Background: The association of Neuromyelitis Optica Spectrum Disorders (NMOSD) with autoimmune disorders including Sjögren’s syndrome (SS), is well recognized. Epstein Barr virus (EBV) has been associated to various neurological entities. We describe a case where EBV infection likely preceded NMOSD in a patient with unrecognized SS. The clinical features, work up and management are described.
Case presentation: A 40-year woman with history of stroke and Guillain-Barre Syndrome (GBS) two years prior, presented with progressive lower extremity weakness and pain. Brain MRI revealed hyperintensities in the cerebellar and parietal lobes consistent with old infarcts, high intensity signal in the white matter and enhancing intramedullary lesion at the level of T2 and the conus medullaris. Cerebrospinal fluid (CSF) revealed no oligoclonal bands. Next day, the patient developed right ankle weakness and urinary incontinence. NMOSD was suspected and pulse steroids initiated. Patient’s weakness resolved. Antinuclear antibodies (ANA), anti-SSA/SSB and Aquaporin 4 antibodies (AQP4Ab) were positive. CSF was positive for EBV. Parotid gland ultrasound revealed non-homogeneous tissue.
Ganciclovir and plasmapheresis were started. The patient’s sensation and motor deficits improved and one month after, she had regained motor power and sphincter control. The patient was discharged on oral prednisone and plans for rituximab infusions.
On follow-up imaging, Spinal MRI showed areas of myelomalacia and complete resolution at the level of T2 and conus medularis lesions respectively. The patient had no additional flares, but did complain of chronic neuropathic pain.
Conclusion: NMOSD commonly coexist with other autoimmune diseases. The association of SS and NMOSD is well recognized. EBV infections can present with neurological manifestations however, EBV has also been linked to the development of autoimmunity. In our case, EBV was detected in CSF and antiviral therapy was initiated in addition to the treatment modalities for NMOSD which led to a full recovery in our patient.
neuromyelitis optica spectrum disorders, sjogren’s syndrome, aquaporin-4 ig g antibodies, epstein-barr virus infection
Sjögren Syndrome (SS) has long been known to involve the central and peripheral nervous systems leading to structural and functional alterations [1-4]. Among the most significant structural derangements in SS patients are white matter lesions that meet the diagnostic criteria of Neuromyelitis Optica Spectrum Disorders (NMOSD). These lesions involve the brain, optic nerves and spinal cord [5,6]. On the other hand, Epstein Barr virus (EBV) infections can also present with neurological manifestations in up to 7.3% of the cases, with long extensive myelitis and acute demyelinating encephalomyelitis being rare [7,8]. Research unveiled that the long observed autoimmunity driven by EBV, is likely explained by an EBV gene product that appears to be central to the interaction between human transcription factors and gene expression patterns triggering autoimmunity [9]. Herein we present the case of middle age woman who had suffered Guillain-Barre Syndrome (GBS) two years prior and presented with progressive weakness. Work-up revealed NMOSD, SS, and EBV in cerebrospinal fluid. Clinical presentation, diagnostic work up, therapeutic plan and outcome will be discussed.
A 40-year-old Guyanese woman presented with a five-week history of progressive bilateral lower extremity weakness and pain. Two years prior, the patient had a right (R) sided cerebrovascular accident (CVA) in the post-operative period of a diaphragmatic hernia repair. One month following the CVA, the patient developed diplopia and R sided ascending paralysis requiring ventilatory support. Guillain-Barre Syndrome (GBS) was suspected and she was treated with intravenous immunoglobulin (IVIG) and steroids. The patient recovered with a residual left-sided tongue deviation and hypoesthesia.
Five weeks prior to the current presentation, the patient developed weakness to flexion of left (L) knee accompanied by “painful tingling” and intermittent cold sensation. Over the weeks, the weakness and pain progressed to include L ankle and hip, immobilizing the patient. The patient traveled to USA in search of medical treatment. There was no additional past medical history. Medications included folic acid and multivitamins and the occasional use of natural tears. Denied smoking, alcohol or illicit drug use. Her mother and grandmother suffered from arthritis. She had one healthy daughter 4 years old.
On examination, her temperature was 36.7°C, pulse of 81 beats per minute, blood pressure 112/75 mmHg, respiratory rate 17 breaths per minute, and her oxygen saturation was 100% on ambient air. The patient appeared comfortable yet distressed, with a normal cardiac, respiratory, and abdominal exam. Neurological examination revealed 3/5 motor strength on L hip flexion and abduction, 1/5 for L knee flexion, 0/5 for L knee extension and L foot dorsiflexion with preserved R sided lower limb motor strength. There was reduced sensation to light touch, pinprick, vibration and joint position throughout the L side of the body including face, upper and lower extremities with complete loss of pinprick sensation in the inner aspect of the L thigh and leg. Reflexes were 2+ for L patellar and Achilles, L plantar was mute. No rashes were noticed.
On day of admission, a head computed tomography (CT) along with brain and spine magnetic resonance imaging (MRI) were performed. The head CT showed no acute bleed or mass effect. Fluid attenuated inversion recovery (FLAIR) images revealed minimal T1 hyperintensities in the R cerebellar and parietal lobes consistent with old infarcts, with high intensity signal within the cerebral white matter, an enhancing 2.0 x 1.0 x 0.8 cm intramedullary lesion in the thoracic spinal cord at the T12 level and a 0.6x0.6 cm enhancing lesion in the distal conus medularis (Figure 1). Laboratory test results obtained on admission are shown in Table 1. Cerebrospinal fluid (CSF) analysis returned with no monoclonal proteins and normal electrophoretogram. Flow cytometry was also negative for lymphoma. Infectious and autoimmune labs were requested.
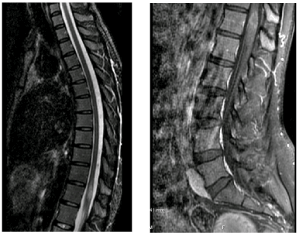
Figure 1. (Left) MRI STIR weighted MRI sequence of the thoracic region showing 2.0 x 1.0 x 0.8cm contrast enhancing intramedullary lesion. (Right) Fat-suppressed T1-weighted MRI sequence showing contrast enhancing lesion at the level of the conus medullaris.
Table 1. Laboratory data
Serum |
Patient |
Reference Range |
WBC (K/uL) |
3.53 |
4.5-10.9 |
RBC (M/uL) |
4.00 |
4.2-5.4 |
Hemoglobin (g/dL) |
12.2 |
12.0-16.0 |
Hematocrit (%) |
34.4 |
37.0-47.0 |
Platelets (K/uL) |
237 |
130-400 |
|
|
|
Sodium (mmol/L) |
142 |
136-146 |
Potassium (mmol/L) |
5.5 |
3.5-5.0 |
Chloride (mmol/L) |
107 |
98-106 |
BUN (mg/dL) |
15 |
6-20 |
Creatinine (mg/dL) |
0.85 |
0.4-1.2 |
Calcium (mg/dL) |
9.5 |
8.4-10.3 |
Total Protein (g/dL) |
7.5 |
6.0-8.5 |
Albumin (g/dL) |
3.6 |
2.8-5.7 |
AST (U/L) |
24 |
10-35 |
ALT (U/L) |
20 |
0-31 |
Alk Phos (U/L) |
71 |
25-125 |
Total Bilirubin |
0.27 |
0.0-1.2 |
Glucose (mg/dL) |
106 |
70-99 |
ESR(mm/h) |
17 |
0-20 |
CRP(mg/dL) |
0.85 |
1.0-4.0 |
TSH (miU/L) |
1.7 |
0.27-4.20 |
Vitamin B12 (pg/mL) |
724 |
211-946 |
Complement C3 (mg/dL) |
115 |
86-184 |
Complement C4 (mg/dL) |
18 |
20-58 |
Methylmalonic Acid (mmol/L) |
0.12 |
<0.40 |
Vitamin D 1,25(OH)2 (pg/mL) |
81.0 |
19.9-79.3 |
Lactate (mmol/L) |
1.4 |
0.5-2.2 |
|
|
|
Cerebrospinal Fluid |
|
|
WBC /cu mm |
69 |
0-5 |
RBC /cu mm |
0 |
<5 |
Lymph (%) |
99 |
40-90 |
Gram Stain |
No organisms seen |
|
Protein (mg/dL) |
46 |
15-45 |
Glucose (mg/dL) |
52 |
40-70 |
Immunoglobulins (IgG, IgA, IgM) |
7.0,1.3,<0.5 |
0.8-7.7, <0.6, <0.5 |
Oligoclonal Bands |
Absent |
Absent |
On hospital day two, the patient developed new R ankle weakness and intermittent loss of urinary and rectal sphincter control. NMOSD was suspected and treatment was initiated with methylprednisolone 1 gm daily for 5 days. Over the initial five days of treatment, the patient’s R sided weakness resolved and the L sided weakness improved. CSF meningo-encephalitis panel returned positive for EBV, anti-nuclear antibody (ANA) titer was 1:160 with a speckled pattern and Anti-SSA/SSB were also positive. Aquaporin 4 antibodies (AQP4 Ab) resulted at a titer of 1:10000 (positive >1:5) (Table 2).
Table 2. Infectious and immunological workup
|
Patient |
Reference Range |
ANA |
1:160 (speckled pattern) |
<1:40 |
Anti-DNA (IU/mL) |
32 |
<100 |
c-ANCA |
7 |
<199 |
p-ANCA |
3 |
<100 |
Anti-SM |
Negative |
Negative |
Anti-RNP |
Negative |
Negative |
Anti-SSA (Ro) |
Positive |
Negative |
Anti-SSB (La) |
Positive |
Negative |
Anti-Centromere |
Negative |
Negative |
Anti-Jo 1 |
Negative |
Negative |
RF Quant (IU/mL) |
6 |
<14 |
Anti-Scl-70 |
Negative |
Negative |
NMO/AQ4 Ab |
1:10000 |
<1:5 |
Angiotensin 1 Converting Enzyme |
36 |
8-52 |
Beta 2 Glycoprotein (IgM/IgA/IgG)(SMU) |
<5.0/7.0/<5.0 |
<20.0 |
Cardiolipin AB IgM (MPL) |
7.2 |
0.0-12.5 |
Lupus Anticoagulant |
Negative |
Negative |
|
|
|
HIV-1/2 Ag/Ab |
Non-reactive |
Non-reactive |
CSF EBV PCR |
Positive |
Negative |
HTLV-I/II AB |
Negative |
Negative |
Mycoplasma AB IGM |
290 |
<910 |
B.Burgdorferi AB Screen |
0.5 |
<0.90 |
Treponemal IgG |
Non-reactive |
Non-reactive |
CSF Viral Culture (HSV. VZV, CMV, Enterovirus) |
Negative |
Negative |
The diagnosis of NMOSD was made; an ultrasound of the parotid glands revealed non-homogenous glandular tissue, supporting SS [10].
The patient was continued on high dose prednisone. Further immunosuppressive agents were held for fear of worsening EBV reinfection. Plasmapheresis was initiated along with Gancyclovir. Patient’s sensation and motor deficits improved.
One month following presentation, the patient was discharged home on prednisone with plans for Rituximab infusions. She had regained motor power in the lower extremities and sphincter control, denied pain or sensation defects.
Patient was seen in clinic two months after discharge, she complained of L knee pain, however was able to perform all activities of daily living. Patient had received the loading doses of Rituximab and was being tapered off high dose prednisone. Significant dysesthesia in the L knee area was noted. Repeat spine MRI showed complete resolution of the conus medularis lesion and a signal alteration with volume loss consistent with myelomalacia at the site of the prior T12 lesion. The patient was started on Gabapentin for neuropathic pain and hydroxychloroquine was added. After this visit, the patient returned to Guyana.
Eight months later, patient was doing well and was continued on hydroxychloroquine and low dose prednisone. Thereafter, the patient was lost to follow-up.
NMOSD is an autoimmune relapsing and demyelinating disorder of the central nervous system (CNS) characterized by optic neuritis (ON) and/or longitudinal extensive transverse myelitis (mainly in the cervical and thoracic segments) [11-13]. Without treatment, the disease is devastating producing blindness and paralysis [11-13]. The disease affects more frequently non-Caucasians, middle-aged females (80-90% of cases reported). Originally considered a subtype of multiple sclerosis (MS) [11-13], NMOSD became a distinct entity in 2004, after the discovery of the antibody against the water channel protein aquaporin-4 (AQP4) [11-13]. Utilization of AQ4 Ab has enhanced diagnostic accuracy and allowed the recognition of other NMOSD presentations, such as area postrema involvement [14].
The International Panel for NMO Diagnosis (IPND) redefined the International Consensus Diagnostic Criteria for NMOSD in 2015 [6]. NMOSD is classified in 3 groups: 1) NMOSD with AQP4 antibody (AQP4-IgG) or seropositive NMOSD, 2) NMOSD without AQP4-IgG, and 3) NMOSD with unknown AQP4-IgG [6]. Our patient has NMOSD with AQP4-IgG or seropositive NMOSD. Similar to other autoimmune diseases, NMOSD’s immune dysregulation likely results from the interaction of a predisposed host (genetic background) with environmental triggers. Mycoplasma pneumoniae, Mycobacterium tuberculosis, Treponema pallidum, Helicobacter pylori and Chlamydia pneumoniae have been linked to the initiation of NMOSD [15-17]. The gut microbiome may also play a role, as Clostridium perfringens was encountered in high numbers among NMOSD patients when compared to healthy and multiple sclerosis cohorts [18,19]. Viral infections such as cytomegalovirus, herpes simplex virus, varicella zoster virus, human immunodeficiency virus, human T lymphotropic virus type 1, and high EBV antibody titers have been also linked to the development of NMO [17,20,21]. Significant elevations in the intrathecal production of immunoglobulin G antibody against Early Antigen (EA) of EBV have been observed in NMO patients [20]. The stages of EBV infection are characterized by antibody formation against different viral antigens; the presence of antibodies against EA indicate active replication of EBV[22].
EBV causes infectious mononucleosis, an acute disease spread by intimate contact, but also is recognized as etiology for cases of GBS, meningoencephalitis, aseptic meningitis, transverse myelitis, optic neuritis and hematologic abnormalities [23,24]. Over 90% of humans are infected with EBV during childhood but remain asymptomatic. EBV resides in B lymphocytes of the infected individual in a latent form [25]. In the B lymphocytes, EBV Nuclear Antigen 2 (EBNA2) regulates latent viral transcription which allows immortalization of the virus and recruitment of transcription factors that bind to regions of both the EBV genome and the host’s own genome [9,26].
However, the link between EBV and autoimmune disease is not well understood. Few studies suggest that EBV triggers SS through cross-reactivity of anti-EBNA1 and anti-viral capside antigens to anti-Ro/La antibodies by molecular mimicry [27,28]. Another hypothesis is the production of anti-apoptotic signals by EBV, which lead to development of ectopic lymphoid structures rich in infected B cells [29]. The elevated number of infected B cells constitutes a reservoir of antigenic and viral particles contributing to the autoimmune process [9]. In a recent study comparing genetic and protein sampling from healthy individuals and patients with autoimmune diseases, Harley et al. identified regulatory gene regions associated with the risk of developing systemic erythematous lupus (SLE) and other autoimmune diseases. These regulatory gene regions also bound EBNA2 and its related transcription factors. Over 44% of the locations on the human genome known to contribute to autoimmune risk were also binding sites for EBNA2 [9]. These findings suggest that EBV infection in cells can drive the activation of these genes and contribute to an individual’s risk of developing autoimmune diseases[9]. Many of the above mechanisms could be implicated in EBV triggering NMOSD, as they could be involved in the pathogenesis of many autoimmune disease. However, NMOSD targets CNS. One possible explanation is that EBV can cause CNS infection which increases the blood brain barrier (BBB) permeability to AQ4-IgG leading to pathogenic changes [30-32]. EBV infection induces systemic increase of IFN-y, TNF-a, IL-6, which can increase BBB permeability [30].
It is well known that other autoimmune diseases may present in association with NMOSD, including SLE and SS [33,34] as our patient. The mechanism by which these autoimmune processes occur together, or if one predisposes the other one, is not known. Recent studies have shown similar pathogenic findings between primary SS and SLE, it has been suggested that primary SS is a mucosal presentation of SLE [35]. Some of these common pathogenic elements are also associated with NMO including: Type I interferon (IFN I), B cells, plasmablasts, plasma cells, and increased levels of B-cell activating factor of the tumor necrosis factor family (BAFF) [36-38]. BAFF is a cytokine that promotes B-cell maturation, proliferation, and survival. It is induced by IFN type I and II providing a link between innate immunity, viral infections as EBV, and autoimmunity. BAFF levels are increased in the CSF of seropositive NMOSD patients [39]. It is possible that NMOSD, SLE and SS are phenotypes of a genetic background susceptible to develop humoral autoimmunity [40]. It has been also proposed that NMOSD is a complication of a systemic rheumatologic disease based on reports of SLE and SS patients without NMOSD and positive AQP4-IgG [40]. In addition, a considerable proportion of patients with SS who develop CNS symptoms were later found to have NMO [1]. In this regard a recent study described the presence of AQP4-IgG in patients with SS and NMOSD but not in SS without NMOSD [41]. The authors concluded that NMOSD is not a CNS manifestation of SS but its own entity [41].
EBV in CSF detected by PCR should not always be interpreted as a cause for neurological manifestations, until anti-EBV IgM or IgG antibodies have been quantified [42,43]. However, in an attempt to summarize the sequence of pathogenic events in our patient, EBV-associated GBS, two years prior could represent initial EBV infection followed by a latency period. During the latent phase, EBV possibly favored SS development culminating in full NMOSD presentation.
In conclusion, we can postulate that positive EBV in our patient’s CSF represented active EBV replication, further supported by CSF mononuclear pleocytosis. The patient’s clinical presentation and laboratory findings warranted the initiation of gancyclovir aiming to inhibit viral replication and arrest the inciting mechanisms of autoimmunity. The excellent clinical response further strengthens our hypothesis of EBV’s pathogenic role in this case. Our case report supports the notion that NMOSD presentations should have CSF scrutinized for concurrent viral infections, given the ubiquitous presence of viral infections in the general population.
This work is supported in part by Dr. Moro O. Salifu's efforts through NIH Grant # S21MD012474.
- Bhattacharyya S, Helfgott SM (2014) Neurologic complications of systemic lupus erythematosus, sjogren syndrome, and rheumatoid arthritis. Semin Neurol 34: 425-36. [Crossref]
- Morreale M, Marchione P, Giacomini P, Pontecorvo S, Marianetti M et al. (2014) Neurological involvement in primary Sjogren syndrome: a focus on central nervous system. PLoS One 9: e84605. [Crossref]
- Ramos-Casals M, Solans R, Rosas J, Camps MT, Gil A, et al. (2008) Primary Sjogren syndrome in Spain: clinical and immunologic expression in 1010 patients. Medicine (Baltimore) 87: 210-9. [Crossref]
- Segal B, Carpenter A, Walk D (2008) Involvement of nervous system pathways in primary Sjögren's syndrome. Rheum Dis Clin North Am 34: 885-906, viii. [Crossref]
2021 Copyright OAT. All rights reserv
- Kahlenberg JM (2011) Neuromyelitis optica spectrum disorder as an initial presentation of primary Sjogren's syndrome. Semin Arthritis Rheum 40: 343-8. [Crossref]
- Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, et al. (2015) International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 85: 177-189. [Crossref]
- Nakamura Y, Nakajima H, Tani H, Hosokawa T, Ishida S et al. (2017) Anti-MOG antibody-positive ADEM following infectious mononucleosis due to a primary EBV infection: a case report. BMC Neurol 17: 76. [Crossef]
- Tselis AC (2014) Epstein-Barr virus infections of the nervous system. Handb Clin Neurol 123: 285-305. [Crossref]
- Harley JB, Chen X, Pujato M, Miller D, Maddox A, et al. (2018) Transcription factors operate across disease loci, with EBNA2 implicated in autoimmunity. Nat Genet 50: 699-707. [Crossref]
- Baldini C, Luciano N, Tarantini G, Pascale R, Sernissi F, et al. (2015) Salivary gland ultrasonography: a highly specific tool for the early diagnosis of primary Sjogren's syndrome. Arthritis Res Ther 17: 146. [Crossref]
- Jarius S, Wildemann B (2013) The history of neuromyelitis optica. J Neuroinflammation 10: 8. [Crossref]
- Cree BA, Goodin DS, Hauser SL (2002) Neuromyelitis optica. Semin Neurol 22: 105-122. [Crossref]
- Matiello M, Jacob A, Wingerchuk DM, Weinshenker BG (2007) Neuromyelitis optica. Curr Opin Neurol 20: 255-260. [Crossref]
- Popescu BF, Lennon VA, Parisi JE, Howe CL, Weigand SD, et al. (2011) Neuromyelitis optica unique area postrema lesions: nausea, vomiting, and pathogenic implications. Neurology 76: 1229-37. [Crossref]
- Koga M, Takahashi T, Kawai M, Fujihara K, Kanda T (2011) A serological analysis of viral and bacterial infections associated with neuromyelitis optica. J Neurol Sci 300: 19-22. [Crossref]
- Sellner J, Hemmer B, Mühlau M (2010) The clinical spectrum and immunobiology of parainfectious neuromyelitis optica (Devic) syndromes. J Autoimmun 34: 371-9. [Crossref]
- Yoshimura S, Isobe N, Matsushita T, Yonekawa T, Masaki K, et al. (2013) Distinct genetic and infectious profiles in Japanese neuromyelitis optica patients according to anti-aquaporin 4 antibody status. J Neurol Neurosurg Psychiatry 84: 29-34. [Crossref]
- Cree BA, Spencer CM, Varrin-Doyer M, Baranzini SE, Zamvil SS (2016) Gut microbiome analysis in neuromyelitis optica reveals overabundance of Clostridium perfringens. Ann Neurol 80: 443-7. [Crossref]
- Varrin-Doyer M, Spencer CM, Schulze-Topphoff U, Nelson PA, Stroud RM, et al. (2012) Aquaporin 4-specific T cells in neuromyelitis optica exhibit a Th17 bias and recognize Clostridium ABC transporter. Ann Neurol 72: 53-64. [Crossref]
- Masuda S, Mori M, Arai K, Uzawa A, Muto M, et al. (2015) Epstein-Barr virus persistence and reactivation in neuromyelitis optica. J Neurol Neurosurg Psychiatry 86: 1137-42. [Crossref]
- Jayarangaiah A, Sehgal R, Epperla N (2014) Sjogren's syndrome and neuromyelitis optica spectrum disorders (NMOSD)--a case report and review of literature. BMC Neurol 14: 200. [Crossref]
- Klutts JS, Ford BA, Perez NR, Gronowski AM (2009) Evidence-based approach for interpretation of Epstein-Barr virus serological patterns. J Clin Microbiol 47: 3204-3210. [Crossref]
- Jacobs BC, Rothbarth PH, van der Meché FG, Herbrink P, Schmitz PI, et al. (1998) The spectrum of antecedent infections in Guillain-Barré syndrome: a case-control study. Neurology 51: 1110-1115. [Crossref]
- Tselis A, Duman R, Storch GA, Lisak RP (1997) Epstein-Barr virus encephalomyelitis diagnosed by polymerase chain reaction: detection of the genome in the CSF. Neurology 48: 1351-5. [Crossref]
- Cohen JI (2000) Epstein-Barr virus infection. N Engl J Med 343: 481-492. [Crossref]
- Wu DY, Kalpana GV, Goff SP, Schubach WH (1996) Epstein-Barr virus nuclear protein 2 (EBNA2) binds to a component of the human SNF-SWI complex, hSNF5/Ini1. J Virol 70: 6020-6028. [Crossref]
- Poole BD, Scofield RH, Harley JB, James JA (2006) Epstein-Barr virus and molecular mimicry in systemic lupus erythematosus. Autoimmunity 39: 63-70. [Crossref]
- Poole BD, Gross T, Maier S, Harley JB, James JA (2008) Lupus-like autoantibody development in rabbits and mice after immunization with EBNA-1 fragments. J Autoimmun 31: 362-371. [Crossref]
- Harley JB, Zoller EE (2014) Editorial: What caused all these troubles, anyway? Epstein-Barr virus in Sjogren's syndrome reevaluated. Arthritis Rheumatol 66: 2328-30. [Crossref]
- Kinoshita M, Nakatsuji Y, Kimura T, Moriya M, Takata K, et al. (2009) Neuromyelitis optica: Passive transfer to rats by human immunoglobulin. Biochem Biophys Res Commun 386: 623-7. [Crossref]
- Lassmann H, Stemberger H, Kitz K, Wisniewski HM (1983) In vivo demyelinating activity of sera from animals with chronic experimental allergic encephalomyelitis. Antibody nature of the demyelinating factor and the role of complement. J Neurol Sci 59: 123-37. [Crossref]
- Saadoun S, Papadopoulos MC, Davies DC, Krishna S, Bell BA (2002) Aquaporin-4 expression is increased in oedematous human brain tumours. J Neurol Neurosurg Psychiatry 72: 262-265. [Crossref]
- Wingerchuk DM (2004) Neuromyelitis optica: current concepts. Front Biosci 9: 834-840. [Crossref]
- Wandinger KP, Stangel M, Witte T, Venables P, Charles P, et al. (2010) Autoantibodies against aquaporin-4 in patients with neuropsychiatric systemic lupus erythematosus and primary Sjogren's syndrome. Arthritis Rheum 62: 1198-200. [Crossref]
- Mariette X, Criswell LA (2018) Primary Sjögren's Syndrome. N Engl J Med 378: 931-939. [Crossref]
- Nocturne G, Mariette X (2013) Advances in understanding the pathogenesis of primary Sjögren's syndrome. Nat Rev Rheumatol 9: 544-556. [Crossref]
- Nocturne G, Mariette X (2018) B cells in the pathogenesis of primary Sjögren syndrome. Nat Rev Rheumatol 14: 133-145. [Crossref]
- Oji S, Nicolussi EM, Kaufmann N, Zeka B, Schanda K, et al. (2016) Experimental Neuromyelitis Optica Induces a Type I Interferon Signature in the Spinal Cord. PLoS One 11: e0151244. [Crossref]
- Wang H, Wang K, Zhong X, Qiu W, Dai Y, et al., Cerebrospinal fluid BAFF and APRIL levels in neuromyelitis optica and multiple sclerosis patients during relapse. J Clin Immunol 32: 1007-11. [Crossref]
- Wingerchuk DM, Weinshenker BG (2012) The emerging relationship between neuromyelitis optica and systemic rheumatologic autoimmune disease. Mult Scler 18: 5-10. [Crossref]
- Birnbaum J, Atri NM, Baer AN, Cimbro R, Montagne J, et al. (2017) Relationship Between Neuromyelitis Optica Spectrum Disorder and Sjögren's Syndrome: Central Nervous System Extraglandular Disease or Unrelated, Co-Occurring Autoimmunity? Arthritis Care Res (Hoboken) 69: 1069-1075. [Crossref]
- Gilden, DH, et al. (2007) Herpesvirus infections of the nervous system. Nat Clin Pract Neurol 3: 82-94.
- Martelius T, Lappalainen M, Palomäki M, Anttila VJ (2011) Clinical characteristics of patients with Epstein Barr virus in cerebrospinal fluid. BMC Infect Dis 11: 281. [Crossref]