarcolemma. Circ Res 94: 1023-1031. [Crossref]
- Rybakova IN, Patel JR, Ervasti JM (2000) The dystrophin complex forms a mechanically strong link between the sarcolemma and costameric actin. J Cell Biol 150: 1209-1214. [Crossref]
- Goodwin FC, Muntoni F (2005) Cardiac involvement in muscular dystrophies: molecular mechanisms. Muscle Nerve 32(5): 577-588. [Crossref]
- Colan SD (2005) Evolving therapeutic strategies for dystrophinopathies: potential for conflict between cardiac and skeletal needs. Circulation 112: 2756-2758. [Crossref]
- Kamogawa Y, Biro S, Maeda M, Setoguchi M, Hirakawa T et al. (2001) Dystrophin-deficient myocardium is vulnerable to pressure overload in vivo. Cardiovasc Res 50: 509-515. [Crossref]
- Wehling-Henricks M, Jordan MC, Roos KP, Deng B, Tidball JG (2005) Cardiomyopathy in dystrophin-deficient hearts is prevented by expression of a neuronal nitric oxide synthase transgene in the myocardium. Hum Mol Genet 14: 1921-1933. [Crossref]
- Williams IA, Allen DG (2007) Intracellular calcium handling in ventricular myocytes from mdx mice. Am J Physiol Heart Circ Physiol 292: H846-H855. [Crossref]
- Cox GA, Cole NM, Matsumura K, Phelps SF, Hauschka SD et al. (1993) Overexpression of dystrophin in transgenic mdx mice eliminates dystrophic symptoms without toxicity. Nature 364: 725-729. [Crossref]
- Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL (1993) Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci USA 90: 3710-3714. [Crossref]
- Yasuda S, Townsend D, Michele DE, Favre EG, Day SM et al. (2005) Dystrophic heart failure blocked by membrane sealant poloxamer. Nature 436: 1025-1029. [Crossref]
- Whitehead NP, Yeung EW, Allen DG (2006) Muscle damage in mdx (dystrophic) mice: role of calcium and reactive oxygen species. Clin Exp Pharmacol Physiol 33: 657-662. [Crossref]
- Romfh A, McNally EM (2010) Cardiac assessment in Duchenne and Becker muscular dystrophies. Curr Heart Fail Rep 7: 212-218. [Crossref]
- Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR et al. (2010) Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol 9: 77-93. [Crossref]
- Thrush PT, Allen HD, Viollet L, Mendell JR (2009) Re-examination of the electrocardiogram in boys with Duchenne muscular dystrophy and correlation with its dilated cardiomyopathy. Am J Cardiol 103: 262-265. [Crossref]
- Perloff JK, Roberts WC, de Leon Jr AC, O'Doherty D (1967) The distinctive electrocardiogram of Duchenne’s progressive muscular dystrophy. An electrocardiographic pathologic correlative study. Am J Med 42: 179-188. [Crossref]
- Sanyal SK, Johnson WW, Thapar MK, Pitner SE (1978) An ultrastructural basis for electrocardiographic alterations associated with Duchenne’s progressive muscular dystrophy. Circulation 57: 1122-1129. [Crossref]
- Markham LW, Michelfelder EC, Border WL, Khoury PR, Spicer RL et al. (2006) Abnormalities of diastolic function precede dilated cardiomyopathy associated with Duchenne muscular dystrophy. J Am Soc Echocardiogr 19: 865-871. [Crossref]
- Frankel KA, Rosser RJ (1976) The pathology of the heart in progressive muscular dystrophy: epimyocardial fibrosis. Human Pathol 7: 375-386. [Crossref]
- Rapezzi C, Leone O, Biagini E, Coccolo F (2007) Echocardiographic clues to diagnosis of dystrophin related dilated cardiomyopathy. Heart 93: 10. [Crossref]
- Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR et al. (2010) Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol 9: 177-189. [Crossref]
- Silva MC, Meira ZM, Giannetti JG, da Silva MM, Campos AF et al. (2007) Myocardial delayed enhancement by magnetic resonance imaging in patients with muscular dystrophy. J Am Coll Cardiol 49: 1874-1879. [Crossref]
- Hagenbuch SC, Gottliebson WM, Wansapura J, Mazur W, Fleck R et al. (2010) Detection of progressive cardiac dysfunction by serial evaluation of circumferential strain in patients with Duchenne muscular dystrophy. Am J Cardiol 105: 1451-1455. [Crossref]
- Hor KN, Wansapura J, Markham LW, Mazur W, Cripe LH et al. (2009) Circumferential strain analysis identifies strata of cardiomyopathy in Duchenne muscular dystrophy: a cardiac magnetic resonance tagging study. J Am Coll Cardiol 53: 1204-1210. [Crossref]
- Puchalski MD, Williams RV, Askovich B, Sower CT, Hor KH et al. (2009) Late gadolinium enhancement: precursor to cardiomyopathy in Duchenne muscular dystrophy? Int J Cardiovasc Imaging 25: 57-63. [Crossref]
- Verhaert D, Richards K, Rafael-Fortney JA, Raman SV (2011) Cardiac involvement in patients with muscular dystrophies: magnetic resonance imaging phenotype and genotypic considerations. Circ Cardiovasc Imaging 4: 67-76. [Crossref]
- Hor KN, Taylor MD, Al-Khalidi HR, Cripe LH, Raman SV et al. (2013) Prevalence and distribution of late gadolinium enhancement in a large population of patients with Duchenne muscular dystrophy: effect of age and left ventricular systolic function. J Cardiovasc Magn Reson 15: 107. [Crossref]
- Tandon A, Villa CR, Hor KN, Jefferies JL, Gao Z et al. (2015) Myocardial fibrosis burden predicts left ventricular ejection fraction and is associated with age and steroid treatment duration in Duchenne muscular dystrophy. J Am Heart Assoc 4: e001338. [Crossref]
- Scully MA, Pandya S, Moxley RT (2013) Review of Phase II and Phase III clinical trials for Duchenne muscular dystrophy. Expert Opin. Orphan Drugs 1: 33-46.
- Goemans N, Buyse G (2014) Current Treatment and Management of Dystrophinopathies. Curr Treat Options Neurol 16: 287. [Crossref]
- Ferlini A, Neri M, Gualandi F (2013) The medical genetics of dystrophinopathies: Molecular genetic diagnosis and its impact on clinical practice. Neuromuscul Disord 23: 4-14. [Crossref]
- Findlay AR, Wein N, Kaminoh Y, Taylor LE, Dunn DM et al. (2015) Clinical phenotypes as predictors of the outcome of skipping around DMD exon 45. Ann Neurol 77: 668-674. [Crossref]
- Shin J, Tajrishi MM, Ogura Y, Kumar A (2013) Wasting mechanisms in muscular dystrophy. Int J Biochem.Cell Biol 45: 2266-2279. [Crossref]
- Falzarano MS, Passarelli C, Ferlini A (2014) Nanoparticle delivery of antisense oligonucleotides and their application in the exon skipping strategy for Duchenne muscular dystrophy. Nucleic Acid Ther 24: 87-100. [Crossref]
- Haas M, Vlcek V, Balabanov P, Salmonson T, Bakchine S (2015) European Medicines Agency review of ataluren for the treatment of ambulant patients aged 5 years and older with Duchenne muscular dystrophy resulting from a nonsense mutation in the dystrophin gene. Neuromuscul Disord 25: 5-13. [Crossref]
- Khadilkar SV, Faldu HD, Patil SB, Singh R (2017) Limb-girdle muscular dystrophies in India: a review. Ann Indian Acad Neurol 20: 87-95. [Crossref]
- Pegoraro E, Hoffman EP (2012) Limb-girdle muscular dystrophy overview. In Gene Reviews [Internet] University of Washington, Seattle.
- Van der Kooi AJ, De Voogt WG, Barth PG, Busch HF, Jennekens FG et al. (1998) The heart in limb girdle muscular dystrophy. Heart 79: 73-77. [Crossref]
- Okere A, Reddy SS, Gupta S, Shinnar M (2013) A cardiomyopathy in a patient with limb girdle muscular dystrophy type 2A. Circ Heart Fail 6: e12-e13. [Crossref]
- Sveen ML, Thune JJ, Køber L, Vissing J (2008) Cardiac involvement in patients with limb-girdle muscular dystrophy type 2 and Becker muscular dystrophy. Arch Neurol 65: 1196-1201. [Crossref]
- Mascarenhas DA, Spodick DH, Chad DA, Gilchrist J, Townes PL et al. (1994) Cardiomyopathy of limb-girdle muscular dystrophy. J Am Coll Cardiol 24: 1328-1333. [Crossref]
- Rosales XQ, Moser SJ, Tran T, McCarthy B, Dunn N et al. (2011) Cardiovascular magnetic resonance of cardiomyopathy in limb girdle muscular dystrophy 2B and 2I. J Cardiovasc Magn Reson 13: 39. [Crossref]
- Lasam G, Lam J (2016) Severe Cardiomyopathy from Limb Girdle Muscular Dystrophy: A Nidus for a Catastrophic Cascade. American Journal of Cardiovascular Disease Research 4: 7-10.
- Pandya S, King WM, Tawil R (2008) Facioscapulohumeral dystrophy. Phys Ther 88: 105-113. [Crossref]
- Padberg GW, Frants RR, Brouwer OF, Wijmenga C, Bakker E et al. (1995) Facioscapulohumeral muscular dystrophy in the Dutch population. Muscle Nerve Suppl 2: S81-S84. [Crossref]
- Flanigan KM, Coffeen CM, Sexton L, Stauffer D, Brunner S et al. (2001) Genetic characterization of a large, historically significant Utah kindred with facioscapulohumeral dystrophy. Neuromuscul Disord 11: 525-529. [Crossref]
- Mostacciuolo ML, Pastorello E, Vazza G, Miorin M, Angelini C et al. (2009) Facioscapulohumeral muscular dystrophy: epidemiological and molecular study in a northeast Italian population sample. Clin Genet 75: 550-555. [Crossref]
- Norwood FL, Harling C, Chinnery PF, Eagle M, Bushby K, Straub V (2009) Prevalence of genetic muscle disease in Northern England: in-depth analysis of a muscle clinic population. Brain 132: 3175-3186. [Crossref]
- Emery AE (2002) The muscular dystrophies. Lancet 359: 687-695. Crossref
- Tawil R, van der Maarel SM, Tapscott SJ (2014) Facioscapulohumeral dystrophy: the path to consensus on pathophysiology. Skelet Muscle 4: 12. [Crossref]
- Frisullo G, Frusciante R, Nociti V, Tasca G, Renna R et al. (2011) CD8(+) T cells in facioscapulohumeral muscular dystrophy patients with inflammatory features at muscle MRI. J Clin Immunol 31: 155-166. [Crossref]
- Tasca G, Pescatori M, Monforte M, Mirabella M, Iannaccone E et al. (2012) Different molecular signatures in magnetic resonance imaging-staged facioscapulohumeral muscular dystrophy muscles. PLoS One 7: e38779. [Crossref]
- Friedman SD, Poliachik SL, Carter GT, Budech CB, Bird TD et al. (2012) The magnetic resonance imaging spectrum of facioscapulohumeral muscular dystrophy. Muscle Nerve 45: 500-506. [Crossref]
- Friedman SD, Poliachik SL, Otto RK, Carter GT, Budech CB et al. (2014) Longitudinal features of stir bright signal in FSHD. Muscle Nerve 2013. [Crossref]
- Janssen BH, Voet NB, Nabuurs CI, Kan HE, de Rooy JW et al. (2014) Distinct disease phases in muscles of facioscapulohumeral dystrophy patients identified by MR detected fat infiltration. PLoS One 9: e85416. [Crossref]
- Emmrich P, Ogunlade V, Gradistanac T, Daneschnejad S, Koch MC et al. (2005) Facioscapulohumeral muscle dystrophy and heart disease. Z Kardiol 94: 348-354. [Crossref]
- Finsterer J, Stöllberger C, Meng G (2005) Cardiac involvement in facioscapulohumeral muscular dystrophy. Cardiology 103: 81-83. [Crossref]
- Tsuji M, Kinoshita M, Imai Y, Kawamoto M, Kohara N (2009) Facioscapulohumeral muscular dystrophy presenting with hypertrophic cardiomyopathy a case study. Neuromuscul Disord 19: 140-142. [Crossref]
- Bache RJ, Sarosi GA (1968) Myotonia atrophica: diagnosis in patients with complete heart block and Stokes-Adams syncope. Arch Intern Med 121: 369-372. [Crossref]
- Turner C, Hilton-Jones D (2010) The myotonic dystrophies: diagnosis and management. J Neurol Neurosurg Psychiatry 81: 358-367. [Crossref]
- McNally EM, Sparano D (2011) Mechanisms and management of the heart in myotonic dystrophy. Heart 97: 1094-1100. [Crossref]
- Wahbi K, Meune C, Becane HM, Laforet P, Bassez G et al. (2009) Left ventricular dysfunction and cardiac arrhythmias are frequent in type 2 myotonic dystrophy: a case control study. Neuromuscul Disord 19: 468-472. [Crossref]
- Bhakta D, Lowe MR, Groh WJ (2004) Prevalence of structural cardiac abnormalities in patients with myotonic dystrophy type I. Am Heart J 147: 224-227. [Crossref]
- Tanaka N, Tanaka H, Takeda M, Niimura T, Kanehisa T et al. (1973) Cardiomyopathy in myotonic dystrophy. Jpn Heart J 14: 202-212. [Crossref]
- Groh WJ, Groh MR, Saha C, Kincaid JC, Simmons Z et al. (2008) Electrocardiographic abnormalities and sudden death in myotonic dystrophy type 1. N Engl J Med 358: 2688-2697. [Crossref]
- Nguyen HH, Wolfe JT 3rd, Holmes DR Jr, Edwards WD (1988) Pathology of the cardiac conduction system in myotonic dystrophy: a study of 12 cases. J Am Coll Cardiol 11: 662-671. [Crossref]
- Merino JL, Carmona JR, Fernandez-Lozano I, Peinado R, Basterra N et al. (1998) Mechanisms of sustained ventricular tachycardia in myotonic dystrophy: implications for catheter ablation. Circulation 98: 541-546. [Crossref]
- Florek RC, Triffon DW, Mann DE, Ringel SP, Reiter MJ (1990) Electrocardiographic abnormalities in patients with myotonic dystrophy. West J Med 153: 24. [Crossref]
- Grigg LE, Chan W, Mond HG, Vohra JK, Downey WF (1985) Ventricular tachycardia and sudden death in myotonic dystrophy: clinical, electrophysiologic and pathologic features. J AM Coll Cardiol 6: 254-256. [Crossref]
- De Ambroggi L, Raisaro A, Marchiano V, Radice S, Meola G (1995) Cardiac involvement in patients with myotonic dystrophy: Characteristic features of magnetic resonance imaging. Eur Heart J 16: 1007-1010. [Crossref]
- Verhaert D, Richards K, Rafael-Fortney JA, Raman SV (2011) Cardiac involvement in patients with muscular dystrophies: magnetic resonance imaging phenotype and genotypic considerations. Circ Cardiovasc Imagin. 4: 67-76. [Crossref]
- Hermans MC, Faber CG, Bekkers SC, de Die-Smulders CE, Gerrits MM et al. (2012) Structural and functional cardiac changes in myotonic dystrophy type 1: a cardiovascular magnetic resonance study. J Cardiovasc Magn Reson 14: 48. [Crossref]
- Turkbey EB, Gai N, Lima JA, Van Der Geest RJ, Wagner KR et al. (2012) Assessment of cardiac involvement in myotonic muscular dystrophy by T1 mapping on magnetic resonance imaging. Heart Rhythm 9: 1691-1697. [Crossref]
- Duboc D, Meune C, Pierre B, Wahbi K, Eymard B et al. (2007) Perindopril preventive treatment on mortality in Duchenne muscular dystrophy: 10 years’ follow-up. Am Heart J 154: 596-602. [Crossref]
- Logigian EL, Martens WB, Moxley RTt, McDermott MP, Dilek N et al. (2010) Mexiletine is an effective antimyotonia treatment in myotonic dystrophy type 1. Neurology 74: 1441-1448. [Crossref]
- Laurent V, Pellieux S, Corcia P, Magro P, Pierre B et al. (2010) Mortality in myotonic dystrophy patients in the area of prophylactic pacing devices. Int J Cardiol 150: 54-58. [Crossref]
- Thanvi BR, Lo TC (2004) Update on myasthenia gravis. Postgrad Med J 80: 690-700. [Crossref]
- Gilhus NE (2016) Myasthenia Gravis. N Engl J Med 375: 2570-2581. [Crossref]
- Hughes BW, De Casillas ML, Kaminski HJ (2004) Pathophysiology of myasthenia gravis. Semin Neurol 24: 21-30. [Crossref]
- Trouth JA, Dabi A, Solieman N, Kurukumbi M, Kalyanam J (2012) Myasthenia gravis: a review. Autoimmune Dis 2012: 874680. [Crossref]
- MacDonald BK, Cockerell OC, Sander JW, Shorvon SD (2000) The incidence and lifetime prevalence of neurological disorders in a prospective community-based study. Brain 123: 665-676. [Crossref]
- Phillips LH, Torner JC, Anderson MS, Cox GM (1992) The epidemiology of myasthenia gravis in central and western Virginia. Neurology 42: 1883-1893. [Crossref]
- Calin C, Savu OA, Dumitru DA, Ghiorghiu IO, Calin AN et al. (2009) Cardiac involvement in myasthenia gravis: is there a specific pattern. Rom J Intern Med 47: 179-189. [Crossref]
- Shivamurthy P, Parker MW (2004) Cardiac manifestations of myasthenia gravis: a systematic review. IJC Metabolic & Endocrine 5: 3-6.
- HooKim K, deRoux S, Igbokwe A, Stanek A, Koo J et al. (2008) IgG anti-cardiomyocyte antibodies in giant cell myocarditis. Ann Clin Lab Sci 38: 83-87. [Crossref]
- Sasaki H, Yano M, Kawano O, Hikosaka Y, Fujii Y (2012) Thymoma associated with fatal myocarditis and polymyositis in a 58-year-old man following treatment with carboplatin and paclitaxel: a case report. Oncol Lett 3: 300-302. [Crossref]
- Kon T, Mori F, Tanji K, Miki Y, Kimura T et al. (2013) Giant cell polymyositis and myocarditis associated with myasthenia gravis and thymoma. Neuropathology 33: 281-287. [Crossref]
- Wong CP, Chia PL (2012) Recurrent takotsubo cardiomyopathy precipitated by myasthenic crisis. Int J Cardiol 155: e11-e12. [Crossref]
- Bansal V, Kansal MM, Rowin J (2011) Broken heart syndrome in myasthenia gravis. Muscle Nerve 44: 990-993. [Crossref]
- Arai M, Ukigai H, Miyata H (2004) A case of transient left ventricular ballooning (“Takotsubo”-shaped cardiomyopathy) developed during plasmapheresis for treatment of myasthenic, crisis. Rinsho Shinkeigaku 44: 207-210. [Crossref]
- Sousa JM, Knobel M, Buchelle G, Sousa JA, Fisher CH et al. (2005) Transient ventricular dysfunction (Takotsubo cardiomyopathy). Arq Bras Cardiol 84: 340-342. [Crossref]
- Antevil JL, Carroll CG, Roberts PF, Johnston MG, Strange RG (2010) Myasthenia gravis: an unexpected cause of respiratory failure and reversible left ventricular dysfunction after cardiac surgery. J Card Surg 25: 662-664. [Crossref]
- Gautier P, Ravan R, Najjar M, Belhakem A, Ferrier N et al. (2011) Takotsubo syndrome during normal human immunoglobolin perfusion. Ann Cardiol Angeiol (Paris) 60: 290-295. [Crossref]
- Shukla G, Gupta S, Goyal V, Singh S, Srivastava A et al. (2013) Abnormal sympathetic hyper-reactivity in patients with myasthenia gravis: a prospective study. Clin Neurol Neurosurg 115: 179-186. [Crossref]
- Limaye K, Vallurupalli S, Lee RW (2016) Myasthenia of the Heart. Am J Med 129: e19-e21. [Crossref]
- Kato T, Hirose S, Kumagai S, Ozaki A, Matsumoto S et al. (2016) Electrocardiography as the first step for the further examination of cardiac involvement in myasthenia gravis. BioMed Res Int 2016. [Crossref]
- Feingold B, Mahle WT, Auerbach S, Clemens P, Domenighetti AA et al. (2017) Management of cardiac involvement associated with neuromuscular diseases: a scientific statement from the American Heart Association. Circulation 136: e200-e231. [Crossref]
- Kolb SJ, Kissel JT (2011) Spinal muscular atrophy: a timely review. Arch Neurol 68: 979-984. [Crossref]
- Arnold WD, Kassar D, Kissel JT (2015) Spinal muscular atrophy: diagnosis and management in a new therapeutic era. Muscle Nerve 51: 157-167. [Crossref]
- Ashrafzadeh F, Sadr-Nabavi A, Asadian N, Akhondian J, Beiraghi Toosi M (2014) Spinal Muscular Atrophy: A Short Review Article. Int J Pediatr 2: 211-215.
- Mercuri E, Finkel RS, Muntoni F, Wirth B, Montes J et al. (2018) Diagnosis and management of spinal muscular atrophy: part 1: recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord 28: 103-115. [Crossref]
- Palladino A, Passamano L, Taglia A, D'Ambrosio P, Scutifero M et al. (2011) Cardiac involvement in patients with spinal muscular atrophies. Acta Myologica 30: 175. [Crossref]
- Distefano G, Sciacca P, Parisi MG, Parano E, Smilari P et al. (1994) Heart involvement in progressive spinal muscular atrophy. A review of the literature and case histories in childhood. Pediatr Med Chir 16: 125-128. [Crossref]
- Wijngaarde CA, Blank AC, Stam M, Wadman RI, Van Den Berg LH et al. (2017) Cardiac pathology in spinal muscular atrophy: a systematic review. Orphanet J Rare Dis 12: 67. [Crossref]
- Tanaka H, Uemura N, Toyama Y, Kudo A, Ohkatsu Y et al. (1976) Cardiac involvement in the Kugelberg-Welander syndrome. Am J Cardiol 38: 528-532. [Crossref]
- Kimura S, Yokota H, Tateda K, Miyamoto K, Yamamoto K et al. (1980) A case of the KugelbergWelander syndrome complicated with cardiac lesions. Jpn Heart J 21: 417-422. [Crossref]
- Elkohen M, Vaksmann G, Elkohen MR, Francart C, Foucher C et al. (1996) Cardiac involvement in Kugelberg-Welander disease. A prospective study of 8 cases. Rev C Arch Mal Coeur Vaiss 89: 611-617. [Crossref]
- Roos M, Sarkozy A, Chierchia GB, De Wilde P, Schmedding E et al. (2009) Malignant ventricular arrhythmia in a case of adult onset of spinal muscular atrophy (Kugelberg-Welander disease). J Cardiovasc Electrophysiol 20: 342-344. [Crossref]
- Takahashi N, Shimada T, Ishibashi Y, Sugamori T, Hirano Y et al. (2006) Cardiac involvement in Kugelberg-Welander disease: a case report and review. Am J Med Sci 332: 354-356. [Crossref]
- Shababi M, Habibi J, Yang HT, Vale SM, Sewell WA et al. (2010) Cardiac defects contribute to the pathology of spinal muscular atrophy models. Hum Mol Genet 19: 4059-4071. [Crossref]
- Heier CR, Satta R, Lutz C, DiDonato CJ (2010) Arrhythmia and cardiac defects are a feature of spinal muscular atrophy model mice. Hum Mol Genet 19: 3906-3918. [Crossref]
- Monani UR, Coovert DD, Burghes AH (2000) Animal models of spinal muscular atrophy. Hum Mol Genet 9: 2451-2457. [Crossref]
- Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR et al. (2001) Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ Res 89: 20-25. [Crossref]
- Bevan AK, Hutchinson KR, Foust KD, Braun L, McGovern VL et al. (2010) Early heart failure in the SMNDelta7 model of spinal muscular atrophy and correction by postnatal scAAV9-SMN delivery. Hum Mol Genet 19: 3895-3905. [Crossref]
- Yasuma F, Kuru S, Konagaya M (2004) Dilated cardiomyopathy in Kugelberg-Welander disease: coexisting sleep disordered breathing and its treatment with continuous positive airway pressure. Intern Med.43: 951-954. [Crossref]
- Ashizawa T, Xia G (2016) Ataxia. Continuum (Minneap Minn) 22: 1208-1226. [Crossref]
- Burk K (2017) Friedreich Ataxia: current status and future prospects. Cerebellum Ataxias 4: 4. [Crossref]
- Cook A, Giunti P (2017) Friedreich’s ataxia: clinical features, pathogenesis and management. Br Med Bull 1: 1-2. [Crossref]
- Vankan P (2013) Prevalence gradients of Friedreich’s Ataxia and R1b haplotype in Europe co-localize, suggesting a common Palaeolithic origin in the Franco-Cantabrian ice age refuge. J Neurochem 126:11-20. [Crossref]
- Weidemann F, Störk S, Liu D, Hu K, Herrmann S et al. (2013) Cardiomyopathy of Friedreich ataxia. J Neurochem 126: 88-93. [Crossref]
- Tsou AY, Paulsen EK, Lagedrost SJ, Perlman SL, Mathews KD et al. (2011) Mortality in Friedreich ataxia. J Neurol Sci. 2011;307:46-49. [Crossref]
- Schultz JC, Hilliard AA, Cooper LT Jr, Rihal CS (2009) Diagnosis and treatment of viral myocarditis. Mayo Clin Proc 84: 1001-1009. [Crossref]
- Jensen MK, Bundgaard H (2012) Cardiomyopathy in Friedreich ataxia: exemplifying the challenges faced by cardiologists in the management of rare diseases. Circulation 125: 1591. [Crossref]
- Weidemann F, Rummey C, Bijnens B, Stork S, Jasaityte R et al. (2012) The heart in Friedreich ataxia: definition of cardiomyopathy, disease severity, and correlation with neurological symptoms. Circulation 125: 1626-1634. [Crossref]
- Koeppen AH, Ramirez RL, Becker AB, Bjork ST, Levi S et al. (2015) The pathogenesis of cardiomyopathy in Friedreich ataxia. PLoS One 10: e0116396. [Crossref]
- Mottram PM, Delatycki MB, Donelan L, Gelman JS, Corben L et al. (2011) Early changes in left ventricular long-axis function in Friedreich ataxia: relation with the FXN gene mutation and cardiac structural change. J Am Soc Echocardiogr 24: 782-789. [Crossref]
- Silva MC, Meira ZM, Gurgel Giannetti J, da Silva MM, Campos AF et al. (2007) Myocardial delayed enhancement by magnetic resonance imaging in patients with muscular dystrophy. J Am Coll Cardiol 49: 1874-1879. [Crossref]
- Weidemann F, Strotmann J (2008) Early detection of Fabry disease: cardiac cases. Clin Ther 30: S46. [Crossref]
- Payne RM, Pride PM, Babbey CM (2011) Cardiomyopathy of Friedreich’s ataxia: use of mouse models to understand human disease and guide therapeutic development. Pediatr Cardiol 32: 366-378. [Crossref]
- Regner SR, Lagedrost SJ, Plappert T et al. (2012) Analysis of echocardiograms in a large heterogeneous cohort of patients with Friedreich ataxia. Am J Cardiol 109: 401-405. [Crossref]
- Dutka DP, Donnelly JE, Nihoyannopoulos P, Oakley CM, Nunez DJ (1999) Marked variation in the cardiomyopathy associated with Friedreich’s ataxia. Heart 81: 141-147. [Crossref]
- Weidemann F, Niemann M, Ertl G, Stork S (2010) The different faces of echocardiographic left ventricular hypertrophy: clues to the aetiology. J Am Soc Echocardiogr 23, 793-801. [Crossref]
- Payne RM, Peverill RE (2012) Cardiomyopathy of Friedreich’s Ataxia (FRDA). Ir J Med Sci 181: 569-570. [Crossref]
- Tsou AY, Paulsen EK, Lagedrost SJ, Perlman SL, Mathews KD et al. (2010) Mortality in Friedreich ataxia. J Neurol Sci 307: 46-49. [Crossref]
- Hart PE, Lodi R, Rajagopalan B et al. (2005) Antioxidant treatment of patients with Friedreich ataxia: four-year follow-up. Arch Neurol 62: 621-626. [Crossref]
- Khan N, Van de Werke I, Ismail F (2010) Neurofibromatosis revisited A pictorial review. SA Journal of Radiology 14: 1.
- Hirsch NP, Murphy A, Radcliffe JJ (2001) Neurofibromatosis: clinical presentations and anaesthetic implications. Br J Anaesth 86: 555-564. [Crossref]
- Gerber PA, Antal AS, Neumann NJ, Homey B, Matuschek C et al. (2009) Neurofibromatosis. Eur J Med Res 14: 102. [Crossref]
- Williams VC, Lucas J, Babcock MA, Gutmann DH, Korf B et al. (2009) Neurofibromatosis type 1 revisited. Pediatrics 123: 124-133. [Crossref]
- İncecik F, Hergüner ÖM, Erdem SA, Altunbaşak Ş (2015) Neurofibromatosis type 1 and cardiac manifestations. Turk Kardiyol Dern Ars 43: 714-716. [Crossref]
- Jurko Jr A, Minarik M, Minarikova E, Nagi AS (2010) Neurofibromatosis associated with hypertrophic cardiomyopathy. Saudi Med J 31: 935-936. [Crossref]
- Fitzpatrick AP, Emanuel RW (1988) Familial neurofibromatosis and hypertrophic cardiomyopathy. Heart 60: 247-251. [Crossref]
- Magrath P, Maforo N, Renella P, Nelson SF, Halnon N et al. (2018) Cardiac MRI biomarkers for Duchenne muscular dystrophy. Biomark Med 12: 1271-1289. [Crossref]
- Schelhorn J, Schoenecker A, Neudorf U, Schemuth H, Nensa F et al. (2015) Cardiac pathologies in female carriers of Duchenne muscular dystrophy assessed by cardiovascular magnetic resonance imaging. Eur Radiol 25: 3066-3072. [Crossref]
- Florian A, Rosch S, Bietenbeck M, Engelen M, Stypmann J et al. (2015) Cardiac involvement in female Duchenne and Becker muscular dystrophy carriers in comparison to their first-degree male relatives: a comparative cardiovascular magnetic resonance study. Eur Heart J Cardiovasc Imaging 17: 326-333. [Crossref]
- Gaur L, Hanna A, Bandettini WP, Fischbeck KH, Arai AE et al. (2016) Upper arm and cardiac magnetic resonance imaging in Duchenne muscular dystrophy. Ann Clin Transl Neurol 3: 948-955. [Crossref]
- Mavrogeni S, Giannakopoulou A, Papavasiliou A, Markousis-Mavrogenis G, Pons R et al. (2017) Cardiac profile of asymptomatic children with Becker and Duchenne muscular dystrophy under treatment with steroids and with/without perindopril. BMC Cardiovasc Disord 17: 197. [Crossref]
- West SC, Wittlieb-Weber CA, Villa CR, Cunningham C, Bock MJ et al. (2018) Utilization of Cardiac MRI in Patients with Duchenne Muscular Dystrophy: A Contemporary Multi-Center Cohort. The Journal of Heart and Lung Transplantation 37: S390.
- Iwase T, Takao S, Akaike M, Adachi K, Sumitomo-Ueda Y et al. (2010) Diagnostic utility of cardiac magnetic resonance for detection of cardiac involvement in female carriers of Duchenne muscular dystrophy. Heart Asia 2: 52-55. [Crossref]
- Giglio V, Puddu PE, Camastra G, Sbarbati S, Della Sala SW et al. (2014) Patterns of late gadolinium enhancement in Duchenne muscular dystrophy carriers. J Cardiovasc Magn Reson 16: 45. [Crossref]
- Lang SM, Shugh S, Mazur W, Sticka JJ, Rattan MS et al. (2015) Myocardial fibrosis and left ventricular dysfunction in Duchenne muscular dystrophy carriers using cardiac magnetic resonance imaging. Pediatr Cardiol 36: 1495-1501. [Crossref]
- Schelhorn J, Schoenecker A, Neudorf U, Schemuth H, Nensa F et al. (2015) Cardiac pathologies in female carriers of Duchenne muscular dystrophy assessed by cardiovascular magnetic resonance imaging. Eur Radiol 25: 3066-3072. [Crossref]
- Mah ML, Cripe L, Slawinski M, Camino E, Al-Zaidy S et al. (2019) Duchenne and Becker muscular dystrophy carriers: evidence of cardiomyopathy by exercise and cardiac magnetic resonance imaging. J Am Coll Cardiol 73: 1455.
- Raman SV, Phatak K, Hoyle JC, Pennell ML, McCarthy B et al. (2010) Impaired myocardial perfusion reserve and fibrosis in Friedreich ataxia: a mitochondrial cardiomyopathy with metabolic syndrome. Eur Heart J 32: 561-567. [Crossref]
- Weidemann F, Liu D, Hu K, Florescu C, Niemann M et al. (2015) The cardiomyopathy in Friedreich's ataxia: new biomarker for staging cardiac involvement. Int J Cardiol 194: 50-57. [Crossref]
- McMurray JJ, Adamopoulos S, Anker SD, Auricchio A, Bohm M, et al. (2012) ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012. Eur J Heart Fail 14: 803-869. [Crossref]
- Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, et al. (2016) 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J 37: 2129-2200. [Crossref]
- Mazur W, Hor KN, Germann JT, Fleck RJ, Al-Khalidi HR et al. (2012) Patterns of left ventricular remodelling in patients with Duchenne muscular dystrophy: a cardiac MRI study of ventricular geometry, global function, and strain. Int J Cardiovasc Imaging 28: 99-107. [Crossref]
- Schulz-Menger J, Bluemke DA, Bremerich J, Flamm SD, Fogel MA, Friedrich MG, Kim RJ, von Knobelsdorff-Brenkenhoff F, Kramer CM, Pennell DJ, Plein S. Standardized image interpretation and post processing in cardiovascular magnetic resonance: Society for Cardiovascular Magnetic Resonance (SCMR) board of trustees task force on standardized post processing. J Cardiovasc Magn Reson 15: 35. [Crossref]
- Vasan RS, Levy D (2000) Defining diastolic heart failure. Circulation 101: 2118-2121. [Crossref]
- Solomon SD, Anavekar N, Skali H, McMurray JJ, Swedberg K et al. (2005) Influence of ejection fraction on cardiovascular outcomes in a broad spectrum of heart failure patients. Circulation 112: 3738-3744. [Crossref]
- McCrohon JA, Moon JC, Prasad SK, McKenna WJ, Lorenz CH et al. (2003) Differentiation of heart failure related to dilated cardiomyopathy and coronary artery disease using gadolinium-enhanced cardiovascular magnetic resonance. Circulation 108: 54-59. [Crossref]
- Quinlivan RM, Lewis P, Marsden P, Dundas R, Robb SA et al. (1996) Cardiac function, metabolism and perfusion in Duchenne and Becker muscular dystrophy. Neuromuscul Disord 6: 237-246. [Crossref]
- Kawai N, Sotobata I, Okada M, Iwase M, Yamamoto S et al. (1985) Evaluation of myocardial involvement in Duchenne’s progressive muscular dystrophy with thallium-201 myocardial perfusion imaging. Jpn Heart J 26: 767-775. [Crossref]
- Haaf P, Garg P, Messroghli DR, Broadbent DA, Greenwood JP et al. (2016) Cardiac T1 mapping and extracellular volume (ECV) in clinical practice: a comprehensive review. J. Cardiovasc Magn Reson 18: 89. [Crossref]