The purpose of this study was to examine the role of dopaminergic mediation in the hypothalamic-pituitary-adrenal (HPA) response challenged by ether stress. First the effects of the D1 selective (SCH-23390) and the D2 selective (haloperidol) dopamine antagonists on ether stress evoked corticosterone responses were tested. The activation of the HPA axis was assessed by measuring plasma corticosterone levels. Low doses of haloperidol (25 nmol/kg, intraperitoneally) failed to affect either basal or stimulated corticosterone secretion, while SCH-23390 elicited moderate inhibition of stimulated release. However, to our surprise higher doses of haloperidol (250 nmol/kg) raised basal secretion and even potentiated the HPA response evoked by ether stress. Therefore to elucidate the role of D2 mediation, the intrinsic activity of the D2 antagonist was tested in further experiments. Haloperidol (from 25 nmol/kg to 2.5 mmol/kg) alone elicited a dose-dependent activation of the HPA system reflected by both corticosterone and ACTH serum levels. This D2-antagonist-evoked HPA activation could be prevented by pre-administration of the CRH antagonist, α-helical CRH9-41. These findings provide unequivocal evidence that D2 receptor can exert inhibitory control over the HPA axis.
hypothalamic-pituitary-adrenal system, dopamine, ether stress, haloperidol, ACTH, corticosterone3.
Although dopaminergic neurons interconnect several brain centers and modulate a plethora of functions, the activities and disorders of three specific systems represent the most important physiologic and pathophysiologic phenomena in monoaminergic neurotransmission [1,2]. The nigrostriatal, mesocorticolimbic and hypothalamic-tuberoinfundibular neural networks predominantly manage inherited, instinctive, neuroendocrine programs (extrapyramidal modules, holokinetic movements, postural reflexes, and hormonal responses), but the modulation and fine-tuning provided by them also have impact even on the most intricate and flexible cognitive processes [3,4].
The complexity of these neural circuits is further multiplied by an arsenal of dopaminergic receptors [5,6], which explains the complex pathology of these systems (schizophrenia, attention-deficit hyperactivity disorder, Parkinson’s disease, Parkinson’s dementia, Lewy body dementia (LBD), various endocrine disorders), and the unpredictable effects and side effects (toxicity and withdrawal symptoms) of dopaminergic pharmacological agents [5-8]. Further, it is well known that, in both endogenous monoaminergic and addictive disorders, the pathological activity of the HPA system can be observed. Most commonly it is represented by either an over-activation or a decreased resilience of the HPA system reflected by resistance to dexamethasone suppression [9]. However, the role of different dopaminergic pathways and receptor classes in the control of the HPA axis is poorly understood. The only aspect, upon which even recent publications agree, is the stimulus dependent role of various dopaminergic pathways in the transmission of the stress response [10].
First, Jezova reported [11] that non-selective dopaminergic agonists (apomorphine, pergolide) evoked significant elevations in ACTH secretion in the rat. According to them, this response could be completely abolished by preadministration of the selective D2 inverse agonist, haloperidol [11]. Moreover, Hennig et al. [12], demonstrated that the human cortisol response to heat stress can be prevented by haloperidol pretreatment.
The next major contribution to the field of dopaminergic control of the HPA axis was the clarification of the central stimulatory activity of cocaine [13-16]. The cocaine-elicited HPA response [13-19] is now generally considered to represent dopaminergic activation [20-21] of corticotropin-releasing hormone (CRH) secretion [16]. However, the specific roles of the D1-like (D1 and D5) and the D2-like families (D2, D3 and D4) of pre- and postsynaptic dopamine receptors in these processes has not yet been clarified. Further, Goebel et al. even reported clear-cut evidence of the central stimulatory action of the D2 antagonist, haloperidol, on corticosterone secretion [23]. This finding is in sharp contrast with that of Jezova [11] and is in complete agreement with several clinical symptoms of dopaminergic deficit. In major depression, bipolar disease, Parkinson’s dementia and, especially, in LBD (LBD completely eradicates the dopaminergic cells in the nervous system) cessation of the circadian rhythm of the HPA axis and overall hypercortisolism are almost unequivocal universal findings [24-27]. Goebel tried to explain this contradiction with a peripheral stimulatory action of prolactin on the adrenal gland [23], however we surmised a dose and challenge dependent role of dopamine in the regulation of the stress response.
Our earlier publications have clearly demonstrated the stressor specificity of the HPA response [28,29], which supports the aforementioned hypothesis. Further, in the case of pharmacologic challenges, the relative contributions of D1 and D2 receptors are still debated and the experimental findings are inconsistent. Zhou et al. [30] found that in the hypothalamus, neither of these receptors influenced CRH expression, while D2 receptors appeared to curb cocaine-binge elicited transcription of proopiomelancocortin (POMC). However, recently Gozzi et al. reported a profound inhibitory activity of D1 receptor antagonism on the orexin positive stress and arousal pathways [31]. Therefore, these contradictory findings necessitate stressor specific evaluation of the role of D1 and D2 receptor families in HPA activation.
Consequently, our experiments first investigated the dopaminergic mediation of ether induced HPA activation by using two selective inhibitors of dopaminergic transmission: the D1 receptor antagonist, SCH-23390, and the D2 dopamine receptor antagonist, haloperidol. However, because we observed a profound additive activity of D2 receptor inhibition on the endocrine response, the mediation of the intrinsic effect was evaluated by measuring plasma levels of adrenocorticotropic hormone (ACTH) and corticosterone and by applying the corticotropin-releasing hormone (CRH) receptor antagonist, α-helical CRF9-41.
Animals
The animals were maintained and handled during the experiments in accordance with the Council Directive of the European Economic Community regarding the protection of animals for experimental and other scientific purposes (86/609/EEC). Male Wistar rats weighing 150-250 g upon arrival were used and they were between 2.5-3 months old at the time of the experiments. The rats were kept in their home cages at a constant room temperature on a standard illumination schedule, with 12-h light and 12-h dark periods (lights on from 6.00 a.m.). Commercial food and tap water were available ad libitum. The rats were allowed a minimum of 1 week to acclimatize before surgery. To minimize the effects of nonspecific stress, the rats were handled daily. All experiments were carried out between 8:00 a.m. and 10:00 a.m.
Surgery
For icv. administration, the rats were implanted, under pentobarbital (Nembutal, Phylaxia-Sanofi, Budapest; 35 mg/kg, intraperitoneal (ip.)) anesthesia, with a stainless steel, Luer cannula (10 mm long) aimed at the right lateral cerebral ventricle. The stereotaxic coordinates were 0.2 mm posterior, 1.7 mm lateral to the bregma, and 3.7 mm deep from the dural surface, according to a rat brain atlas [32]. Cannulas were secured to the skull with dental cement and acrylate. The rats were used for studies after a recovery period of at least 5 days. After the experiments to verify the permeability of the cannulas, methylene blue was injected into each decapitated head and the brains were dissected. Only the data obtained from animals which manifested a diffusion of methylene blue into all the ventricles were used for statistical analysis
Treatments
The animals were subjected to pretreatment with either the D1 receptor selective dopamine blocker, SCH-23390 (Tocris, Bristol, UK), or the D2 dopamine receptor antagonist, haloperidol (Richter, Budapest, Hungary). All compounds were dissolved in 0.9% saline. The initial concentrations of the antagonists were doses we found to be most effective in previous experiments [33,34], but which per se did not affect hormone levels. Different doses of haloperidol (from 25 nmol/kg to 2.5 mmol/kg) and SCH-23390 (25-250 nmol/kg) were dissolved in 0.5 ml saline and administered ip. to conscious rats. The same volume of saline was administered to the controls ip. To evaluate the mediation of the intrinsic activity of haloperidol, combined treatment with a CRH antagonist was performed. Thirty minutes prior to haloperidol treatment, 5 nmol/kg CRH antagonist, α-helical CRH9-41 (Bachem, Germany) was administered icv. in a volume of 2 μl.
Ether stress
Thirty minutes after the above treatments the animals were exposed to ether stress in the following manner. Rats were removed from their cages and placed individually for 1.5 min in a large closed jar containing a diethyl-ether (Reanal, Hungary) dampened paper towel at the bottom. Unconsciousness always occurred within this period of breathing the ether-saturated atmosphere [28,29].
Determination of plasma corticosterone
Thirty min after the ether stress, the rats were sacrificed by cervical dislocation and decapitation, and approximately 3 ml of trunk blood was collected in heparinized tubes for corticosterone assay. The plasma corticosterone concentration was measured by fluorescence assay [36,37].
Determination of plasma ACTH
The most effective doses of haloperidol found in the corticosterone or saline alone (control animals) assays were injected ip. into conscious rats. Fifteen min after this treatment, the animals were sacrificed and 2 ml of blood was collected in EDTA-containing tubes. The ACTH concentrations of the samples were determined by a solid-phase two-site sequential chemiluminescent immunometric assay (Immulite 2000, Diagnostics Products Corporation, Los Angeles, USA). The analytical sensitivity of the assay is 5 pg/ml, whereas the intra-assay and inter-assay precisions are 6.8 % and 8.2 %, respectively.
Statistical analysis
Values are presented as means ± S.E.M. Statistical analysis of the data was performed by general linear model (GLM). The differences between groups were examined by Tukey’s post hoc comparison test, and a probability level of 0.05 or less was accepted as indicating a statistically significant difference. Data reductions and statistical analyses were performed by SigmaPlot 12.0 (Systat Software, Inc., Chicago, IL) and IBM SPSS Statistics 20.0 (IBM Corporation, Armonk, NY).
As expected, ether stress caused a marked increase in plasma corticosterone (Figure 1; F(1, 106)=134.97, p<0.01 vs. control). Pretreatment with antagonists also proved to be effective (Figure 1; F(4, 106)=25.102, p<0.01). According to Tukey’s post hoc comparisons, SCH-23390 slightly attenuated the HPA response at the highest dose (Figure 1; p<0.01 for 250 nmol SCH-23390 vs. saline). Haloperidol pretreatment in a dose of 25 nmol/kg showed a tendency to increase the basal but did not influence the ether induced corticosterone secretion. In contrast, a higher dose of haloperidol (250 nmol/kg) elevated the basal plasma corticosterone level and the same pretreatment even augmented the ether induced response (Figure 1; p<0.05 vs. saline for 25 nmol/kg haloperidol and p<0.01 vs. saline for 250 nmol/kg).
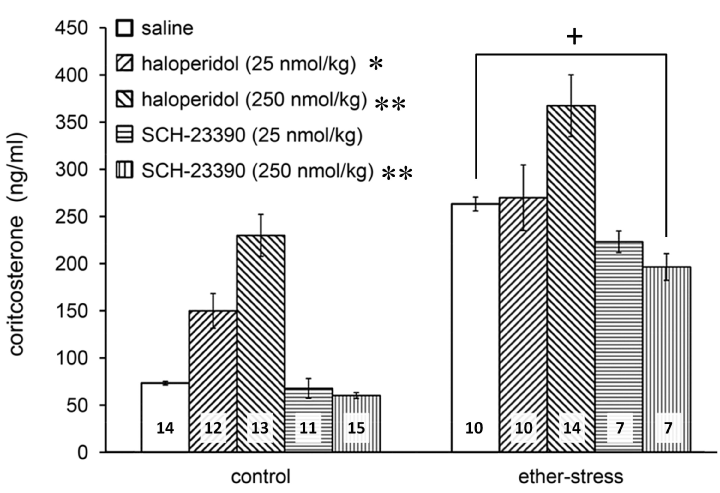
Figure 1. Effects of the D1 antagonist, SCH-23390, and D2 antagonist, haloperidol, on ether stress evoked corticosterone release. Data are expressed as means ± S.E.M. Numbers within bars are the numbers of animals used. Symbols: *=p<0.05 vs. saline, **=P<0.01 vs. saline, +=p<0.01 ether vs. control.
In further experiments we established that the stimulatory effect of haloperidol is dose-dependent on both the ACTH (Figure 2; F(3, 21)=3.2, p<0.05 for 2.5 mmol/kg haloperidol vs. saline with Tukey’s post hoc test) and corticosterone responses (Figure 2; F(3, 21)=18.85, p<0.01 for both 250 nmol/kg haloperidol vs. saline and 2.5 mmol/kg haloperidol vs. saline with Tukey’s post hoc test).
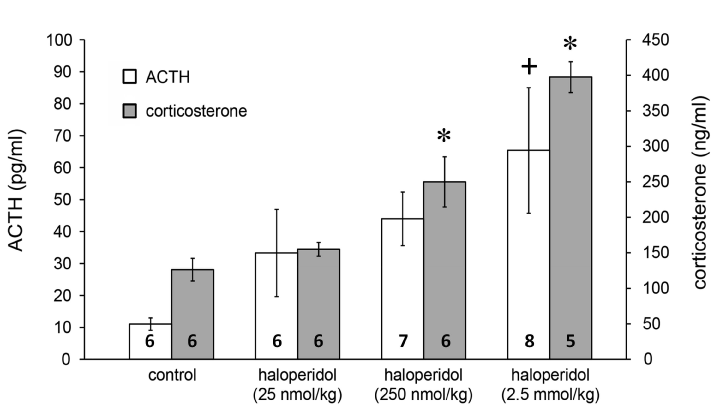
Figure 2. Effects of haloperidol on ACTH and corticosterone release. Data are expressed as means ± S.E.M. Numbers within bars are the numbers of animals used. Symbols: *=p<0.01 vs. saline; +=p<0.05 vs. saline.
In the third set of experiments the D2 antagonist evoked response (Figure 3; F(2, 48)= 80.901, p<0.01 vs. saline) could be partially attenuated by pretreatment with the CRH antagonist α-helical CRH9-41 (Figure 3; F(1, 48)=28.171, P<0.01 vs. saline). The revealed interaction between the two treatments (F(2, 48)=11.0, P<0.01 vs. saline) suggested specificity of the CRH antagonist.
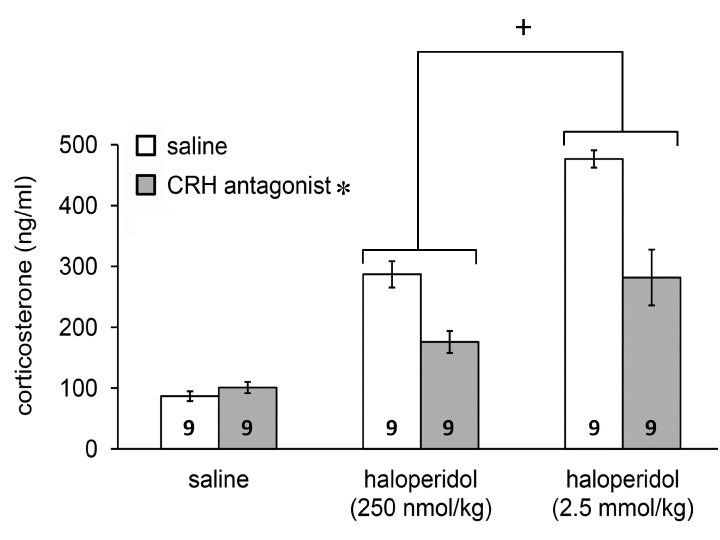
Figure 3. Inhibitory action of the CRH antagonist, α-helical CRH9-41, pretreatment on haloperidol-evoked corticosterone release. Data are expressed as means ± S.E.M. Numbers within bars are the numbers of animals used. Symbols: *=p<0.01 for CRH antagonist vs. saline; +=p<0.05 for haloperidol vs. saline.
The results obtained from our experiments with ether stress and selective dopaminergic antagonists refine the proposed oversimplified model of dopaminergic mediation in the control of the HPA axis [11,38,39]. Selective D1 inhibition by SCH-23390 did not have a significant impact on basal secretion and appeared slightly antagonistic on stimulated activity of the HPA axis, which supports the observations of Zhou et al. regarding cocaine challenge [30]. However, to our surprise, haloperidol at higher doses even augmented the ether evoked corticosterone response. Because the available literature reflects conflicting data regarding the action of antipsychotics on the HPA axis [23,30,40], in separate, dose-response experiments, the intrinsic activity of D2 antagonism on the HPA system was investigated. It was not only the inconsistent literature but the increasingly recognized importance of the CRH positive neural networks in the development of dopaminergic system-related disorders [23,35,41-45] that motivated us to investigate this question. Nevertheless, our results unambiguously revealed that the dopamine D2 receptor blockade by haloperidol per se increased both ACTH and corticosterone secretion and the response could also be inhibited by pretreatment with the CRH antagonist, α-helical CRH9-41. These findings argue for two important conclusions. First, in the regulation of the HPA axis the activity of D2 receptors predominate and that of the D1 family is negligible. Second, the D2-family exerts tonic inhibitory control over HPA stimulation and blockade of the receptors leads to profound augmentation of the stress response. Our findings somewhat correspond to those of Zhou but unlike cocaine-binge [23] ether stress appears to rely on CRH, not POMC, transcription. Taking into account the effectiveness of haloperidol and, that the tuberoinfundibular system mainly expresses D2 and D3 receptors [46-49] it appears that the D2-like family exerts tonic inhibitory control over HPA activation in the hypothalamus. Earlier studies clearly demonstrated that ether stimulates directly the hypothalamic CRH secretion [28,29], because it proved effective despite complete hypothalamic deafferentation [50-52] or olfactory bulbectomy [53]. Therefore, haloperidol apparently interrupts centrally, in the hypothalamus the feedback regulation exerted by presynaptic D2 autoreceptors on dopamine synthesis and release. Further, at the same time, it may also disinhibit the action of D2/D3 receptors on postsynaptic adenyl-cyclase which, in turn, increase the level of cAMP [1,46]. Since, in our studies, the CRH antagonist attenuated the haloperidol-evoked response it seems that this effect, to some extent, is independent of the direct action of dopaminergic neurons of the arcuate nucleus on POMC-positive cells of the pituitary. Further, not completely excluding the potential influence of some regions of the mesolimbic system, such as the nucleus accumbens [49], we propose that it is the dopaminergic and peptidergic inputs from the arcuate nucleus [54] that may play significant roles in the central processing of ether-induced HPA activation [28,29]. This way, our experiments provided in vivo, functional evidence for D2 mediated disinhibition of CRH and ACTH secretion. This has not been previously clarified, unlike the dopaminergic control of prolactin secretion, which is also inhibited by a D2 receptor pathway [55,56]. Nevertheless, our findings may reflect ether-challenge specific features of HPA signaling.
Therefore, decreased activity in the dopaminergic system may lead to a two-pronged endocrine response in the form of CRH and prolactin secretion. This may favor the integrity of the individual (HPA response, self-maintenance) at the expense of inhibition of gonadotropins (reproduction) by prolactin [56] in a stressful environment. Perhaps kisspeptin and other RFamides may play a crucial, integrative role in these processes influencing dopamine and then, in turn, CRH and prolactin release [57-60]. Moreover, dopamine receptor down-regulation, and consequent hyperprolactinemia may also aggravate HPA stimulation [23].
Nonetheless, further pharmacodynamic studies would be necessary to clarify this bewilderingly complex picture and the individual contributions of hypothalamically expressed D2 and D4 receptors. Moreover, serotonin and noradrenaline and those pharmacologic agents, which influence their levels (monoamine oxidase inhibitors, tricyclics and selective serotonin reuptake inhibitors), play equally ambiguous [9,61,62] roles in the regulation of the HPA axis and mood disorders [61-64]. To clarify the mediation of their activity, and to shed light on their similarly unpredictable and dangerous side-effect spectrum [64,65] further studies are also required.
This work was supported by OTKA grants T 022230 and T 006084, by Hungarian Ministry of Social Welfare Grants T-02-670/96, FKFP 0091/1997, TÁMOP 4.2.2-A-11/KONV-2012-0052 and KTIA_13_NAP-A-III/8. The authors of the manuscript have no conflicts of interest to declare.
- Meltzer HY (1982) “Dopamine autoreceptor stimulation: clinical significance”. Pharmacol Biochem Behav 17 Suppl 1: 1-10. [Crossref]
- Lieberman JA1 (2004) Dopamine partial agonists: a new class of antipsychotic. CNS Drugs 18: 251-267. [Crossref]
- George O, Koob GF (2010) Individual differences in prefrontal cortex function and the transition from drug use to drug dependence. Neurosci Biobehav Rev 35: 232-247. [Crossref]
- Trainor BC (2011) Stress responses and the mesolimbic dopamine system: social contexts and sex differences. Horm Behav 60: 457-469. [Crossref]
- Kebabian JW (1978) “Multiple classes of dopamine receptors in mammalian central nervous system: the involvement of dopamine-sensitive adenylyl cyclase”. Life Sci 23(5): 479-483.
- Beaulieu JM, Espinoza S, Gainetdinov RR (2015) Dopamine receptors - IUPHAR Review 13. Br J Pharmacol 172: 1-23. [Crossref]
- Haddad PM, Dursun SM (2008) Neurological complications of psychiatric drugs: clinical features and management. Hum Psychopharmacol 23 Suppl 1: 15-26. [Crossref]
- Hensler JG, Artigas F, Bortolozzi A, Daws LC, De Deurwaerdère P, et al. (2013) Catecholamine/Serotonin interactions: systems thinking for brain function and disease. Adv Pharmacol 68: 167-197. [Crossref]
2021 Copyright OAT. All rights reserv
- Holsboer F, Müller OA, Winter K, Doerr HG, Sippell WG (1983) “Effect of serotonin uptake inhibition by zimelidine on hypothalamic-pituitary-adrenal activity”. Psychopharmacology (Berl) 80(1): 85-87. [Crossref]
- Koob GF (2008) A role for brain stress systems in addiction. Neuron 59: 11-34. [Crossref]
- Jezová D, Jurcovicová J, Vigas M, Murgas K, Labrie F (1985) Increase in plasma ACTH after dopaminergic stimulation in rats. Psychopharmacology (Berl) 85: 201-203. [Crossref]
- Hennig J, Rzepka U, Mai B, Netter P (1995) Suppression of HPA-axis activity by haloperidol after experimentally induced heat stress. Prog Neuropsychopharmacol Biol Psychiatry 19: 603-614. [Crossref]
- Moldow RL, Fischman AJ (1987) Cocaine induced secretion of ACTH, beta-endorphin, and corticosterone. Peptides 8: 819-822. [Crossref]
- Rivier C, Vale W (1987) “Cocaine stimulates adrenocorticotropin (ACTH) secretion through a corticotropin-releasing factor (CRF)-mediated mechanism”. Brain Res 422(2): 403-406. [Crossref]
- Sarnyai Z, Bíró E, Penke B, Telegdy G (1992) “The cocaine-induced elevation of plasma corticosterone is mediated by endogenous corticotropin-releasing factor (CRF) in rats”. Brain Res 589(1): 154-156. [Crossref]
- Sarnyai Z (1998) Neurobiology of stress and cocaine addiction. Studies on corticotropin-releasing factor in rats, monkeys, and humans. Ann N Y Acad Sci 851: 371-387. [Crossref]
- Stanley SA, Small CJ, Murphy KG, Rayes E, Abbott CR, et al. (2001) Actions of cocaine- and amphetamine-regulated transcript (CART) peptide on regulation of appetite and hypothalamo-pituitary axes in vitro and in vivo in male rats. Brain Res 893: 186-194. [Crossref]
- Chmielowska M1, Baranowska-Bik A, Baranowska B, Wolinska-Witort E, Martynska L, et al. (2011) “The influence of cocaine-amphetamine regulated transcript (CART) on pituitary hormones, corticosterone and leptin levels in starved rats”. Neuro Endocrinol Lett 32(1): 82-89. [Crossref]
- Skrapits K, Borsay BÁ, Herczeg L, Ciofi P, Bloom SR, et al. (2014) “Colocalization of cocaine- and amphetamine-regulated transcript with kisspeptin and neurokinin B in the human infundibular region”. PLoS One 9(8): e103977. [Crossref]
- Ross SB, Renyi AL (1967) Inhibition of the uptake of tritiated catecholamines by antidepressant and related agents. Eur J Pharmacol 2: 181-186. [Crossref]
- Ritz MC, Lamb RJ, Goldberg SR, Kuhar MJ (1987) Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science 237: 1219-1223. [Crossref]
- Ritz MC, Cone EJ, Kuhar MJ (1990) “Cocaine inhibition of ligand binding at dopamine, norepinephrine and serotonin transporters: a structure-activity study”. Life Sci 46(9): 635-645. [Crossref]
- Goebel S, Dietrich M, Jarry H, Wuttke W (1992) “Indirect evidence to suggest that prolactin mediates the adrenal action of haloperidol to stimulate aldosterone and corticosterone secretion in rats”. Endocrinology 130(2): 914-919. [Crossref]
- Breen DP, Vuono R, Nawarathna U2, Fisher K, Shneerson JM2, et al. (2014) Sleep and circadian rhythm regulation in early Parkinson disease. JAMA Neurol 71: 589-595. [Crossref]
- Kitajima Y, Hori K, Konishi K, Tani M, Tomioka H, et al. (2015) A Review of the Role of Anticholinergic Activity in Lewy Body Disease and Delirium. Neurodegener Dis 15: 162-167. [Crossref]
- Melo MC, Abreu RL2, Linhares Neto VB2, de Bruin PF3, de Bruin VM3 (2016) Chronotype and circadian rhythm in bipolar disorder: A systematic review. Sleep Med Rev. [Crossref]
- Melo MC, Garcia RF2, Linhares Neto VB2, Sá MB2, de Mesquita LM2, et al. (2016) Sleep and circadian alterations in people at risk for bipolar disorder: A systematic review. J Psychiatr Res 83: 211-219. [Crossref]
- Jászberényi M, Bujdosó E, Telegdy G (1998) Effects of C-type natriuretic peptide on pituitary-adrenal activation in rats. Neuroreport 9: 2601-2603. [Crossref]
- Jászberényi M, Bujdosó E, Telegdy G (2000) Effects of brain natriuretic peptide on pituitary-adrenal activation in rats. Life Sci 66: 1655-1661. [Crossref]
- Zhou Y, Spangler R, Yuferov VP, Schlussmann SD, Ho A, et al. (2004) “Effects of selective D1- or D2-like dopamine receptor antagonists with acute “binge” pattern cocaine on corticotropin-releasing hormone and proopiomelanocortin mRNA levels in the hypothalamus”. Brain Res Mol Brain Res 130(1-2): 61-67. [Crossref]
- Gozzi A, Lepore S, Vicentini E, Merlo-Pich E, Bifone A (2013) “Differential effect of orexin-1 and CRF-1 antagonism on stress circuits: a fMRI study in the rat with the pharmacological stressor Yohimbine”. Neuropsychopharmacology 38(11): 2120-2130. [Crossref]
- Pellegrino LJ, Cushman AJ (1967) A stereotaxic atlas of the rat brain. New York, Appleton-Century-Crofts.
- Bujdosó E, Jászberényi M, Tömböly C, Tóth G, Telegdy G (2001) Effects of endomorphin-1 on open-field behavior and on the hypothalamic-pituitary-adrenal system. Endocrine 14: 221-224. [Crossref]
- Bujdosó E, Jászberényi M, Gardi J, Földesi I, Telegdy G (2003) The involvement of dopamine and nitric oxide in the endocrine and behavioural action of endomorphin-1. Neuroscience 120: 261-268. [Crossref]
- Bagosi Z, Jászberényi M, Szabó G, Telegdy G (2008) The effects of CRF and the urocortins on [3H]GABA release from the rat amygdala--an in vitro superfusion study. Brain Res Bull 75: 15-17. [Crossref]
- Zenker N, Bernstein De (1958) The estimation of small amounts of corticosterone in rat plasma. J Biol Chem 231: 695-701. [Crossref]
- Purves HD, Sirett NE (1965) Assay of corticotrophin in dexamethasone-treated rats. Endocrinology 77: 366-374. [Crossref]
- Campbell A, Baldessarini RJ, Cremens C, Teicher MH, Marsh E, et al. (1989) Bromocriptine antagonizes behavioral effects of cocaine in the rat. Neuropsychopharmacology 2: 209-224. [Crossref]
- Borowsky B, Kuhn CM (1991) Monoamine mediation of cocaine-induced hypothalamo-pituitary-adrenal activation. J Pharmacol Exp Ther 256: 204-210. [Crossref]
- Fuller, R. W. and H. D. Snoddy (1981) “Elevation of serum corticosterone concentrations in rats by pergolide and other dopamine agonists”. Endocrinology 109(4): 1026-1032. [Crossref]
- Bagosi Z, Jászberényi M, Bujdosó E, Telegdy G (2006) “The effects of corticoptropin-releasing factor and the urocortins on striatal dopamine release induced by electrical stimulation - An in vitro superfusion study”. Neurochem Res 31(2): 209-213. [Crossref]
- Refojo D, Holsboer F (2009) CRH signaling. Molecular specificity for drug targeting in the CNS. Ann N Y Acad Sci 1179: 106-119. [Crossref]
- Bonfiglio JJ, Inda C, Refojo D, Holsboer F, Arzt E, et al. (2011) “The corticotropin-releasing hormone network and the hypothalamic-pituitary-adrenal axis: molecular and cellular mechanisms involved”. Neuroendocrinology 94(1): 12-20. [Crossref]
- Koob GF, Buck CL, Cohen A, Edwards S, Park PE, et al. (2014) Addiction as a stress surfeit disorder. Neuropharmacology 76 Pt B: 370-382. [Crossref]
- Schatzberg AF (2015) “Anna-Monika Award Lecture, DGPPN Kongress, 2013: the role of the hypothalamic-pituitary-adrenal (HPA) axis in the pathogenesis of psychotic major depression”. World J Biol Psychiatry 16(1): 2-11. [Crossref]
- Beaulieu JM, Gainetdinov RR (2011) The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev 63: 182-217. [Crossref]
- Liang SL, Pan JT (2012) “An endogenous dopaminergic tone acting on dopamine D3 receptors may be involved in diurnal changes of tuberoinfundibular dopaminergic neuron activity and prolactin secretion in estrogen-primed ovariectomized rats”. Brain Res Bull 87(2-3): 334-339. [Crossref]
- Ares-Santos S, Granado N, Moratalla R (2013) The role of dopamine receptors in the neurotoxicity of methamphetamine. J Intern Med 273: 437-453. [Crossref]
- Liang SL, Hsu SC (2014) “Involvement of dopamine D2 receptor in the diurnal changes of tuberoinfundibular dopaminergic neuron activity and prolactin secretion in female rats”. J Biomed Sci 21: 37.
- Matsuda K, Duyck C, Kendall Jw Jr, Greer Ma (1964) Pathways By Which Traumatic Stress And Ether Induce Increased Acth Released In The Rat. Endocrinology 74: 981-985. [Crossref]
- Greer MA, Rockie C (1968) Inhibition by pentobarbital of ether-induced ACTH secretion in the rat. Endocrinology 83: 1247-1252. [Crossref]
- Feldman S, Conforti N (1981) “Effects of hypothalamic deafferentations on adrenocortical responses in the rat following hippocampal stimulation”. Exp Brain Res 44(2): 232-234. [Crossref]
- Marcilhac A, Anglade G, Hery F, Siaud P (1999) Effects of bilateral olfactory bulbectomy on the anterior pituitary corticotropic cell activity in male rats. Horm Metab Res 31: 399-401. [Crossref]
- van-Hover C, Li C (2015) “Stress-activated afferent inputs into the anterior parvicellular part of the paraventricular nucleus of the hypothalamus: Insights into urocortin 3 neuron activation”. Brain Res 1611: 29-43. [Crossref]
- Fitzgerald P, Dinan TG (2008) Prolactin and dopamine: what is the connection? A review article. J Psychopharmacol 22: 12-19. [Crossref]
- Grattan DR (2015) 60 Years of neuroendocrinology: The hypothalamo-prolactin axis. J Endocrinol 226: T101-122. [Crossref]
- Jászberényi M, Bagosi Z, Thurzó B, Földesi I, Szabó G, et al. (2009) Endocrine, behavioral and autonomic effects of neuropeptide AF. Horm Behav 56: 24-34. [Crossref]
- Csabafi K, Jászberényi M, Bagosi Z, Lipták N, Telegdy G (2013) “Effects of kisspeptin-13 on the hypothalamic-pituitary-adrenal axis, thermoregulation, anxiety and locomotor activity in rats”. Behav Brain Res 241: 56-61. [Crossref]
- Jászberényi M, Bagosi Z, Csabafi K, Palotai M, Telegdy G2 (2014) The actions of neuropeptide SF on the hypothalamic-pituitary-adrenal axis and behavior in rats. Regul Pept 188: 46-51. [Crossref]
- Ribeiro AB, Leite CM, Kalil B, Franci CR, Anselmo-Franci JA, et al. (2015) “Kisspeptin regulates tuberoinfundibular dopaminergic neurones and prolactin secretion in an oestradiol-dependent manner in male and female rats”. J Neuroendocrinol 27(2): 88-99. [Crossref]
- Vermes I, Dull G, Telegdy G, Lissák K (1972) Possible role of serotonin in the monoamines-induced inhibition of the stress mechanism in the rat. Acta Physiol Acad Sci Hung 42: 219-223. [Crossref]
- Kirilly E, Gonda X, Bagdy G (2015) [Antidepressants, stressors and the serotonin 1A receptor]. Neuropsychopharmacol Hung 17: 81-89. [Crossref]
- Hayes PE, Ettigi P (1983) Dexamethasone suppression test in diagnosis of depressive illness. Clin Pharm 2: 538-545. [Crossref]
- Haberzettl R, Bert B, Fink H, Fox MA (2013) Animal models of the serotonin syndrome: a systematic review. Behav Brain Res 256: 328-345. [Crossref]
- Dosi R, Ambaliya A (2014) “Serotonin syndrome versus neuroleptic malignant syndrome: a challenging clinical quandary”. BMJ Case Rep.