Abstract
Diabetes mellitus remains with an ever-increasing prevalence, indefectibly associated to progressive and irreversible complications. Diabetic lower extremities ulcerations contribute to amputations, disability, and mortality. Ulcers result from a wound healing failure characterized by proliferative arrest, apoptosis, and senescence of granulation tissue-producing cells. Diabetic wounds are also distinguished by an inflamed, toxic, and degradative environment, acting as deterrents for local growth factors availability and receptors’ physiology. The emergence of growth factors caused expectation as biological modifiers for wounds repair arrest. The clinical introduction of growth factors was precocious when critical pieces of chronicity pathophysiology and growth factors pharmacology remained elusive. Mounting observations indicated that topical administration of these agents failed by the effect of local proteolysis, narrow bioavailability window, inadequate local kinetics/diffusion, and a regenerating polymicrobial biofilm. As an alternative to circumvent these pharmacodynamics obstacles as to preserve EGF biological capabilities, we developed a series of experiments which provided the rationale and fundamentals for an intra-ulcer infiltrative delivery route. The clinical development program has included from a proof-of-concept to post-marketing studies in poor-prognosis ischemic, neuropathic and neuroischemic wounds. Along 18 years of clinical progress more than 259 000 patients were treated. As demonstrated by pharmacovigilance studies, aside from the success in the primary healing, the infiltrated EGF accounted for a reduction of amputation risks, negligible rates of annual recurrence, and prolonging survival of the healed patients. This pharmacological intervention is added to conventional treatments and surgical procedures. Infiltrated EGF has proved to reverse wound cells arrest being efficacious and safe for long terms of follow up.
Key-words
diabetic complications, diabetic foot ulcers, EGF, amputation
Brief reflections on diabetes and the wound healing failure
Since the seminal contribution of Banting and Best diabetes treatment was revolutionized. Hereafter, insulin therapy eliminated ketoacidosis as a principal cause of death among diabetics who enjoyed a longer lifespan. However, traditional insulin therapy combined with emerging novel approaches did not translate into a significant reduction of major complications that nowadays lead to morbidity and mortality [1]. Type 2 Diabetes Mellitus (T2-DM) is a heterogeneous and complex process comprising multiple pathogenic factors [2] and multi-organs complications’ that remain as a challenge for scientists and clinicians. T2-DM has progressively expanded as a pandemic condition accounting for 90% to 95% of all the diabetic population [3,4].
Diabetic foot ulceration (DFU) is one of the most frightened diabetic complications, leading to amputation-disability, social exclusion and early mortality [5]. The lifetime incidence of foot ulcers has been estimated to reach up to 34% of subjects with diabetes whereas diabetes-related lower extremity complications affect about 159 million people worldwide [6,7]. Accordingly, diabetic population still contributes to 80% of all non-traumatic lower extremities amputations around the world [8].
An illuminating review by Armstrong and coworkers prompted for the first time to distinguish the ulcer healing process not as the mere route for limb salvage, but as the foremost alternative to prevent early mortality. The 5-year relative mortality rate after limb amputation in diabetics rises to 68% only preceded by lung cancer [9].
The molecular pillars governing diabetic wounds chronification appear to reside on three major events: precocious senescence, proliferative refractoriness, and apoptosis [10]. Notoriously, these traits seem to be related to a threshold of senescent cells which may be secondary to a substantial deficit of growth factors (GFs) [11]. An alluring hypothesis is that the role of GFs within the diabetic wound microenvironment may even include the prevention and/or the reversion of a cellular senescence program [12].
The diabetic wound biochemical milieu and local growth factors availability
Conceptually, diabetic wounds exhibit a singular and complex networking of inflammatory cytokines, local proteases, cytotoxic reactive oxygen and nitrogen species, and a polymicrobial biofilm. All together contribute to the stagnant phenotype. Diabetes predisposes to inflammation which is more a condition than a transient reaction while impairment in its resolution also perpetuates the inflammatory induration [13]. Converging evidences suggest that fibroblasts, endothelial cells, and keratinocytes are significantly impinged by the “hard-to-forget” hyperglycemic stress, which eventually dictates a particular epigenome/transcriptome, that somehow recreates a senescent landscape by the replicative refractoriness and the pro-inflammatory senescence-associated secretory phenotype (SASP) [11]. In this regard, a continued activation of the nuclear factor-kappaB (NF-κB)-p65 and downstream effectors play a definitive role in the inflammatory response. Germane for the cytotoxicity within the diabetic wounds environment is the disproportionate generation of reactive oxygen species (ROS), and the accumulation of advanced glycoxidation-end products (AGEs) which warrant a relentless state of multicellular cytotoxic aggression. The NF-κB-related “response-to-injury” is also mechanistically linked to the orchestration of a procatabolic/prodegradative phenotype which appear largely mediated by the substantial upregulation of matrix metalloproteinases (MMPs) [14]. The crosstalk of these molecular aspects is summarized in Figure 1.
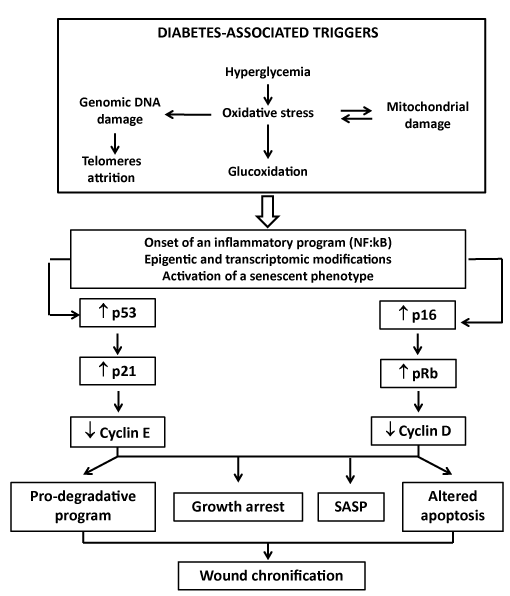
Figure 1. Putative mechanism linking wound chronification in diabetes
The high-level oxidative stress induced by hyperglycemia and other molecular disturbances associated to diabetes have the potential to foster premature senescence. Diabetic microenvironment is therefore permissive to senescence. Senescence is therefore a terminal mitotic fate in which cells lose the capability to proliferate in response to growth factors. We deem that diabetes-related molecular disorders can induce both telomeres attrition and replicative senescence by direct DNA damage – i.e., reactive oxygen species. Mitochondrial DNA is also targeted by oxygen which progressively perpetuates both mitochondrial damage and free radicals spillover. Thereafter it seems that epigenetic forces upregulate p53-p21 and/or p16Ink4a-pRb overexpression which implement the senescence program. Cells become arrested, pro-apoptogenic, and display and abnormal morphology. Beyond this, the pro-inflammatory response known as senescence-associated secretory phenotype is also instrumented. This SASP phenotype that includes the secretion of pro-inflammatory cytokines and proteases which contribute to poor granulation tissue growth.
Myriad of evidences document the negative impact of an uncontrolled release and activation of MMPs for the local bioavailability and bioactivity of the GFs and their receptors [15-17]. Since the senescent phenotype depends on the downregulation of proliferation regulatory-positive genes (i.e., c-myc, c-fos, CDC2, and cyclins) ordinarily upregulated by GFs [18]; the early idea that GFs could reverse this arrest within the repair process appeared thoughtfully justified [19,20].
Back along the history, it is likely that the first approach to recombinant human GFs topical administration dates back to almost 40 years ago which fostered an alternative for torpid healing wounds. Nonetheless, the initial expectations with these “magic bullets” vanished away in about a 10-year period. To our understanding, two main factors quenched such excitement: (a)-the inputs from basic science that associated GFs to malignant cells promotion and progression [21] and (b)-the setback that stemmed from clinical trials in which the topical administration of epidermal growth factor (EGF) failed to enhance the healing process of chronic wounds [22] and, unexpectedly, of acute, controlled, and purposely induced wounds in healthy volunteers [23].
These disappointments warned about the need for additional research in GFs physiology and pharmacology as in the understanding of the wound milieu biochemistry [24]. Thereafter, numerous investigations plagued the literature supporting the need to modify wound local factors to ensure an appropriate GFs pharmacodynamics response. Others claimed the need of GFs combinations as the optimal tool to restore the healing trajectory in chronic wounds [25]. The debut of GFs in the clinical arena seemed premature in relation to the basic science knowledge supporting its molecular pharmacology in the context of chronic wounds [26]. This statement is likely based on the classic observation by Davidson's group in 1985 in which they demonstrated that EGF wound healing enhancement was promoted under a prolonged, sustained, slow release system [27]. This innovation assimilated the classic concept that EGF required a constant exposure to its receptor. It means that the EGF-related wound healing properties could happen if the receptors are exposed for at least 8 to 12 hours when these are steadily occupied and a mitogenic signal is eventually transduced [27,28].
Conclusively diabetic chronic wounds microenvironment is hostile for local GFs stability, chemical integrity, bioavailability, and ultimately to their physiological role as major drivers of the healing process. Within this environment the receptors steady expression and signaling ability are impaired [29,30]. These thoughts lead us to support the tenet that diabetic wound cells are embedded in a GFs-negative balance which would presuppose reduced cytoprotective reserves and proliferative capabilities, and an obvious inclination to a precocious senescent phenotype [31,32].
Biological fundamentals for EGF administration to diabetic wounds
EGF is perhaps the most widely studied growth factor. Since the 60’s interpretation that its exogenous administration timely reprogrammed chronologically-specified biological events, this polypeptide has been used to repair multiple forms of wounds in both peripheral and internal tissues and organs [33]. Unquestionably, EGF is endowed with the sufficient biological competence for potentially reversing wounds’ chronicity [34]. The first evidence suggesting a role for EGF in tissue repair derived from Stanley Cohen in the early 1960’s (Prof. Stanley Cohen–unpublished observations, personal communication) in rabbits with corneal burns that received eye drops based on natural EGF. Two major intracellular pathways are activated by the EGF-Receptor that invokes the two most important biological actions of EGF in tissue repair: cell proliferation and cell survival. It means that EGF-Receptor agonistic stimulation may shift toward mitogenic and pro-survival programs that concertedly translate into an increase in cell population number and stress tolerance. For the mitogenic response, the RAS–RAF–MEK–MAPK pathway that controls cell-cycle progression from the G1 phase to the S phase is important, whereas the PI3K–Akt pathway activates a cascade of anti-apoptotic and cytoprotective mediators thus rescuing injured cells [33].
EGF receptor is expressed in most human cell types including those which play critical roles for wound repair such as fibroblasts, endothelial cells and keratinocytes (undifferentiated, marginal, leading edge, hair follicles, sweat ducts and sebaceous glands). Aside from the classic mitogenic, motogenic and cytoprotective actions for healing events [35-37], EGF biological spectrum involve a potential senolytic effect. Experimental evidences support the hypothesis that EGF aborts cellular senescence programs in response to certain forms of DNA damage [38]. Likewise, EGF showed to activate the expression of human telomerase reverse transcriptase in cultured cells, via Ets-2, a cancer-specific transcription factor that appears to depend on EGFR-mediated Erk and Akt activation [39]. Experimental studies suggest that EGF is able to enhance cell survival and tissue replenishment before otherwise lethal scenarios, by controlling oxidative stress and mitigating cellular senescence [40]. Chronic wounds irrespective to their etiology are a rich source of pro-oxidant metabolites with a negative local and systemic impact including cellular senescence [41,42]. In the context of free radicals over production we showed that diabetic ulcerated subjects behave as a unique pathological group since they exhibit an exacerbation of the oxidative stress arm along with a concomitant deterioration of the antioxidant reserves, as compared to non-ulcerated diabetic individuals. EGF administration for 3-4 weeks contributed to restore circulating levels of numerous redox status markers up to values close to those of non-ulcerated diabetic patients and non-diabetic subjects. Hence, we consider that EGF is a key factor in indirectly attenuating premature senescence, apoptosis and proliferative arrest. Furthermore, EGF intra-ulcer administration also tended to restore the systemic balance between MMP-9/TIMP-1, suggesting the recovery between pro-degradative and pro-synthetic forces, [43] which may denote an attenuation of the SASP-associated phenotype [44]. Finally, a 2015 groundbreaking study revealed for the first time that the EGF exerts a potent anti-senescence activity in certain stem and differentiated cells. The study showed that cell cultures depleted of EGF orchestrated a senescencent phenotype with enlarged morphology, elevated SA-β-gal activity, decreased proliferation, reduced Rb phosphorylation and elevated p21 expression. Therefore, these results advise that cultured cells may depend on EGF as a mechanism to escape from senescence and ensure proliferation, thus placing EGF as a central driver in preserving mitogenic competence and suppressing senescence [45,46].
Diabetes is associated to a particular deficit of circulating [47] and salivary levels of EGF [48]. Of major relevance is the evidence that EGF and other GFs that are essential for cell proliferation are missing or reduced in chronic wounds including diabetic foot ulcers [49-51]. The low concentrations of several key GFs and the ensued reduced mitogenic activity incited to theorize that exogenous GFs therapy to chronic wounds would act as a sort of “replacement therapy”. Nevertheless a major subsisting hurdle is how to target the GFs message in a timely manner to a sensitive/responsive cellular stratum within the chronic wound bed [17].
The EGF replacement therapy via intralesional infiltration
During the early 90’s we accrued the experience of EGF local infiltration into rats' hind limbs denervated upon sciatic nerve full-thickness transection. In addition to significantly assisting in neurological restoration, the treatment enhanced limbs peripheral soft tissues survival by delaying or preventing the onset of plantar ulcers and toes necrosis [52]. These experiments rendered an important lesson: locally injected EGF could stimulate the survival and repair of cutaneous and adjacent soft tissues in a context of circulatory neurogenic deterioration. We subsequently showed in a variety of pathological models that single or repeated EGF systemic or local injections exerted “clear-cut” cytoprotective and proliferative responses supporting the intrinsic ability of EGF at supraphysiological concentrations to unleash biological events required for tissue repair [53,54]. Notwithstanding from the above message, EGF intralesional infiltration associated to wound debridement, appears to overcome the enlisted obstacles confronted upon its topical application:
Local GFs degradation
A relevant requisite for topically applied GF pharmacodynamics is local bioavailability and pharmacokinetic. Different laboratories demonstrated the degradative action of locally secreted proteases in chronic wounds against GFs and their cellular receptors [55,56]. Moreover, proteolytic effect was also observed using a synthetic fluorescent substrate with an EGF-like molecule sequence when incubated with sterile exudate, obtained from full-thickness controlled acute wounds in Yorkshire pigs under laboratory conditions [57]. The understanding of the pathogenic significance of the diabetic wounds biofilm rendered explanation on why topically administered GFs may have failed in healing some of these lesions [58].
Limited diffusion of topically administered GFs
One of the greatest advances in GFs investigation was offered by Cross and Roberts in 1999. They demonstrated that EGF only penetrated slightly into the upper granulating layers of the wound site, showing an exponential decline in solute concentration with tissue depth in the wound and underlying area. Diffusion limitations into the wound bed-deep layers, appeared to be critical irrespective to the phase of the healing process. This limited absorption kinetic also contributed to explain why topically administered GFs failed in the clinical arena [59].
EGF receptor expression is limited on the wound surface cells
Immunohistochemistry studies (Figure 2) with specific antibodies showed that in neuropathic granulation tissue the EGF receptor is not detected on the wound surface cells layer but in deeper strata of fibroblasts. This is particularly relevant for the catalytic domain of tyrosine-1197 residue, which is involved in cell survival, motility, and proliferation. On the contrary, the wound surface cells exhibit far more abundant expression of prohibitin, a cell cycle arrest protein. These findings suggest that topical administration pharmacodynamics is questionable by two major limiting factors: limited diffusion to deeper wound layers, and the lack of EGF-Receptor on the wound surface cells [60].
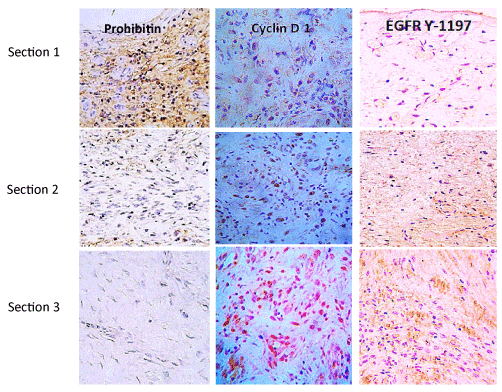
Figure 2. Cell proliferation cycle effectors in granulation tissue of neuropathic diabetic foot ulcers
Three sections (≈2-3mm) length are immunohistochemically distinguished in biopsies materials by immunolabeling with antibodies against prohibitin as a cell cycle negative regulator; Cyclin D1 as a master switch controlling G1–S transition in response to growth factor stimulation. The active EGF-receptor phosphorylated in tyrosine 1197 as a critical substrate for multiple physiological functions of the receptor is shown. Prohibitin is far more abundant on section 1 corresponding to the most superficial stratum of the wound. Conversely, Cyclin D1 and EGFR Y-1197 appear marginally labeled. It is noticeable that proliferative Cyclin D1 and the phosphorylated form of the EGFR are by large abundantly detected in section 3 – deepest wound layer where no prohibitin is detected. From Jorge Berlanga-Acosta et al., Biomed Res International. Volume 2017, Article ID 2923759. https://doi.org/10.1155/2017/2923759 (with permission).
Limited interaction with cell receptors
125I-EGF formulated in a semisolid vehicle was rapidly cleared from the application site, probably by protease-driven cleavage and receptor-mediated endocytosis. Mean residence time values suggested that over 60% of the amount administered could have disappeared as early as two hours after administration. These evidences reinforce previous paradigmatic findings which provided the elementary principles for an EGF-mediated cellular mitogenic response [27,29,61,62].
Evidences supporting the intralesional infiltrative administration
In a brief manner, injecting EGF down into the base and contours of the wounds, including the dermo-epidermal junction, appears to (1) reduce its local degradation, (2) jump over the diffusion limiting barriers, and (3) ensure its bioavailability for a prolonged interaction with the receptor, in a deep fibroblasts-populated stratum along the longitudinal axis of granulation tissue. A time-point immunoelectron microscopy kinetic study addressed to characterize the intracellular trafficking of the EGFR in ulcers-collected fibroblasts, showed that locally infiltrated EGF into Wagner’s 3 and 4 neuropathic ulcers resulted in (a) dramatic increase of the EGF-Receptor expression 15 minutes after the EGF infiltration as compared to “time zero” (T0: prior to the intervention); this evidence suggests the induction of the receptor by the high-affinity ligand EGF; (b) immediate endocytosis of the EGFR; (c) translocation and biodistribution to different cytoplasmic organelles from 15 minutes to 24 hours after the infiltration; (d) nuclear translocation of the receptor and its binding to DNA which appeared to last from minute 45 to 24 hours after the treatment; (e) a concomitant activation of the proliferating cell nuclear antigen, PCNA (a cell cycle promoting protein) gene transcription, since a burst of this protein was detected following EGF intervention which appeared evident even at hour 24th after the treatment; (f) a significant and intriguing accumulation of the receptor in mitochondria which lasted for 24 hours after the infiltration; (g) significant accumulation of the receptor bound to collagen fibers within the extracellular matrix [63]. It is noteworthy for the above described findings that classic studies support the notion that EGF makes association complex with extracellular matrix proteins, thus enhancing cell proliferation and migration via a sort of natural slow deliver system [64]. All together these data suggest that EGF delivery via infiltration stimulates the EGF receptor in a manner that meets the biological requisites for cell proliferation.
Clinical authentication of EGF intralesional infiltrative administration
Increasing and convergent experimental in vivo evidences supported the hypothesis that EGF parenteral administration elicited broader tissue’s healing and cytoprotective responses relative to its topical administration. Accordingly, during 2001-2002 we initiated a pilot study that included 29 poor-prognostic ischemic and neuropathic patients/wounds (Wagner stages 3 and 4 ulcers and non-healing amputation residual bases) that received intralesional EGF thrice a week after sharp debridement and standard medical interventions. The main idea was to stimulate granulation tissue growth, especially in areas where bone and tendons were exposed. By infiltrating around the dermo-epidermal junction wound contraction and reepithelialization appeared to be resumed. From these 29 individuals, 17 re-epithelialized their lesions in about 5 to 8 weeks and were spared from amputation. Wound bed tissue biopsies collected before and after 3 to 4 weeks of treatment showed the pharmacodynamics of locally infiltrated EGF by promoting the secretion of extracellular matrix ingredients as the local homing of granulation productive cells. For the particular case of ischemic wounds, a remarkable angiogenic response was observed along with attenuation in apoptotic bodies and inflammatory infiltration [65]. This seminal study ultimately validated the concept of local infiltration versus topical administration. For the severity of wounds treated in these patients the topical route would be useless (Figure 3 offers an idea of reserved prognosis-treated wounds). This study also shed hopes on the local infiltration as a creditable to reduce recurrences. The efficacy demonstrated in these patients/wounds paved the way for a progressive clinical development which culminated in a nationwide, double-blind, placebo-controlled phase III clinical trial, and the insertion of this type of intervention within the integral program for diabetic foot ulcers care [66]. A nationwide pharmacovigilance study including more than 2000 patients, confirmed the clinical usefulness of this delivery route to trigger and sustain the healing process as demonstrated during the clinical trials. In terms of figures, it was shown that infiltrated EGF elicited 75% full granulation response, 61% of wound closure, and 71% reduction of amputation-relative risk, as well as positive benefit-risk balance. Of paramount clinical and social relevance is that recurrences were reported as an exceptional event (approximately 5%) upon a 12-month follow-up period [67,68] which had been anticipated since the proof-of-concept trial. Irrespective to the primary healing efficacy for different therapeutic modalities, recurrence rates remain disproportionately high. Recent investigations disclose that roughly 40% of patients have a recurrence within 1 year after ulcer healing, almost 60% within 3 years, and 65% within 5 years [69]. Other international groups who have introduced EGF intralesional infiltration in the daily practice converge to report that EGF infiltration triggers an exceptional healing response with low amputation rates [70-72].

Figure 3. Clinical evidences of the effect of EGF in both neuropathic and neurosichemic wounds
On the safety of intralesional infiltrated EGF
Human recombinant EGF was subjected to clinical assessment as early as 1989 within the field of tissue repair [73]. EGF, like most growth factors stimulates cell proliferation, migration, extracellular matrix synthesis, cell survival, phenotypic reprogramming, and local angiogenesis during wound repair. The fact that early basic researches demonstrated that these events are also induced by GFs upon the malignant transformation process [74] ignited concern, particularly when some GFs were identified in the conditioned culture medium of different cancer cell lines [75,76] and when homology was observed among some GFs receptors-coding genes and viral oncogenes [77]. In most human epithelial tumors EGF receptor is amplified or overexpressed and its signaling system deregulated with a “gain-of-function” profile [78,79]. In parallel to EGF investigation in tumor biology its clinical use expanded, including systemic repeated administrations with successful outcomes for the patients [80]. EGF has been orally [81], systemically [82,83], transrectally [83], and topically administered including to burn wounds which are a well-known substrate for squamous carcinomas [84]. To the best of our knowledge the first systemic intervention with EGF was conducted in 1975 in patients affected by Zollinger-Ellison disease [81] whereas in the most recent clinical use EGF is locally infiltrated into recalcitrant diabetic foot wounds for a period of 5 to 8 weeks [31,65,84].
Compelling studies demonstrate that EGF overexposure even in pharmacological concentrations does not initiate malignant transformation, and that its “promoting” role is controversial. By the contrary EGF may induce apoptosis in some experimental epithelial tumors [21,85,86]. In contrast to platelet-derived growth factor (PDGF), EGF is not an oncogene-derived product and does not appear to be endowed with the ability to irreversibly transform cells [85,86]. These notions are supported by the evidences derived from different transgenic models which definitively support that innate overexposure to EGF does not provoke tumors in the animals with the exception of a liver-targeted construct that provoked hepatocarcinogenesis.
Along these almost 30 years of EGF clinical use, no evidence has ever been identified to indicate that any of these therapeutic and repeated interventions with EGF at pharmacological concentrations, and by any of the above mentioned routes is associated to carcinogenesis. Some of these studies have included follow-up periods of 6-12 months up to 4 years; the later representing an appropriate biological window for EGF-mediated long-term adverse reactions [67,68]. Likewise, prior to EGF or another peptide growth factor administration, a careful patient selection should be performed based in the personal and family background, and on the basis of the benefit/risk balance. In fact, this rule is ordinarily applied in clinical practice for many approved drugs.
Concluding remarks
Skin cells cannot hide out from the diabetic biochemical derangements and an imprinting is left in fibroblasts, vascular cells and keratinocytes. Not less significant is the damage induced by hyperglycemia and its associate by-products on the mesenchymal-derived stem cells. Diabetic subjects cutaneous cells exhibit an abnormal behavior and a short replicative life span mirroring a senescencent phenotype even under placed under ideal in vitro culture conditions. This replicative refractoriness that remains in the cellular memory is one of the major grounds for wound chronification. Diabetic wound is conceptually inflamed, catabolic, and an additional source of circulating pro-inflammatory cytokines and oxidative radicals, establishing a self-perpetuating loop. More recently, the picture has been further completed with the recent identification and characterization of dozen of miRNA released from the diabetic wound realm. The microenvironment of these wounds has proved to be hostile for growth factors and their receptors in terms of physicochemical integrity and physiology. This growth factors deficit has been associated to the early onset of cellular senescence.
The discovery of growth factors opened a new era in wound healing biology and planted hopes for the treatment of recalcitrant, hard-to-heal wounds. Despite the initial promise for optimal wound management and despite long years of research efforts, growth factors have not conquered definitive praise in the clinical armamentarium. For the particular case of EGF, it was the first in line used by topical administration in acute and chronic wounds. In the aftermaths, the results were either controversial or neutral in both basic and clinical studies. Despite these outcomes, there is no question about its intrinsic biological potency in mitogenic commitment for most epithelial and mesenchymal-derived cells. Critical is also its role during embryonic development – thus, the acquisition of a differentiated phenotype is on the list of its biological attributes. The experiments based on the local infiltration of EGF in rats injured hind limbs were seminal and graphic in showing the big gap existing between the topical and the infiltrative delivery routes in terms of a clear-cut tissue healing response. These experiments translated up today in more than two hundred thousand diabetic patients successfully treated in terms of healing, low recurrence rates, reductions of amputations, and prolonged survival.
This intralesional infiltrative procedure seems to ensure EGF bioactivity for prolonged times, thus reinforcing the concept that the spatio-temporal control of EGF availability and local residence time, are relevant requisites for their clinical success as a regenerative alternative. Conclusively, we deem that growth factors are biologically “empowered” molecules with broad pharmacological potential in regenerative medicine; we just need to learn more from them and to know how, when, and where to deliver their messages to the cells.
References
- Brownlee M (2005) The pathobiology of diabetic complications. A unifying mechanism. Diabetes 54: 1615-1625.
- Kahn SE, Cooper ME, Del Prato S (2014) Pathophysiology and treatment of type 2 diabetes: perspectives on the past, present, and future. Lancet 383: 1068-1083. [Crossref]
- Shaw JE, Sicree RA, Zimmet PZ (2010) Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract 87: 4-14. [Crossref]
- International Diabetes Federation (2017) IDF Diabetes Atlas. London.
- Costa RHR, Cardoso NA, Procopio RL, Navarro TP, Dardik A, et al. (2017) Diabetic foot ulcer carries high amputation and mortality rates, particularly in the presence of advanced age, peripheral artery disease and anemia. Diabetes Metab Syndr 11: S583-S587.
- Game FL, Apelqvist J, Attinger C, Hartemann A, Hinchliffe RJ, et al. (2016) Effectiveness of interventions to enhance healing of chronic ulcers of the foot in diabetes: a systematic review. Diabetes Metab Res Rev 32: 154-168.
- Lazzarini PA, Pacella RE, Armstrong DG, van Netten JJ (2018) Diabetes-related lower- extremity complications are a leading cause of the global burden of disability. Diabet Med 35: 1297-1299.
- Singh N, Armstrong DG, Lipsky BA (2005) Preventing foot ulcers in patients with diabetes. JAMA 293: 217-228. [Crossref]
- Armstrong DG, Wrobel J, Robbins JM (2007) Guest editorial: Are diabetes-related wounds and amputations worse than cancer? Int Wound J 4: 286-287. [Crossref]
- Berlanga-Acosta J, Schultz GS, López-Mola E, Guillen-Nieto G, García-Siverio M, et al. (2013) Glucose toxic effects on granulation tissue productive cells: The diabetics' impaired healing. Biomed Res Int.
- Telgenhoff D, Shroot B (2005) Cellular senescence mechanisms in chronic wound healing. Cell Death Differ 12: 695-698.
- Berlanga-Acosta J, Armstrong DG, Schultz GS, Herrera-Martinez L (2014) Chronic wounds with emphasis in diabetic foot ulcers. Biomed Res Int 2014: 890352. [Crossref]
- Acosta JB, del Barco DG, Vera DC, Savigne W, Lopez-Saura P, et al. (2008) The pro-inflammatory environment in recalcitrant diabetic foot wounds. Int Wound J 5: 530-539. [Crossref]
- Lui Y, Min D, Bolton T, Nube V, Twigg SM, et al. (2009) Increased matrix metalloproteinase-9 predicts poor wound healing in diabetic foot ulcers. Diabetes Care 32: 117-119.
- Serra MB, Barroso WA, da Silva NN, Silva SDN, et al. (2017) From Inflammation to Current and Alternative Therapies Involved in Wound Healing. Int J Inflam 2017: 3406215. [Crossref]
- Caley MP, Martins VL, O'Toole EA (2015) Metalloproteinases and Wound Healing. Adv Wound Care (New Rochelle) 4: 225-234. [Crossref]
- Krishnaswamy VR, Mintz D, Sagi I (2017) Matrix metalloproteinases: the sculptors of chronic cutaneous wounds. Biochim Biophys Acta Mol Cell Res 1864: 2220-2227.
- Esposito F, Ammendola R, Faraonio R, Russo T, Cimino F (2004) Redox control of signal transduction, gene expression and cellular senescence. Neurochem Res 29: 617-628. [Crossref]
- Papanas N, Maltezos E (2008) Becaplermin gel in the treatment of diabetic neuropathic foot ulcers. Clin Interv Aging 3: 233-240. [Crossref]
- Muñoz-Espín D, Serrano M (2014) Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 15: 482-496. [Crossref]
- Berlanga-Acosta J, 2021 Copyright OAT. All rights reserv AG, Martín-Machado J, Guillén-Nieto G (2011) Epidermal growth factor (EGF) and platelet-derived growth factor (PDGF) as tissue healing agents: clarifying concerns about their possible role in malignant transformation and tumor progression. J Carcinogene Mutagene 2: 100-115.
- Berlanga-Acosta J, Fernández-Montequín J, Valdés-Pérez C, Savigne-Gutierrez W, Mendoza-Marí Y, et al. (2017) Diabetic foot ulcers and epidermal growth factor: revisiting the local delivery route for a successful outcome. Biomed Res Int 29: 237-250.
- Cohen K, Crossland MC, Garret A, Diegelmann RF (1995) Topical application of epidermal growth factor onto partial-thickness wounds in human volunteers does not enhance reepithelialization. Plast Reconstr Surg 96: 251-254.
- Falanga V (1992) Growth factors and chronic wounds: the need to understand the microenvironment. J Dermatol 19: 667-672. [Crossref]
- Bennett NT, Schultz GS (1993) Growth factors and wound healing: biochemical properties of growth factors and their receptors. Am J Surg 165: 728-737. [Crossref]
- Falanga V (2005) Wound healing and its impairment in the diabetic foot. Lancet 366: 1736-1743. [Crossref]
- Buckley A, Davidson JM, Kamerath CD, Wolt TB, Woodward SC (1985) Sustained release of epidermal growth factor accelerates wound repair. Proc Natl Acad Sci U S A 82: 7340-7344. [Crossref]
- Knauer DJ, Wiley HS, Cunningham DD (1984) Relationship between epidermal growth factor receptor occupancy and mitogenic response. Quantitative analysis using a steady state model system. J Biol Chem 259: 5623-5631. [Crossref]
- Portero-Otín M, Pamplona R, Belmunt MJ, Ruiz MC, Prat J, et al. (2002) Advanced glycation end product precursors impair epidermal growth factor recep tor signaling. Diabetes 51: 1535-1542.
- Barrientos S, Brem H, Stojadinovic O, Tomic-Canic M (2014) Clinical application of growth factors and cytokines in wound healing. Wound Repair Regen 22: 569-578.
- Berlanga-Acosta J, Mendoza-Marí Y, Martínez MD, Valdés-Pérez C, Ojalvo AG, et al. (2013) Clinical application of growth factors and cytokines in wound healing. Int Wound J 10: 232-236.
- Tsourdi E, Barthel A, Rietzsch H, Reichel A, Bornstein SR (2013) Current aspects in the pathophysiology and treatment of chronic wounds in diabetes mellitus. Biomed Res Int 2013: 385641. [Crossref]
- Berlanga-Acosta J, Gavilondo-Cowley J, López-Saura P, González-López T, Castro-Santana MD, et al. (2009) Epidermal growth factor in clinical practice–a review of its biological actions, clinical indications and safety implications. Int Wound J 6: 331-346.
- Tiaka EK, Papanas N, Manolakis AC, Georgiadis GS (2012) Epidermal growth factor in the treatment of diabetic foot ulcers: an update. Perspect Vasc Surg Endovasc Ther 24: 37-44. [Crossref]
- Werner S, Grose R (2003) Regulation of wound healing by growth factors and cytokines. Physiol Rev 83: 835-870. [Crossref]
- Pastore S, Mascia F, Mariani V, Girolomoni G (2008) The epidermal growth factor receptor system in skin repair and inflammation. J Invest Dermatol 128: 1365-1374. [Crossref]
- Bodnar RJ (2013) Epidermal Growth Factor and Epidermal Growth Factor Receptor: The Yin and Yang in the Treatment of Cutaneous Wounds and Cancer. Adv Wound Care (New Rochelle) 2: 24-29. [Crossref]
- Wang M, Morsbach F, Sander D, Georghiu L, Nanda ABC (2011) EGF receptor inhibition radiosensitizers NSCLC cells by inducing senescence in cells sustaining DNA double-strand breaks. Cancer Res 71: 6261-6269.
- Hsu CP, Lee LW, Tang SC, Hsin IL, Lin YW, et al. (2015) Epidermal growth factor activates telomerase activity by direct binding of Ets-2 to hTERT promoter in lung cancer cells. Tumour Biol 36: 5389-5398.
- Hu ZH, Chen CL, Yang JS, Zhou ZL, Song ZM, et al. (2014) PI3K-mediated glioprotective effect of epidermal growth factor under oxidative stress conditions. Int J Ophthalmol 7: 413-420.
- Gordillo GM, Sen CK (2003) Revisiting the essential role of oxygen in wound healing. Am J Surg 186: 259-263. [Crossref]
- Soneja A, Drews M, Malinski T (2005) Role of nitric oxide, nitroxidative and oxidative stress in wound healing. Pharmacol Rep 57:108-19.
- Ojalvo AG, Berlanga-Acosta J, Marí YM, Mayola MF, Pérez CV, et al. (2017) Healing enhancement of diabetic wounds by locally infiltrated epidermal growth factor is associated with systemic oxidative stress reduction. Int Wound J 14: 214-225.
- Demaria M, Desprez PY, Campisi J, Velarde MC (2015) Cell Autonomous and Non-Autonomous Effects of Senescent Cells in the Skin. J Invest Dermatol 135: 1722-1726. [Crossref]
- Alexander PB, Yuan P, Yang L, Sun T, Chen R, et al. (2015) EGF promotes mammalian cell growth by suppressing cellular senescence. Cell Res 25: 135-138.
- Ai G, Shao X, Meng M, Song L, Qiu J, et al. (2017) Epidermal growth factor promotes proliferation and maintains multipotency of continuous cultured adipose stem cells via activating STAT signal pathway in vitro. Medicine 96: e7607.
- Astaneie F, Afshari M, Mojtahedi A, Mostafalou S, Zamani MJ, et al. (2005) Total antioxidant capacity and levels of epidermal growth factor and nitric oxide in blood and saliva of insulin-dependent diabetic patients. Arch Med Res 36: 376-381. [Crossref]
- Oxford GE, Tayari L, Barfoot MD, Peck AB, Tanaka Y, et al. (2000) Salivary EGF levels reduced in diabetic patients. J Diabetes Complications 14: 140-145.
- Galkowska H, Wojewodzka U, Olszewski WL (2006) Chemokines, cytokines, and growth factors in keratinocytes and dermal endothelial cells in the margin of chronic diabetic foot ulcers. Wound Repair Regen 14: 558-565.
- Lobmann R, Schultz GS, Lehnert H (2005) Proteases and the diabetic foot syndrome: mechanisms and therapeutic implications. Diabetes Care 28: 461-471.
- Dinh T, Tecilazich F, Kafanas A, Doupis J, Gnardellis C, et al. (2012) Mechanisms involved in the development and healing of diabetic foot ulceration. Diabetes 61: 2937-2947. [Crossref]
- Prats P, Castañeda O, Berlanga-Acosta J, Falcón V, Rodríguez V, et al. (1998) El factor de crecimiento epidérmico en lesiones del sistema nervioso periférico. Rev Mex Cienc Farm 29: 17-23.
- Berlanga-Acosta J, Caballero E, Prats P, López-Saura P, Playford RJ (1999) The role of the epidermal growth factor in cell and tissue protection. Medicina Clínica 113: 222-229.
- Berlanga-Acosta J, Mella-Lizama C (1998) Some physiological considerations of epidermal growth factor in relation to its pharmacological applications. Biotecnología Aplicada 15: 141-148.
- Gibson D, Schultz GS (2009) Chronic wound diagnostic for matrix metalloproteinase. Wound Healing Southern Africa 2: 68-70.
- Mathew S, Ravisanker V, Potluri T, Suchithra T (2015) Delayed diabetic wound healing: A focus on bacterial proteases in chronic wound and foot ulcer. Int J Curr Res Rev 7: 36-43.
- Berlanga-Acosta J, Lodos J, Reyes O, Infante JF, Caballero E, et al. (1998) Epidermal growth factor stimulated re-epithelialization in pigs: The possible role of acute wound proteases. Biotecnología Aplicada 15: 83-87.
- Dowd SE, Wolcott RD, Sun Y, McKeehan T, Smith E, et al. (2008) Polymicrobial nature of chronic diabetic foot ulcer biofilm infections determined using bacterial tag encoded FLX amplicon pyrosequencing (bTEFAP). PLoS One 3: e3326.
- Cross SE, Roberts MS (1999) Defining a model to predict the distribution of topically applied growth factors and other solutes in excisional full-thickness wounds. J Invest Dermatol 112: 36-41. [Crossref]
- Berlanga-Acosta J (2011) Diabetic lower extremity wounds: the rationale for growth factors-based infiltration treatment. Int Wound J 8: 612-620.
- Taylor JM, Cohen S, Mitchell WM (1970) Epidermal growth factor: high and low molecular weight forms. Proc Natl Acad Sci U S A 67: 164-171. [Crossref]
- Wiley HS, Cunninghan DD (1981) A steady state model for analyzing the cellular binding, internalization and degradation of polypeptide ligands. Cell 25:433-440.
- Cama VF, Mayola MF, Marí YM, Rivero NA, Ojalvo AG, et al (2016). Epidermal growth factor based therapy promotes intracellular trafficking and accumulation of its receptor in the nucleus of fibroblasts from diabetic foot ulcers. J Diabetic Complication Med 1: 110-111.
- Hollier B, Harkin DG, Leavesley D, Upton Z (2005) Responses of keratinocytes to substrate-bound vitronectin: growth factor complexes. Exp Cell Res 305: 221-232. [Crossref]
- Berlanga-Acosta J, Savigne-Gutiérrez W, Valdés-Pérez C, Franco-Pérez N, lba JS, et al. (2006) Epidermal growth factor intralesional infiltrations can prevent amputation in patients with advanced diabetic foot wounds. Int Wound J 3: 232-239.
- Fernández-Montequín J, Valenzuela-Silva CM, Díaz OG, Savigne-Gutierrez W, Sancho-Soutelo N, et al. (2009) Intra-lesional injections of recombinant human epidermal growth factor promote granulation and healing in advanced diabetic foot ulcers: Multicenter, randomised, placebo-controlled, double-blind study. Int Wound J 6: 432-443.
- Yera-Alos IB, Alonso-Carbonell L, Valenzuela-Silva CM, Tuero-Iglesias AD, Moreira-Martínez M, et al. (2013) Active post-marketing surveillance of the intralesional administration of human recombinant epidermal growth factor in diabetic foot ulcers. BMC Pharmacol Toxicol 14: 40-44.
- López-Saura P, Yera-Alos IB, Valenzuela-Silva CM, González-Díaz O, del Río-Martín A (2013) Medical practice confirms clinical trial results of the use of intralesional human recombinant epidermal growth factor in advanced diabetic foot ulcers. Adv Pharmacoepidem Drug Safety 2: 128.
- Armstrong DG, Boulton AJM1, Bus SA1 (2017) Diabetic Foot Ulcers and Their Recurrence. N Engl J Med 376: 2367-2375. [Crossref]
- Gómez-Villa R, Aguilar-Rebolledo F, Lozano-Platonoff A, Teran-Soto JM, Fabián-Victoriano MR, et al. (2014) Efficacy of intralesional recombinant human epidermal growth factor in diabetic foot ulcers in Mexican patients: A randomized double-blinded controlled trial. Wound Repair Regen 22: 497-503.
- Ertugrul BM, Buke C, Ersoy OS, Ay B, Demrez DS, et al. (2015) Intralesional epidermal growth factor for diabetic foot wounds: the first cases in Turkey. Diabet Foot Ankle 6: 28419.
- Ertugrul BM, Lipski BA, Guvenc U (2017) Turkish intralesional epidermal growth factor study group for diabetic foot wounds. An assessment of intralesional epidermal growth factor for treating diabetic foot wounds. J Am Podiatr Med Assoc 107: 17-29.
- Brown GL, Nanney LB, Griffen J, Cramer AB, Yancey JM, et al. (1989) Enhancement of wound healing by topical treatment with epidermal growth factor. N Engl J Med 321: 76-79. [Crossref]
- Lindsey S, Langhans SA1 (2015) Epidermal growth factor signaling in transformed cells. Int Rev Cell Mol Biol 314: 1-41. [Crossref]
- de Larco JE, Todaro GJ (1978) Growth factors from murine sarcoma virus-transformed cells. Proc Natl Acad Sci U S A 75: 4001-4005. [Crossref]
- Moses HL, Roberts AB, Derynck R3 (2016) The Discovery and Early Days of TGF-β: A Historical Perspective. Cold Spring Harb Perspect Biol 8. [Crossref]
- Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, et al. (2006) Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 366: 2-16. [Crossref]
- Danielsen AJ, Maihle NJ (2002) Ligand-independent oncogenic transformation by the EGF receptor requires kinase domain catalytic activity. Exp Cell Res 275: 9-16. [Crossref]
- Normanno N, Maiello MR, De Luca A (2003) Epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs): simple drugs with a complex mechanism of action? J Cell Physiol 194: 13-19. [Crossref]
- Guglietta A, Sullivan PB (1995) Clinical applications of epidermal growth factor. Eur J Gastroenterol Hepatol 7: 945-950.
- Palomino A, Hernández-Bernal F, Haedo W, Franco S, Más JA, et al. (2000) A multicenter, randomized, double-blind clinical trial examining the effect of oral human recombinant epidermal growth factor on the healing of duodenal ulcers. Scand J Gastroenterol 35: 1016-1022.
- Sinha A, Nightingale J, West KP, Berlanga-Acosta J, Playford RJ (2003) Epidermal growth factor enemas with oral mesalamine for mild-to-moderate left-sided ulcerative colitis or proctitis. N Engl J Med 349: 350-357. [Crossref]
- Sabin SR, Goldstein G, Rosenthal HG, Haynes KK (2004) Aggressive squamous cell carcinoma originating as a Marjolin's ulcer. Dermatol Surg 30: 229-230. [Crossref]
- Fernández-Montequín J, Infante-Cristiá E, Valenzuela-Silva CM, Franco-Pérez N (2007) Intralesional injections of Citoprot-P® (recombinant human epidermal growth factor) in advanced diabetic foot ulcers with risk of amputation. Int Wound J 4: 333-343.
- Guillén IA, Camacho H, Fernández ME, Palenzuela DO, Pérez L, et al. (2012) Effect of human epidermal growth factor on the tumor cell line A431: in vivo analysis of tumor growth inhibition and gene expression. Biotecnología Aplicada 29: 155-161.
- Camacho H, Fernández ME, Guillén IA, Pérez L, Fernández JR, et al. (2013) Analysis of cell proliferation and gene expression profiles in epidermal growth factor-treated tumor cell lines. Minerva Biotecnológica 25: 43-54.