Evidence associating endocrinopathies such as diabetes, thyroid and growth hormone disorders with cardiomyopathies (CMs) and heart failure (HF) has been long-established. Initially, endocrine disorders were considered an independent risk factor for the development of HF. However, increasing diagnosis of endocrine disorders in patients with CM suggest the involvement of hormones in the pathogenesis of CMs. Moreover, the success of hormonal replacement therapies in a sub-population of patients diagnosed with dilated cardiomyopathy (DCM) provides additional evidence for the role of hormones in the pathophysiology of CMs. In addition, identifying endocrine disorders able to cause CM is important since early initiation of hormonal replacement therapy or surgery can normalize hormonal levels and potentially reverse cardiac dysfunction. However, specific guidelines for the diagnosis and management of endocrine CM are lacking. This has been partly occasioned by a paucity of clinical trials specific to endocrine CM attributed to a disproportionate focus of CM literature on the long-established morphofunctional forms of CM, particularly dilated, hypertrophic and restrictive CMs. Thus, this paper reviews current published data on endocrine CM. In particular, the paper discusses specific endocrine disorders capable of causing CM including their pathophysiology diagnosis and treatment.
endocrine cardiomyopathy, endocrine dysfunction, diabetes, thyroid dysfunction, growth hormone dysfunction
Abbreviations:
ACTH: Adrenocorticotropic Hormone; AF: Atrial Fibrillation; AGE: Advanced Glycation End-products; AHA: American Heart Association; BP: Blood Pressure; CAD: Coronary Artery Disease; CM: Cardiomyopathy; CVD: Cardiovascular; DCM: Dilated Cardiomyopathy; DM: Diabetes Mellitus; GH: Growth Hormone; GHD: Growth Hormone Deficiency; GHRH: Growth Hormone Releasing Hormone; HF: Heart Failure; IGF-I: Insulin‑like Growth Factor I; LGE: Late Gadolinium Enhancement; LV: Left Ventricular; LVEF: Left Ventricular Ejection Fraction; LVH: Left ventricular Hypertrophy; MRI: Magnetic Resonance Imaging; NO: Nitric Oxide; PAH : Pulmonary Artery Hypertension; RAAS: Renin-Angiotensin-Aldosterone System; SMC: Smooth Muscle Cells; T1DM: Type 1 Diabetes Mellitus; T2DM: Type 2 Diabetes Mellitus; TH: Thyroid Hormone; TSH: Thyroid Stimulating Hormone
A growing evidence of endocrinopathies in patients diagnosed with dilated cardiomyopathy (DCM) implicates hormonal involvement in the pathogenesis of some types of cardiomyopathy (CM). A recent cross-sectional observational study enrolling 20 female DCM patients with depressed systolic function reported significantly elevated insulin‑like growth factor I (IGF-I) compared to healthy controls, 40% with thyroid stimulating hormone (TSH) above normal level, and 15% with thyroxine hormones below normal levels [1]. Similarly, a 3-month preliminary study of growth hormone (GH) therapy in seven patients with idiopathic DCM with moderate to severe HF reported a significant increase in LV wall mass (275±11 g to 326±12 g (p < 0.05), LVEF (34±1.5 to 47±1.9% (p < 0.05), and exercise duration (6.5±0.5 to 8.9±0.9 min (p < 0.05) suggesting improved hemodynamics, exercise capacity and myocardial energy metabolism [2]. A meta-analysis of 14 trials associated GH therapy in HF patients with a significant increase in exercise duration, decreased New York Heart Association (NYHA) functional class, increased maximum oxygen consumption, and increased LV mass and wall thickness [3]. Recently, the 2016 American Heart Association (AHA) scientific statement on current diagnostic and treatment strategies for specific DCM also lists some known endocrinopathies such as thyroid disorders and growth hormones disorders as aetiologies of DCM [3]. Even with the growing evidence, specific data on endocrine CM is sporadic undermining a specific understanding of endocrine CM. This review paper synthesizes published evidence on endocrine CM with a particular focus on endocrine diseases that can cause CM including their pathophysiology, clinical presentation, diagnosis and clinical management.
Endocrine system is a collection of several glands: hypothalamus, pituitary, pineal, thyroid, parathyroid, thymus, pancreas, adrenal, ovaries and testes. The endocrine system monitors and sustains basic human functions through the release of a cascade of hormones that send chemical messages to other parts of the body enabling actions to start or stop as well as to speed up or slow down. The basic functions regulated by the endocrine system include heart rate, blood pressure, cognitive processes, appetite, energy storage and utilization levels, tissue growth and rejuvenation, sleep, sexuality, bone health, fertility, overall body metabolism, masculinity and femininity, and virtually every aspect of continued life. Table 1 lists the major endocrine glands and the hormones they produce alongside the function of these hormones.
Table 1. Glands and the hormones secreted and their function
Glands |
Hormone(s) |
Function |
Adrenal |
Adrenaline
Cortisol
Aldosterone
Androgens |
Influence the body’s metabolism, blood chemicals and body characteristics as well as influence the part of the nervous system that is involved in the response and defence against stress. |
Hypothalamus & Pituitary |
Adrenocorticotrophic hormone
Thyroid-stimulating hormone
Luteinising hormone
Follicle-stimulating hormone
Prolactin
Growth hormone
Melanocyte-stimulating hormone
Antidiuretic hormone
Vasopressin.
Oxytocin. |
Activates and controls part of the nervous system that controls involuntary boy function, the hormonal system, and many body functions such as regulating sleep and stimulating appetite. |
Ovaries & Testicles |
Oestrogen and Testosterone |
Influence female and male characteristics respectively |
Pancreas |
Insulin |
Controls the use of glucose by the body |
Parathyroid Glands |
Regulate |
Maintains the calcium levels in the body. |
Pineal Body |
Melatonin |
Involved in daily biological cycles. |
Thymus Gland |
Not named |
Plays a role in the body’s immune system. |
Thyroid Gland |
Thyroid |
Stimulate body heat production, bone growth and body’s metabolism. |
Impaired endocrine function in any of the glands would have an adverse effect on the function of the dependent organ system. For glands that produce hormones targeting the cardiovascular system, any dysfunction can result in pathological effects on the structure and function of the heart that may result in CM. Depending on the causative hormone, the AHA classifies endocrine dysfunction as an aetiology of DCM or HCM. The 2016 European Society of Cardiology (ESC) working group on myocardial and pericardial diseases defines DCM as a myocardial disorder characterized by left ventricular (LV) or biventricular dilation and global systolic dysfunction in the absence of chronic abnormal loading conditions such as hypertension and valvular disorders or coronary artery disease (CAD) in sufficient quantities to cause global systolic impairment [5]. Both AHA and ECS define HCM as a myocardial disorder characterized by increased LV wall thickness not solely explained by flow-limiting CAD or abnormal loading conditions capable of causing hypertrophy [6,7]. Taking these definitions together, endocrine CM can be defined as a heart muscle disease secondary to endocrine dysfunction and characterised by progressive dilation of the LV, and sometimes the right ventricle (RV), which may or may not be accompanied by compensatory wall thickening (hypertrophy).
Thyroid dysfunction
The thyroid is butterfly-shaped gland located immediately below the larynx on each side of and anterior of the trachea. It is one of the largest endocrine glands weighing 15-20 g in adults [8]. The thyroid gland secretes the thyroid hormone (TH), which consists of two hormones: (i) thyroxine (T4) and (ii) triiodothyronine (T3), which account for 93% and 7% of total secretion respectively but T3 is eventually converted to T4 in peripheral tissues. The regulation of the secretion of TH is through the thyroid-stimulating hormone (TSH) produced by the anterior pituitary gland [8]. TH that exerts a direct cellular effect on almost all tissues of the body. The heart is very sensitive to TH changes and the most prominent and common symptoms of thyroid dysfunction are those that result from the effects of TH on the heart and cardiovascular (CVD) system [9-16]. Hyperthyroidism and hypothyroidism produce significant changes in cardiac contractility, cardiac output, myocardial oxygen consumption, blood pressure and systemic vascular resistance [17-21]. TH influences cardiac performance by genomic and non-genomic effects. However, genomic effects (consisting of T3 linking to nuclear receptors that bind to thyroid responsive elements in the promoter of target genes) mediates many of the physiological effects [12].
The TH regulates cardiac performance by acting on both the heart and the vascular system. Several experimental and clinical studies have demonstrated the relationship between TH and CVD [22-25] later confirmed by significant changes in cardiac structure and function in patients with persistence subclinical thyroid dysfunction [26-29]. T3 is the biologically active form of TH in the heart mostly generated by T4 in peripheral tissues. The heart is vulnerable to changes in the levels of T3 because it is essential in the preservation of cardiac morphology and function [22]. The TH influences both diastolic and systolic cardiac function as well as ventricular contractility through inducing changes in hemodynamic conditions secondary to effects of TH on peripheral vascular tone. In particular, T3 increases the force and speed of systolic contraction and the speed of diastolic relaxation via its effect on myosin and isoforms and calcium-handling proteins [13,30,31]. T3 also decreases vascular resistance including coronary vascular tone and increases coronary arteriolar angiogenesis as well as promotes physiological and pathological hypertrophies [31]. Hyperthyroidism and hypothyroidism can both lead to CVD injury that may progress to HF although DCM related only to thyroid disorder is only present in a small proportion of patients with thyroid dysfunction [12].
Hypothyroidism: Hypothyroidism is an endocrine dysfunction due to abnormally low levels of circulating TH due to under-secretion from the thyroid gland. The disorder afflicts 4-10% of the population and the prevalence of subclinical hypothyroidism is about 10% [12,32,33]. In hypothyroidism, low levels of TH result in elevated TSH while in subclinical hypothyroidism TH levels are normal but with low or undetectable TSH levels [34]. TH exert a variety of effect on the CVD system influencing cardiac function (Figure 1). Hypothyroidism leads to decreased cardiac output due to impaired relaxation of vascular smooth muscles and reduced availability of endothelial nitric oxide (NO). These events produce a cascade effect of increased arterial stiffness that elevates systemic vascular resistance. At the molecular level, these alterations causes a reduction in the expression of sarcoplasmic reticulum Ca2+-ATPase and increased expression of phospholamban that inhibits ATPase. TH also affects the renin-angiotensin-aldosterone system (RAAS). T3 stimulates the synthesis of renin substrates in the liver. In a hypothyroid state, diastolic blood pressure (BP) increases, pulse pressure narrows and renin levels decrease resulting in diastolic hypertension that is often sodium sensitive [4]. TH also regulates pacemaker-related genes through transcription as well as the beta-adrenergic system in cardiomyocytes. These mechanisms increases heart rate in the presence of TH and decreases in hyperthyroid state [34,35].
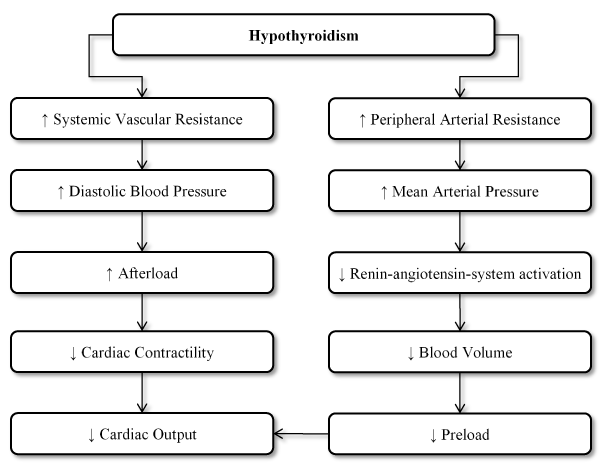
Figure 1. Effect of hypothyroidism on the heart
The changes induced by hypothyroid state may effect cardiac contractility that is diastolic in nature impairing myocardial relaxation, which is consistent with previous findings that diastolic dysfunction is the most common finding in hyperthyroid patients [36-38]. Associated diastolic hypertension or co-occurring CAD can further impair myocardial diastolic function [39]. On cardiac echocardiography, relaxation is impaired in both overt and subclinical hypothyroidism. Prolongation of the isovolumetric relaxation time and the reduction in the E/A (early to late ventricular filling velocities) ratio demonstrates early impaired relaxation in subclinical hypothyroidism [40]. Reduced E/A ratio signifies diastolic dysfunction from impaired relaxation consequently resulting in low cardiac output state with decreased heart rate and stroke volume [34]. In advanced HF secondary to myocardial infarction, conversion of T4 to T3 reduces and may affect myocardial contractility and remodelling [39]. Hyperthyroid patients often exhibit prolonged QT interval due to prolonged ventricular action potential, various degrees of atrioventricular (AV) block and low QRS complexes [13,41].
Thyroid hormone replacement is an established therapy for hypothyroidism. The most commonly prescribed TH replacement is pure synthetic T4 [42]. T4 therapy reverses CVS changes caused by hypothyroidism [43,44]. Older hypothyroid patients or those with known or suspected ischaemic heart disease or cardiac dysfunction should initially start with 25-50% of anticipated replacement dose and increase stepwise ay 6-8 week interval. In ischaemic heart disease patients, after the initiation of TH therapy, new or worsening angina or acute myocardial infarction was rare and more patients had improved anginal symptoms demonstrating potential benefits of TH therapy in improving myocardial oxygen consumption and lowering systemic vascular resistance [4]. However, whether patients with subclinical hypothyroidism should be treated with TH therapy remains controversial, but from a cardiac standpoint therapy offers some benefits but with minimal risk [44]. In Case reports of hypothyroid DCM patients treated with TH replacement for 5-12 months, patients achieved euthyroid state, normalized LVEF and LV size suggesting DCM secondary to hypothyroidism is reversible with restoration of normal thyroid function [36-38].
Hyperthyroidism: Hyperthyroidism is an endocrine dysfunction due to excessive endogenous TH production and thyrotoxicosis, the condition resulting from excess TH whether endogenous (hyperthyroidism) or exogenous (TH treatment). Hyperthyroidism is associated with palpitations tachycardia, increased exercise tolerance, dyspnoea on exertion, widened pulse presses, and sometimes, atrial fibrillation (AF) [45]. Hemodynamic effect of hyperthyroidism include elevated systemic vascular resistance, increasing resting heart rate and LV contractile function and enhanced isovolumic ventricular relaxation [23]. Reduced systemic vascular resistance stimulates the production of renin with activation of the angiotensin axis resulting in renal sodium reabsorption and increased blood volume. Increased secretion of erythropoietin due to hyperthyroidism adds to the circulating blood volume, which can increase up to 25%. Hyperthyroidism therefore results in an increased preload, decreased afterload and a cardiac output that is increased by up to 300% from a euthyroid state [46]. Although hyperthyroidism induces a fall in systemic arterial pressure, pulmonary artery hypertension (PAH) is increasingly being recognized in hyperthyroid patients. PAH could be the result of increased pulmonary blood flow that is unaccompanied by the same decrease in pulmonary vascular resistance that occurs in the systemic circulation. PAH, may in turn lead to increased load on the RV, leading to RV dilatation and an increase in right atrial and central venous pressures [4].
Hemodynamic alterations caused by hyperthyroidism decrease myocardial contractile reserve, precluding further increases in ejection fraction and cardiac output on exertion, altogether predisposing the patients to HF. Thus, hyperthyroid patients can manifest signs and symptoms of HF in the absence of cardiac injury, a state sometimes referred to as high-output heart failure characterized with paradoxical features of enhanced cardiac output and contractility due to excessive TH. True HF manifests as decreased contractility, abnormal diastolic compliance and pulmonary congestion, all of which can be consequences of severe and chronic hyperthyroidism, tachycardia and AF [12,29,35]. Based on the pathological changes associated with hyperthyroidism, thyrotoxic CM can be defined as progressive myocardial injury due to toxic effects of excessive TH leading to altered myocyte energy production, intracellular metabolism and myofibril function [45]. The major manifestations of thyrotoxic CM are LV hypertrophy, heart rhythm disturbances primarily AF, dilated heart chambers, HF and diastolic dysfunction [47]. Hyperthyroid patients with high-output HF can manifest with symptoms such as exertional dyspnoea, fatigue, fluid retention with peripheral oedema, pleural effusion, hepatic congestion and PAH [18]. Sinus tachycardia is the most common rhythm disturbance recorded in almost all hyperthyroid patients. Increased resting heart rate is characteristics of hyperthyroidism. However, AF is the most commonly identified in patients with thyrotoxicosis ranging from 2-20% [47].
The initial treatment of patients with cardiac-associated symptoms and signs of hyperthyroidism ranging from sinus tachycardia and exertional dyspnoea to HF should include beta-blocker therapy targeting to lower the heart rate to near normal [13]. The therapy targets to improve the tachycardia-mediated component of ventricular dysfunction while the direct inotropic effect of thyroid hormone could persist [48,49]. Beta-blocker therapy has been associated with rapid improvement in cardiac, neuro-muscular and psychological manifestation of hyperthyroidism. Standard therapy for hyperthyroidism can then be pursued with iodine 131 alone or in combination with an anti-thyroid drug as appropriate. Treatment for hyperthyroid patients with clinical signs and symptoms of HF should be based on guideline-recommended therapies for HF [4]. However, the use of amiodarone is controversial in hyperthyroid patients. Amiodarone, an effective antiarrhythmic drug used in the treatment of atrial and ventricular cardiac rhythm disturbance. The drug inhibits the conversion of T4 to T3 and the iodine released from the amiodarone metabolism can directly inhibit thyroid gland function, and if the effect persists, can cause amiodarone induced-hyperthyroidism [47].
Growth hormone dysfunction: Growth hormone (GH) also known as somatotropin is a peptide hormone synthesized and secreted by the pituitary gland. GH stimulates growth, cell production and cell regeneration in humans and thus it is important for human development [50]. GH secretion occurs throughout life although the amount decreases with advancing age. Peak levels of GH production usually occur in the evening when individuals are asleep. IGF-I mediates most of the effects of GH. GH also helps with lipolysis (fat breakdown), stimulates protein synthesis and helps the body in sodium and water retention. Excessive production of GH can result in gigantism if it occurs before puberty and acromegaly if it occurs after puberty. Abnormally low GH production may cause short stature or rare cases of dwarfism, although most cases are due to genetic mutation [50].
Besides their role in human development, GH and IGF-1 hormones can affect cardiac function because they appear to be physiological modulators of the structure and function of the myocardium [50,51]. GH activates the growth of cardiac cells but does not alter the collagen content of the myocardium or the density of the capillary as well as induces physiological ventricular remodelling where growth response may enhance ventricular contractility [52,53]. GH may reduce energy cost and improve thermodynamic efficiency of contractile apparatus and together with IGF-1 may exert stimulatory effect on myocardial contractile function possible with the mediation of changes in intracellular calcium handling. Moreover, cases of impaired cardiac growth and function manifests in patients with GH deficiency have been reported [54,55]. Both excessive (acromegaly) and deficiency of GH have been associated with myocardial dysfunction and the development of CM. Administration of GH to patients with GH deficiency may cause an increase in ventricular wall thickness and normalization of cardiac performance while acromegaly may induce cardiac hypertrophy and hyperkinetic syndrome and increase cardiac output and reduced vascular resistance [56,57].
Acromegaly (Excess GH): Cardiomyopathy in acromegaly involve nearly all aspects of the CVD – cardiomyocyte and intercellular myocardial composition, systolic and diastolic ventricular function, valvular heart disease and heart electrical disturbances [58-60]. Acromegaly is relatively rare, with an estimated prevalence of 60 cases per million and an incidence of 3-4 new cases per million annually in the United State [61]. Acromegaly has a slow progression with diagnosis made in the fourth to tenth year after onset at an average age of 40 years in both sexes although younger patients tend to possess more aggressive tumours and have higher GH concentrations [61,62]. A variety of signals including GH releasing hormone (GHRH) stimulates GH secretion, and somatostatin signalling suppresses secretion. Pituitary tumour and in rare cases excess GHRH originating from neuro-endocrine tumour or hypothalamic tumour causes acromegaly [63]. Increased GH levels stimulates the liver to synthesize increased IGF-1, which contributes to somatic manifestations of acromegaly [64]. The GH/IGF-1 axis induces three major aspects of the CVD system: cardiomyocyte growth, cardiac contractile function and vascular function (Figure 2).
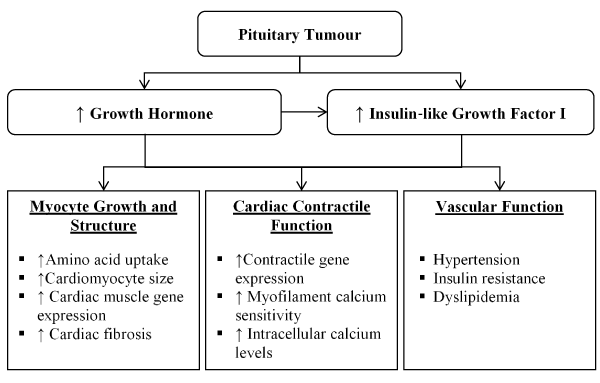
Figure 2. Pathogenesis of acromegalic cardiomyopathy
The influence of increased GH/IGF-1 levels in regulation of cardiomyocyte growth manifests as an increase in amino acid uptake (protein synthesis), cardiomyocyte size and cardiac muscle gene expression. The increased levels of the two hormones also stimulates cardiac growth leading to a characteristics biventricular hypertrophic response. Abnormally increased expression of IGF-1 independently increases the transcription of major cardia muscle specific genes including troponin 1, myosin light chain, α-actin, and IFG-1-binding protein resulting in fibrosis and sarcomerogenesis [60,65]. IGF-1 particularly promotes collagen synthesis by fibroblasts while GH increases the rate of cardiac collagen accumulation. This interstitial remodelling impairs ventricular relaxation eventually leading to initial diastolic dysfunction and subsequently systolic dysfunction [50,65]. The inhibitory influence on IGF-1 on apoptosis prevents cardiomyocyte loss complementing to the development of acromegalic heart when present in excess. The myocardium and endocardium possess both GH and IGF-1 receptors but also can produces IGF-1 because of GH-mediated autocrine and paracrine response thereby exacerbating the effects of GH-producing pituitary tumour [50]. Elevated levels of GH and IGF-1 affects cardiac contractile function in a similar manner to its effect on cardiomyocyte growth and structure. IGF-1 promotes the transcription of certain contractile genes and within the cardiomyocyte cells and GH/IGF-1 axis elevates intracellular calcium levels [66]. Vascular comorbidities associated with acromegaly include hypertension, insulin resistance (pancreases produces sufficient insulin but the body is unable to use it) and dyslipidaemia. Hypertension manifests in about 30-45% of acromegaly cases resulting from anti-natriuretic activity of GH body fluid expansion, increased arterial stiffness and endothelial dysfunction although the precise mechanism remains unclear [67]. GH/IGF-1 axis in insulin resistance causes reversible hyperglycaemia with normalization of GH levels. GH also acts as a regulator of lipolysis, leading to dyslipidaemia [65].
Clinically, acromegalic CM manifests in three stages. The first or hyperkinetic stage is typical in young patients with a short disease duration characterized by biventricular hypertrophy with increased contractility and systolic output [48,68]. The second stage is characterized with more significant hypertrophy associated with abnormalities in diastolic filling at rest and impaired cardiac performance during exercise. The third and end-stage of untreated acromegaly late in the disease course is characterized by impaired systolic and diastolic performance with low cardiac output and overt HF [69]. The end stage acromegalic CM is not easily distinguishable from idiopathic DCM because morphofunctional and histological features are not diagnostic of acromegalic CM [4]. Moreover, heart diseases due to acromegaly can be demonstrated by the evidence of pharmacological suppression of GH production leading to significant regression of cardiac hypertrophy and improvement of cardiac dysfunction [4]. Cardiac arrhythmias and sudden cardiac death are the most common causes of increased mortality in acromegaly patients. Beat-to-beat QT variability is elevated as well as late potentials that correlate with ventricular tachyarrhythmias [59].
The target of clinical management of acromegalic CM is curing or minimizing the parent condition – acromegaly and secondarily to treat cardiac signs and symptoms of HF using the standard HF medications [4]. In the greater majority of patients, acromegaly is due to pituitary adenoma, trans-sphenoidal resection is the most cost-effective and permanent treatment [70]. Surgical controlling of GH levels has been successful in improving prognosis – reduction of serum GH to < 1µg/L projects a mortality rate almost identical to age-matched controls. Although IGF-1 levels is not an independent prognostic indicator of mortality in acromegalic patients, normalization of serum IGF-1 weakly correlates with a reduction in mortality rate [71]. Despite the benefits of surgical and pharmacological therapy normalizing HG/IGF-1 levels significantly decreases LV hypertrophy (LVH), in some patients cardiac systolic function may remain unaffected [72,73]. The use of traditional HF medication such as angiotensin receptor blockers (ARBs), beta-adrenergic antagonists, diuretics, and calcium channel blockers have normalized blood pressure and increased LVEF as well as end-diastolic and end-systolic volumes in acromegalic patients [74,75].
GH deficiency: Growth hormone deficiency (GHD) is an endocrine disorder in the setting of the failure of pituitary gland to produce sufficient GH due to a primary pituitary lesion or a problem with the hypothalamus. In children, GHD results in growth failure and short stature whereas in adults GHD causes a decrease in muscle mass and increased fat mass and increased mortality from CVD causes [76]. Although cardiac involvement in acromegaly has been long-established, the evidence of GHD causing CVD impairment has only been recently described. Beyond the effect on growth, deficiency in the serum levels of GH and IGF-1 have been associated with relevant effects on cardiac development and performance [69]. Long-standing GHD in adults has been associated with changes in bone turnover and body composition, impaired lipid profile, reduced exercise capacity and abnormalities in cardiac function [77-79]. Epidemiological data suggests that adults with hypopituitarism have reduced life expectancy compared to healthy controls, with more than 2-fold increase in mortality rate for CVD disease [80].
Pathological effect of GHD on CVD functions appears to be due to a direct effect by GH/IGF-1 but also to an indirect mechanism exerted by unfavourable lipid profile, hypercoagulability and atherosclerosis as well as decreased muscle performance, pulmonary capacity and endothelial function [81]. However, most of the current evidence for GHD-associated CM results from improvement of cardiac function on GHD patients receiving GH replacement therapy. Several studies reveal that GHD adults who are not receiving GH replacement therapy have reduced LV mass with impaired myocardial contractility indices and cardiac output and decreased exercise capacity [50,55,82]. Some studies suggest GHD with a childhood onset has more severe cardiac dysfunction that adult onset due to substantially reduced levels GH/IGF-1 during cardiac growth and development of the heart but these findings lack consistency across studies. Short-term placebo studies report GH replacement therapy in adult GHD patients has an anabolic effect on cardiac structure resulting in improved diastolic and systolic function [82-84].
Research evidence so far associate low-dose individualized GH replacement therapy with improved cardiac function and reduced risk of developing cardiac hypertrophy, and together with increased muscle strength, could explain improved exercise capacity in GH-deficient adults on GH replacement therapy. In contrast, inappropriately high dose of GH introduces a risk of an unwanted increment in LV mass, particularly in elderly GHD patients during chronic treatment [85,86]. About one to two thirds of HF patients are also affected by GH/IGF-1 deficiency [87,88]. In these patients, smaller short-term trials on GH replacement therapy demonstrate improvement in various parameters of cardiac structure, LVEF, functional status and exercise performance [89,90]. However, larger long-term randomized placebo controlled clinical trials are warranted to confirm these findings and to evaluate the beneficial effect of GH replacement therapy on clinical outcomes in patients with systolic HF.
Diabetes mellitus (DM), classified into Type 1 (T1DM) and Type 2 (T2DM), is an autoimmune disorder characterized by the destruction of all the insulin-producing cells in the pancreases (T1DM) or inadequate production of insulin to control glucose levels (T2DM) although about 7% of T2DM patients may lose the ability to synthesize insulin and consequently require insulin therapy [91,92]. DM affects the heart through various mechanisms leading to the development of functional and structural changes in the myocardium in the absence of CAD, hypertension and significant valvular disease, a disorder known as diabetic CM [91]. In fact, HF affects 19-26% of DM patients [93-95]. The Framingham Heart Study found the incidence of HF is significantly increased in DM patients compared with age-matched controls independent of obesity, hypertension, dyslipidaemia and coronary heart disease [96]. Typical manifestations of diabetic CM are impaired diastolic and systolic function, and reduced contractile reserves [92,97].
The main metabolic abnormalities in DM patients are hyperglycaemia, hyperinsulinemia, and insulin resistance. Hyperglycaemia plays a pivotal role in the development of diabetic CM although hyperglycaemia, inflammation, and hyperlipidaemia may also stimulate the production of nitrogen species or reactive oxygen species (ROS) that cause most diabetic complications including diabetic CM and diabetic neuropathy [98]. Multifactorial mechanics are involved in the pathophysiology of diabetic CM including insulin resistance, microvascular impairment, subcellular component abnormalities, metabolic disturbances, cardiac autonomic dysfunction, alterations in the renin-angiotensin-aldosterone system (RAAS), and maladaptive immune responses.
Hyperglycaemia is a hallmark feature of untreated DM. Persistent hyperglycaemia increases glucose metabolism resulting in increased oxidative stress through the development of reactive oxygen species (ROS) from the mitochondria [99]. Oxidative stress leads to reduced myocardial contractility eventually inducing myocardial fibrosis [100]. Oxidative stress and ROS can speed up cardiomyocyte apoptosis and cellular DNA damage in turn activating DNA reparative enzymes, which redirects glucose metabolism from its primary glycolytic pathway to an alternative biochemical pathway. The change in metabolic pathway may lead to the development of various mediators leading to hyperglycaemia-induced cellular injury. Oxidative stress induced by persistent hyperglycaemia increases the amount of advanced glycation end-products (AGE) [98]. AGE is able to form covalent cross-links with various extra- and intra-cellular proteins including elastin and collagen to impair relaxation and increase myocardial stiffness [98,101]. Besides hyperglycaemia, insulin resistance and hyperinsulinemia are typical pathological abnormalities in T2DM patients and in pre-diabetic conditions. Cardiomyocyte hypertrophy in DM patients is modulated at the transcriptional level [102]. Hyperinsulinemia causes epigenetic and genetic alterations that activate multiple transcription factors that regulate extracellular and cellular protein expression. The activation of these transcription factors results in the accumulation of extracellular matrix proteins and cardiomyocyte hypertrophy leading to focal myocardial fibrosis in DM patients [102].
Impaired myocardial microcirculation is a frequent pathologic feature in patients with T2DM, insulin resistance and diabetic CM [103,104]. Impaired coronary microvasculature is a consequence of reduced levels of bioavailable NO. Nitric oxide activates kinases and guanylyl cyclase required for coronary relaxation [105,106]. Under condition of reduced insulin sensitivity, NO degradation increases while NO production reduces. Decreased blood supply due to microcirculatory dysfunction affect the vasa vasorum in DM further damages the medium and small arterioles of the diabetic heart. Other pathological vascular changes causing coronary microvascular ischemia in DM include perivascular fibrosis and interstitial changes, formation of micro-aneurysms, in small vessels and thickening of the capillary basement membrane. Ischemia contributes to myocardial fibrosis, stiffness and dysfunction in diabetic CM [98]. Hyperinsulinemia and insulin resistance contribute to vessel stiffness [107]. Hyperinsulinemia promotes differentiation of smooth muscle cells (SMC) to an osteoblast-like phenotype promoting stiffness while elevated insulin levels enhance stiffness by increasing osteocalcin expression, alkaline phosphate activity and formation of mineralized nodules in vascular SMC. Impaired vascular SMC and endothelial cell function are related to an increased risk of developing CAD in DM patients [108,109]. Other mechanisms contributing to the development of diabetic CM include subcellular component abnormality, altered lipid metabolism, RAAS activation, cardiac autonomic neuropathy and maladaptive immune response [92,98].
Significant pathological changes in the function and structure of the myocardium as a result of diabetic CM leading to pathological consequences. Myocardial changes mostly manifest in the early stages of the disease, and cardiac contractility ensues due to these alterations [110]. Later cardiac microcirculatory dysfunction, interstitial and perivascular fibrosis, and ventricular myocardial hypertrophy manifests. Impaired diastolic dysfunction is the initial abnormality in diabetic CM patients while systolic dysfunction develops only in the later stages of the disease [110,111]. Diabetic CM0induced ventricular fibrosis and hypertrophy are the major causes of diastolic dysfunction. When systolic dysfunction manifests, cardiac output decreases progressively with disease severity [98]. Echocardiography is useful for assessing LV hypertrophy (2D), mitral inflow for diastolic function, and tissue Doppler imaging for diastolic and systolic function. Cardiac MRI may also be useful for assessing LV hypertrophy and MRI late gadolinium enhancement (LGE) for diastolic and systolic function.
Diagnosis for diabetic CM rests on serological tests and non-invasive cardiac imaging. Serological tests for alterations in the levels of serum/plasma cardiac biomarkers that may indicate some myocardial structural and metabolic functions. Elevated levels of matrix metalloproteinase (MMPs) and reduced levels of tissue inhibitors of MMPs are associated with myocardial fibrosis. Serum aminoterminal propeptide of type III is a proposed early index of LV dysfunction in insulin resistance obese subjects [112]. Cardiac troponins (T, N and I) indicated myocardial injury in inflammatory or ischemic disease but their use is unclear in adult diabetic CM [113]. Finally, improved understanding of the pathophysiology of diabetic CM has provided improved management options. These options include lifestyle modification (Weight loss, limitation of fat and total energy intake, and regular physical activity), diabetic control (better glycaemic control), management of co-existing hypertension and CAD is present (vasoactive medication), lipid lowering therapies and management of HF (traditional HF medications and therapies) [98].
Pheochromocytoma
Pheochromocytoma is a rare neuroendocrine catecholamine-secreting tumour arising from chromaffin cells of the adrenal medulla or extra-adrenal sympathetic paraganglia [114,115]. They are rare tumours prevalent in 0.1% to 0.6% in hypertensive patients with an annual incidence of 2-8 cases per million people. About 20% are malignant and 10-20% are familial [116,117]. The disease has a gradual progression with the mean age of diagnosis at 40 years [116]. Pheochromocytoma leads to the secretion of excessive amounts of catecholamines (adrenaline or noradrenalin) leading to severe paroxysms of hypertension. On rare occasions, pheochromocytoma can also secrete other hormones such as calcitonin, neuropeptide and adrenocorticotropic hormone (ACTH) [115]. The classic triad of symptoms is headache, palpitations and diaphoresis although pheochromocytoma can be asymptomatic for years [116-119]. However, patients may also present with multiple non-specific symptoms (nausea, vomiting, weight loss, shortness of breath, chest pain, tremor, hypertension, and pulmonary oedema) including life-threatening complications of which 20% are CVD complications [120-123].
Chronic or acute catecholamine intoxication has been implicated as the cause of structural myocardial alterations. Pheochromocytoma can lead to different types of CM with the same life-threatening initial clinical presentation but with a different recovery rate. In a systematic review of 145 case reports of Pheochromocytoma-associated CM, Batisse-Lignier et al. [120] reported 49 cases of Takotsubo CM and 96 of catecholamine-induced CM. The exact pathophysiology of pheochromocytoma-associated CM is unclear since the current evidence-based is case reports. However, Batisse-Lignier et al. [120] suggests adrenergic cardiotoxicity as a possible pathophysiologic mechanism. Acute catecholaminergic stress in takotsubo CM overwhelms myocardial beta-receptors leading to stunning of the LV and slight histological apoptosis. In the case of chronic adrenergic exposure, the heart is able to adapt. Recent studies demonstrate that GRK2 and beta-arrestin regulates the secretion of catecholamine in the adrenal medulla suggesting a GRK2/beta-arrestin-mediated cross-talk between adrenal gland and the heart. Similarly, protein kinase A is able to promote desensitization and downregulation of beta-adrenergic receptors in cardiomyocyte. Despite this regulation, chronic catecholamine administration causes interstitial fibrosis, promotes cardiac apoptosis, and induces contractile dysfunction via LV dilatation. In catecholamine-induced CM, repetitive myocardial aggression because of longer catecholaminergic explain may explain LV hypertrophy, extended histological fibrosis and extracellular matrix collagen turnover all contributing to lower recovery rate. Significant fibrosis has been associated with poorer prognosis. Thus, a delay in diagnosis may lead to a deterioration in systolic and diastolic function to influence recovery rate and the type of CM [120].
Clinical presentation of pheochromocytoma-associated CM consists of cardiac symptoms of dyspnoea, chest pain or weakness, and specific pheochromocytoma symptoms of headaches, palpitations, sweating, and digestive signs in early stages of the disease. Diagnosis is based on demonstration of elevated catecholamines levels and evidence of cardiac dysfunction. Batisse-Lignier et al. [120] reports in 144 cases, common ECG abnormalities were sinus tachycardia (63.8%), ST-segment elevation (37.6%), and T-wave elevation (32.6% as well as a trend towards a higher incidence of AF. Chest radiography may reveal cardiomegaly in 56.6% of cases. In 133 cases, common findings on transthoracic echocardiography were LV dilatation (50.4%), hypertrophic cardiopathy (10.5%) [120].
The mainstay clinical management strategy for pheochromocytoma-associated CM is early intervention in the form of surgery. There are reports of complete reversibility of myocardial dysfunction after surgical intervention [124]. Myocardial function improves as early as eight days after surgery [125]. Stable patients may be treated with diuretics, ACE-I and beta-blockers, which is the standard HF therapy. Anticoagulation therapy can be considered in hypokinetic patients until restoration of myocardial contractile function. Systolic function typically recovers in 1-4 weeks [116]. Biochemical screening is recommended for patients to assess for metastatic disease and recurrence or delayed appearance of rumours. However, recurrence in more likely in familial pheochromocytoma-associated CM or patients with right adrenal tumours and extra-adrenal tumours. Patients at risk of recurrence may take beta-blocker or a dual therapy of alpha- and beta-blocker indefinitely [116].
Cushing's syndrome
Cushing's syndrome results from hypercortisolim due to adrenocorticotrophic hormone (ACTH) producing pituitary/ectopic tumours or cortisol synthesizing adrenal tumours [126]. The syndrome has five-fold excess mortality with a reported prevalence of 0.7-2.4 per million and a mean onset of 36 years [127]. Cushing’s syndrome is characterized by truncal obesity, moon face, weakness, amenorrhea, and hirsutism. Hypertension is the most frequent CVD complication of Cushing’s syndrome [128]. Other clinical signs and symptoms include LV hypertrophy, cardiomegaly, diastolic HF and myocardial ischemia [129]. However, CM as a complication of Cushing’s syndrome is very rare. Several case reports have evaluated cardiac changes in patients with Cushing’s syndrome [129-133]. These case reports reveal changes in the LV structural characteristics, particularly, asymmetrical wall thickening, suggest cardiac remodelling. These cardiac changes correlate significantly with disease duration but not with blood pressure or the levels of urinary cortisol [129]. Remarkable improvement in cardiac function following therapeutic reduction of glucocorticoid levels suggests that CM is related to hypercortisolim but the exact mechanism in uncertain. For this reason, the Endocrine Cardiomyopathy in Cushing Syndrome: Response to Cyclic GMP PDE5 inhibitOrs (ERGO) trial was designed.
The pathophysiologic mechanisms of cardiac remodelling and decompensated HF possibly involves complex mechanism including the effects of hypertension and activation of neurohormonal factors such as alpha-adrenergic and RAAS [134,135]. Experimental models suggest the effect of noradrenaline, angiotensin II and aldosterone are increased with hypercortisolim [136-138]. Consequently, excess cortisol itself may potentially explain the association between CM, cardiac failure and Cushing’s syndrome [129]. The saturation of 11-beta-hydroxysteroid dehydrogenase type 2 enzyme resulting in mineralocorticoid receptor activation by cortisol has been suggested as a contributory factor to the development of CM. Distinctive diagnostic features are lacking. However, on echocardiography or cardiac MRI, patients with Cushing’s syndrome frequently manifest with significant subclinical biventricular and left atria dysfunction, and increased LV mass, in the absence of dense myocardial fibrosis. Successful treatment of cortisol excess has been shown to improve systolic ventricular and atrial performance leading to regression of the LV mass and myocardial thickness. Since structural and functional abnormalities of the myocardium may manifest early in Cushing’s syndrome, there is a need for active investigation to guide strategies designed to prevent CM and overt HF.
Meta-analysis of diagnosis
Endocrine dysfunction is an important cause of CM due to excessive or reduced secretion of certain hormones. Definitive diagnosis during the onset of the disease is clinically relevant because endocrine CM is potentially reversible through early initiation of treatment targeting the dysfunctional hormone before irreversible cardiac damage has manifested. However, specific data on both diagnosis and management of endocrine CM are lacking since endocrine CM has been classified as a less common aetiology of other long-established morphofunctional forms of cardiomyopathy, particularly DCM and HCM [4-7]. Data from prospective randomized clinical trials are sporadic, and thus, most of the available evidence supporting the diagnosis and clinical management of the different types of endocrine CM comes from case reports and expert consensus guidelines. The objective of this meta-analysis is to review diagnostic features of endocrine cardiomyopathy.
The search for relevant studies were identified through MEDLINE, PubMed and Google scholar searches of literature from inception to the present. The key terms employed in the search included a combination of the following: (i) endocrine disorders (thyroid disorder: hyperthyroidism, hypothyroidism, and thyrotoxicosis); growth hormone dysfunction (acromegaly and growth hormone deficiency), diabetes mellitus, Pheochromocytoma and Cushing’s syndrome; and (ii) diagnostic features (electrocardiogram, echocardiogram and cardiac magnetic resonance). Studies were included is they enrolled patients with endocrine disorders and signs and symptoms of cardiac dysfunction, were evaluated using ECG and other non-invasive tests for cardiac involvement, and documented findings of abnormalities in cardiac structure and/or function. Studies that evaluated diagnostic features and outcomes other than changes in cardiac structure and function, case reports and series with only two patients, conference papers (they are not final and are usually subject to changes) and review articles were excluded.
Data for analysis was extracted under three main categories: (i) study characteristics (author, year and design); (ii) patient characteristics (number enrolled, mean age and gender proportion), and diagnostic; and (iii) treatment characteristics (endocrine disorder, diagnostic test method and outcomes) (Table 2). Study variables were described using standard summary statistics. Categorical variables were expressed as frequencies and percentages, continuous variables as mean and standard deviation or range, and dichotomous data as weighted mean and 95% confidence intervals (CI). The I2 statistics was used to quantify the percentage of total variation across studies that is not due to chance. Fixed or random effect model was used when I2 ≥ 50% or I2 < 50% respectively and a p-value < 0.05 was considered statistically significant.
Table 2. Summary of study, patient and outcome characteristics of included studies
1st Author |
Year |
Patient No. |
Mean Age |
Male (n) |
Endocrine Disorder |
Diagnostic Method |
Summary of the Main Findings: Endocrine dysfunction patients compared to healthy controls |
Mercuro [139] |
2000 |
18 |
48±15 |
10 |
Acromegaly |
Echo |
↑ LVMI (143.8±23 vs. 97.5±17); ↑ IVRT (114.1±9.8 vs. 76.1±12.7); ↓ E/A ratio (1.2±0.4 vs. 1.6±0.5) |
Damjanovic [140] |
2002 |
102 |
22-71 |
44 |
Acromegaly |
Echo |
↑ LVMI (230±56 vs. 118±40), ↓ LVEF (42±17 vs. 66±9) and ↑ cardiac index (4.3±1.8 vs. 3.5±0.8) |
Herrmann [141] |
2002 |
13 |
|
|
Acromegaly |
Echo |
↓ E/A ratio (0.71±0.26 vs. 1.0±0.1); ↑ DT (209±19 vs. 179±22) |
Van der Klaauw [142] |
2008 |
37 |
54±14 |
15 |
Acromegaly |
Echo |
↓ LVEF (67±11 vs. 69±11); ↑ LVESD (34±6 vs. 31±6); = LVEDD (51±7 vs. 51±6) |
Guo [143] |
2015 |
108 |
41.2±12.7 |
44 |
Acromegaly |
Echo |
↓ E/A ratio (1.2±0.3 vs. 1.3±0.3); ↑ LVEDD (49.9±5.0 vs. 46.6±4.1); ↑ LVESD (31.0±4.3 vs. 28.9±3.3); ↑ LVEF (67.3±5.6 vs. 68.1±5.2) |
Orosz [144] |
2015 |
30 |
55.7±10.4 |
7 |
Acromegaly |
Echo |
↓ LVEF (67.2±6.9 vs. 70.6±5.4); ↑ LVEDD (52.6±5.4 vs. 48.0±3.9); ↑ LVESD (32.3±5.2 vs. 29.1±4.4) |
Hoffman [145] |
1960 |
123 |
|
24 |
Thyrotoxicosis |
ECG |
Sinus tachycardia (37.4%); ST-T changes (22.8%); AF (12.2%) |
Osman [146] |
2007 |
393 |
49.0 (36-63) |
81 |
Hyperthyroidism |
ECG |
ECG changes: AF (6%), LVH (2%), RBBB (2%), LBBB (0.5%), pathological Q-wave (2%) |
Satpathy [147] |
2013 |
28 |
|
12 |
Hyperthyroidism |
ECG |
Hyperthyroidism: sinus tachycardia (60.7%), LVH (42.9%), Prolonged QTc interval (28.6%); Hypothyroidism: sinus bradycardia (27.3%), prolonged QTc interval (18.2%), low voltage complexes ((18.2%)) |
44 |
|
8 |
Hypothyroidism |
|
Anakwue [148] |
2015 |
50 |
|
|
Thyrotoxicosis |
Echo |
LV enhanced systolic function (30%), enhanced diastolic function (34%), diastolic dysfunction (34%), HF with preserved EF (10%), HF with reduced EF (6%); LVH (34%) |
Goyal [149] |
2016 |
24 |
33.9 |
5 |
Hyperthyroidism |
ECG |
ECG abnormality: Hypothyroidism – sinus bradycardia (61.5%), ST-T changes (26.9%); hyperthyroidism – sinus tachycardia (79.2%), AF (12.5%), ST-T changes (8.3%). |
26 |
|
7 |
Hypothyroidism |
ECG |
Widya [150] |
2013 |
78 |
56.5±5.6 |
78 |
T2DM |
CMRI |
Impaired indices of RV diastolic function: peak filling rate (315±63 vs. 356±90 mL/s; p<0.01) and peak deceleration gradient (2.3±0.8 vs. 2.8±0.8 mL/s2 X 10-3; p<0.0.01) of the early filling phase |
Garg [151] |
2015 |
100 |
41-50 |
65 |
T2DM |
ECG/Echo |
Prevalence of CM by ECG (63%); by Echo (62%) |
Pham [152] |
2015 |
656 |
59.7±8.7 |
359 |
T2DM |
Echo |
Echo: LVH (31.9%); LV dilatation (7.3%); systolic dysfunction (3.8%); LVEDD (47.5±5.3); LVESD (29.5±5.1); LVEF (67.1±9.0%) |
Jørgensen [153] |
2016 |
1030 |
65.5 |
679 |
T2DM |
Echo |
Echo abnormalities (49.8%); diastolic dysfunction (19.4%); LVH (21.0%); LA enlargement (19.6). |
Zakria [154] |
2017 |
55 |
|
|
T2DM |
Echo |
LVEF (63.6±1.1); LVEDD (50±6) and LVESD (32±7) |
Park [155] |
2011 |
36 |
49.8±15.8 |
21 |
Pheochromocytoma |
ECG, Echo |
Echo: Normal systolic function (62.1%); LVH with normal systolic function (27.6%); LV systolic function (10.3%). Diastolic function - relaxation abnormality (75.9%). ECG: normal (52.8%), LVH (33.3%) and sinus tachycardia (8.3%) |
Kamenicky [67] |
2014 |
18 |
40.5 (24.2-51.5) |
5 |
Cushing |
CMRI |
↑ LVMI (53.6 vs. 59.1 - 49.1) and ↑ LVEF (64.3 vs 55.0 - 57.8); ↑ Basal, mid-ventricular and apical wall thickness (p<0.001) |
CI: Cardiac Index; CMRI: Cardiac Magnetic Resonance Imaging; E/A: Transmitral Early/Late Diastolic Velocity; ECG: Electrocardiogram; Echo: Echocardiography; IVRT: Isovolumic Relaxation Time; LBBB: Left Bundle Branch Block; LVEDD: Left Ventricular End-Diastolic Diameter; LVESD: Left Ventricular Systolic Diameter; LVH: Left Ventricular Hypertrophy; LVMI: Left Ventricular Mass Index; PWTDI: Pulse Wave Tissue Doppler Imaging; RV: Right Ventricular; RBBB: Right Bundle Branch Block; T2DM: Type 2 Diabetes
Study characteristics
Out of 762 studies yielded in the search, only 18 studies that satisfied the inclusion/exclusion criteria were included in the final analysis [67,139-155]. The studies were published between 1960 and 2017. In total, the 18 studies enrolled a population of 2,851 patients with various endocrine disorders, and signs and symptoms of cardiac dysfunction. The mean age was 50.35 years (range 33.9 to 65.5) and with an almost equal gender representation (men = 51.4%; women = 48.6%) reported in 15 studies [67,139,140,142-147,149-153,155]. Ten studies included 701 control patients [67,139-144,146,149,150]. The underlying endocrine disorders investigated were acromegaly in six studies [139-144], thyroid disorders in five studies [145-149], in which two studies [147,149] divided patients into hyperthyroidism and hypothyroidism, diabetes mellitus in five studies [150-154] and one study each investigated Cushing syndrome [67] and Pheochromocytoma [155]. Diagnostic methods used in the studies to assess cardiac function included echocardiography in 12 studies [139-144, 148,151-155], ECG in seven studies [145-147,149,150,151,155] and cardiac MRI in two studies [67,150].
Diagnostic outcomes
In the present analysis, the evaluated diagnostic methods for cardiac dysfunction were ECG and non-invasive imaging (echocardiography and cardiac MRI). Only studies enrolling patients with thyroid dysfunction provided data on ECG changes. The most common ECG abnormalities are ST-T changes in 57 out of 245 patients in three studies translating into an event rate (ER) of 24.2% (95% CI: 19.1 to 30.1) [145,147,149] (Figure 3). LV hypertrophy in 24/471 (ER: 9.6%; 95% CI: 1.4 to 44.3) [146,147,149] (Figure 4). Sinus tachycardia in 82/175 hyperthyroid patients (ER: 58.5%; 95% CI: 32.5 to 80.5) [145,147,149] (Figure 5). Sinus bradycardia in 28/70 hypothyroid patients (ER: 43.2%; 95% CI: 15.5 to 75.9) [147,149] (Figure 6). Prolonged QTc interval in 17/98 (ER: 17.8%; 95% CI: 8.0 to 35.0) [147,149] (Figure 7). Atrial Fibrillation in 48/568 (ER: 11.2%; 95% CI: 6.2 to 19.4) [145,146,147,149] (Figure 8).
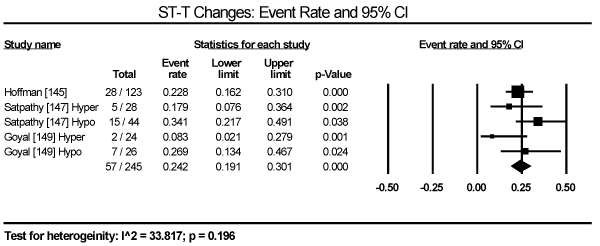
Figure 3. Event rate for ST-T changes
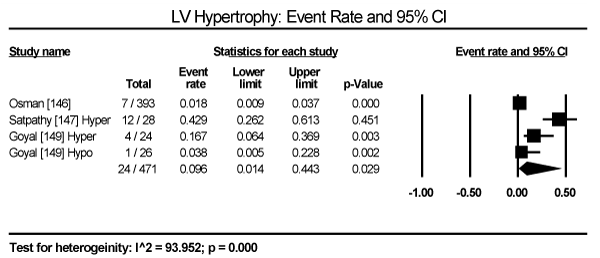
Figure 4. Event rate for LV hypertrophy
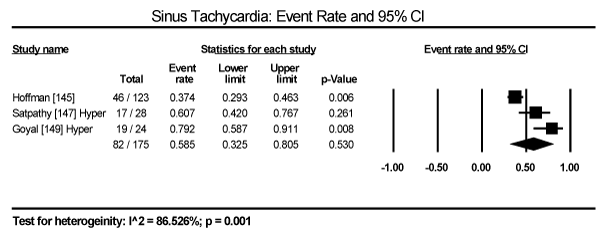
Figure 5. Event rate for sinus tachycardia
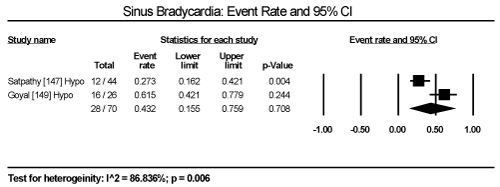
Figure 6. Event rate for sinus bradycardia
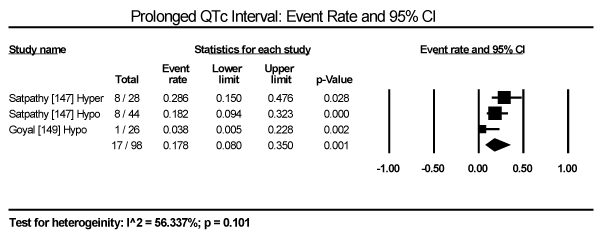
Figure 7. Event rate for prolonged QTc interval
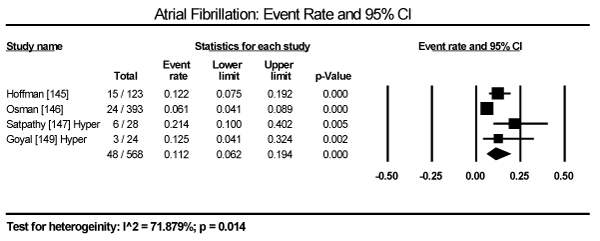
Figure 8. Event rate for atrial fibrillation
Echocardiographic findings revealed significant differences between treatment group and control group in both systolic and diastolic parameters. Data pooled from five studies showed a longer LV-end diastolic dimension (LVEDD) by a weighted mean difference (WMD) of 6.69 mm (95% CI: 1.34 to 12.03; p = 0.014) [140,142-144,148] (Figure 9). Longer LV end-systolic diameter (WMD: 2.34 mm; 95% CI: 1.45 to 3.32; p = 0.000) [142-144] (Figure 10). Higher LV mass index (WMD: 66.06 g/m2 (95% CI: 30.23 to 101.90; p=0.000) [139,140,148] (Figure 11). Reduced LVEF (WMD: -2.41; 95% CI: -7.23 to 2.42; p=0.329) [139,140,142,143,144,148,150] (Figure 12). Pooled data fron three studies revealed changes on diastolic function by a significantly decreased transmitral early/late diastolic velocity (E/A) ratio (WMD: -0.23; 95% CI: -0.39 to -0.05; p=0.010) [139,141,143] (Figure 13). In addition, one study reported diastolic dysfunction with significantly increased isovolumic relaxation time (IVRT) time in acromegalic patients compared to healthy controls (WMD: 114.1±9.8 vs. 76.1±12.7; p<0.05) [139]. In two studies [140,148] that enrolled patients with acromegaly and thyrotoxicosis, cardiac index (CI) was significantly increased compared to healthy age-matched controls (WMD: 0.887; 95% CI: 0.657 to 1.117; p=0.000) and decreased deceleration time (DT) compared to healthy age-matched controls (mean difference: 209±19 vs. 179±22; p<0.05). Other important findings include in diabetic patients were impaired RV diastolic function: peak filling rate (315±63 vs. 356±90 mL/s; p<0.01) and peak deceleration gradient (2.3±0.8 vs. 2.8±0.8 mL/s2 X 10-3; p<0.0.01) of the early filling phase [150].
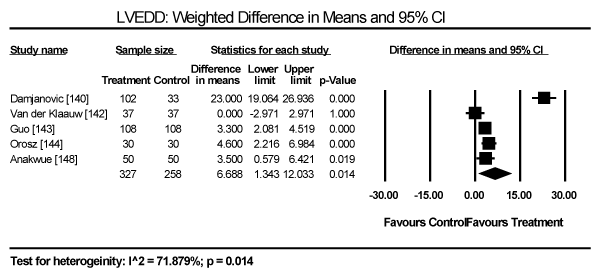
Figure 9. Weighted mean difference for LV end-diastolic diameter
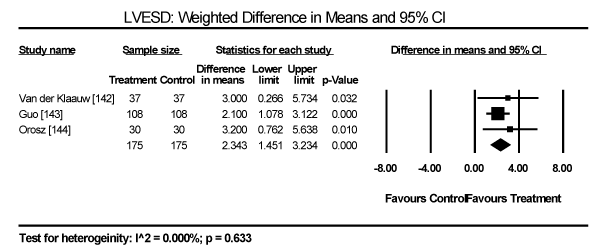
Figure 10. Weighted mean difference for LV end-systolic diameter
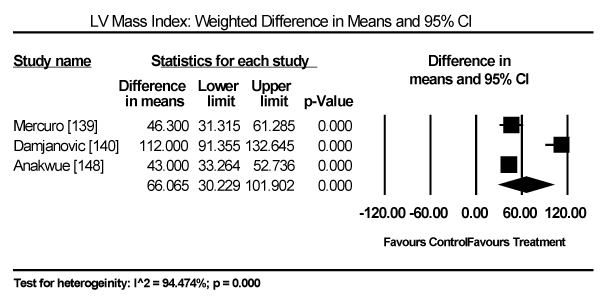
Figure 11. Weighted mean difference for LV mass index
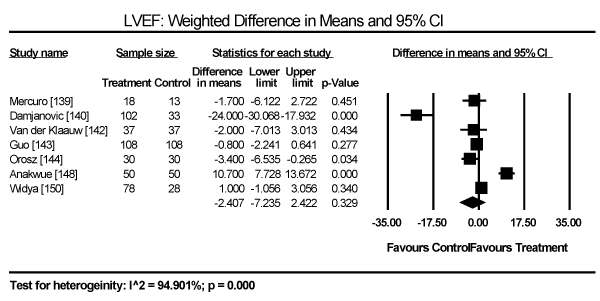
Figure 12. Weighted mean difference for LV ejection fraction
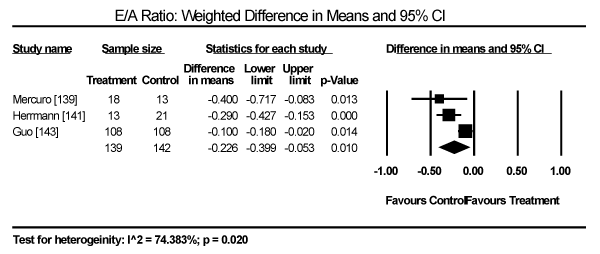
Figure 13. Weighted mean difference for E/A ratio 34
This meta-analysis evaluated current evidence for the diagnosis of myocardial involvement in patients with various endocrine disorders. Specifically, this analysis assessed the prevalence of cardiac abnormalities on ECG and/or echocardiography or cardiac MRI. However, the search for relevant studies revealed that a majority of studies on endocrine dysfunction did not specifically focus on cardiomyopathy but general cardiac dysfunction. Moreover, there was a lack of randomized clinical trials and consequently most of the included studies adopted an open label non-randomized design. The present analysis is disease-specific, informed by a wide range of clinical presentation and pathophysiologic mechanisms in different types of endocrine disorders that can cause CM. Pooled analysis finds that patients with thyroid dysfunction (hyperthyroidism, hypothyroidism or thyrotoxicosis) have a significantly higher prevalence of ECG abnormalities specifically ST-T changes, LVH and prolonged QT interval. In particular, hyperthyroid patients have a higher prevalence of sinus tachycardia and AF while hypothyroid patients have higher prevalence of sinus bradycardia. The bulk of the evidence in the present analysis on systolic dysfunction assessed by echocardiography came from acromegalic patients. Compared to age-matched healthy controls, patients with acromegaly and thyrotoxicosis had significantly longer mean measures of LVEDD and LVMI, and significantly reduced LVEF. Compared to healthy age-matched controls, the evidence of diastolic dysfunction were significantly reduced E/A ratio and significantly increased isovolumic relaxation time. Patients with diabetic CM had significantly impaired RV function demonstrated by decreased peak filling rate and peak deceleration gradient.
The present meta-analysis complements findings from expert consensus reports. The AHA and ESC scientific statement on diagnosis and management of CMs recommended disease-specific diagnostic criteria and treatment because of heterogeneity of the underlying endocrine disorders and variations in pathogenic and pathophysiologic mechanisms. The diagnosis of endocrine disorder or abnormal levels of the causative hormone predate the diagnosis of myocardial dysfunction [4-6]. In the present analysis, the bulk of the evidence comes from thyroid and growth hormone disorders that have different pathophysiologic mechanisms. For instance, hyperthyroid patients usually exhibit increased resting heart rate, blood volume, stroke volume, myocardial contractility and ejection fraction. In contrast, hypothyroid patients exhibit lower heart rate and weakened myocardial contractility and relaxation with prolonged systolic and early diastolic times [12]. On the other hand, acromegalic CM causes pathological changes in myocyte growth and structure, cardiac contractility and vascular function, which manifests as bi-ventricular hypertrophy, diastolic and systolic dysfunction, and valvular regurgitation [59]. Endocrine disorders as aetiologies of CMs have not received adequate attention since they are always researched and discussed as an uncommon form of DCM or HCM. However, since endocrine CM are potentially reversible with treatment of the underlying endocrine disorder, it is important for clinicians to test for hormonal involvement in DCM or HCM patients.
Finally, despite a meta-analysis for treatment for endocrine CM being clinically relevant, each endocrine disorder has significant variations in pathophysiologic mechanisms as well as in clinical presentation. Consequently, current treatment guidelines are disease-specific, making a meta-analysis too cumbersome to be clinically relevant. A majority of the studies included in the present meta-analysis enrolled acromegalic and diabetic patients, and those with thyroid dysfunction. Data on other endocrine disorders such as pheochromocytoma and Cushing’s syndrome that can cause CMs are lacking. The current evidence is based on case reports and lacks reliable evidence fron randomized clinical trials to support development of specific diagnostic and management criteria. A prospective randomized trial for Cushing’s syndrome, the Endocrine Cardiomyopathy in Cushing Syndrome: Response to Cyclic GMP PDE5 inhibitOrs (ERGO) trial is ongoing. Long-term clinical trials for other endocrine CMs are warranted to develop a precise understanding of clinical presentation, pathogenesis, diagnosis and management of endocrine CMs.
Endocrine CM is a myocardial disorder caused by excessive or deficient production of certain hormones characterized by progressive LV (and sometimes RV) dilatation with or without compensatory wall thickening. Endocrine disorders associated with the development of CM include thyroid dysfunction (hyperthyroidism or hypothyroidism), growth hormone (GH) dysfunction (acromegaly or GH deficiency), diabetes mellitus (insufficient insulin), pheochromocytoma (excessive catecholamine) and Cushing's syndrome (excessive cortisol). Pathophysiologic mechanisms vary widely depending on the underlying endocrine disorder. Thyroid hormone increases the force and speed of systolic contraction and diastolic relaxation, increases vascular resistance and ventricular hypertrophy. Growth hormone activates cardiac cell growth without altering collagen content of the myocardium and enhances contractility. Insulin deficiency causes hyperglycaemia in turn causes microvascular impairment, subcellular component abnormalities, metabolic disturbances, alters cardiac autonomic dysfunction and the renin-angiotensin-aldosterone system. The exact pathophysiologic mechanisms of excessive catecholamine and cortisol remains unclear. Diagnosis of endocrine CM rests on demonstrating excessive or deficiency of certain hormones and the evidence of cardiac dysfunction through laboratory tests, ECG tests and echocardiography or cardiac MRI. The primary target of clinical management is to treat the underlying endocrine dysfunction through hormonal replacement therapy or surgical excision of the dysfunctional gland. Management of cardiac dysfunction lacks specific guidelines and depends on therapy developed for heart failure. Further, clinical trials on endocrine CM are warranted to understand pathophysiology and in turn improve diagnosis and management.
- Jain A, Ramakrishanan S, Khadgawat R (2015) Endocrine abnormalities in dilated cardiomyopathy. J Pract Cardiovasc Sci 1: 247-251.
- Fazio S, Sabatini D, Capaldo B, Vigorito C, Giordano A, Guida R, et al. (1996) A preliminary study of growth hormone in the treatment of dilated cardiomyopathy. N Engl J Med 334: 809‑814. [Crossref]
- Tritos NA, Danias PG (2008). Growth hormone therapy in congestive heart failure due to left ventricular systolic dysfunction: A meta‑analysis. Endocr Pract 14: 40-49. [Crossref]
- Bozkurt B, Colvin M, Cook J, Cooper LT, Deswal A et al. (2016) Current diagnostic and treatment strategies for specific dilated cardiomyopathies: a scientific statement from the American Heart Association. Circulation 134: e579-e646. [Crossref]
- Pinto YM, Elliott PM, Arbustin E, Adler Y, Anastasakis A et al. (2016) Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J 37: 1850-1858. [Crossref]
- Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA et al. (2011). 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy. Circulation 124: 2761-2796 [Crossref]
- Elliott, P. M., Anastasakis, A., Borger, M. A., Borggrefe, M., Cecchi, F., Charron, P., ... & McKenna, W. J. (2014). 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy. Eur Heart J 35: 2733-2779. [Crossref]
- Arthur JR, Beckett GJ (1999) Thyroid function. Br Med Bull 55: 658-668. [Crossref]
- Bhardwaj R (2009) Hypothyroidism Presenting as Dilated Cardiomyopathy. Med J Armed Forces India 65: 284-286. [Crossref]
- Biondi B, Klein I (2004) Hypothyroidism as a risk factor for cardiovascular disease. Endocrine 24: 1-13. [Crossref]
- Dillmann WH (2002) Cellular action of thyroid hormone on the heart. Thyroid 12: 447-452. [Crossref]
- Klein I, Danzi S (2007) Thyroid disease and the heart. Circulation 116: 1725-1735. [Crossref]
- Klein I, Ojamaa K (2001) Thyroid hormone and the cardiovascular system. N Engl J Med 344: 501-509. [Crossref]
- Savinova OV, Liu Y, Aasen GA, Mao K, Weltman NY, et al. (2011) Thyroid hormone promotes remodeling of coronary resistance vessels. PLoS One 6: e25054. [Crossref]
- Tang YD, Kuzman JA, Said S, Anderson BE, Wang X, et al. (2005) Low thyroid function leads to cardiac atrophy with chamber dilatation, impaired myocardial blood flow, loss of arterioles, and severe systolic dysfunction. Circulation 112: 3122-3130. [Crossref]
- Hajje G, Saliba Y, Itani T, Moubarak M, Aftimos G et al. (2014) Hypothyroidism and its rapid correction alter cardiac remodeling. PLoS One 9: e109753. [Crossref]
- Weltman NY, Wang D, Redetzke RA, Gerdes AM (2012) Longstanding hyperthyroidism is associated with normal or enhanced intrinsic cardiomyocyte function despite decline in global cardiac function. PLoS One 7: e46655. [Crossref]
- Biondi B (2012) Mechanisms in endocrinology: Heart failure and thyroid dysfunction. Eur J Endocrinol 167: 609-618. [Crossref]
- Gencer B, Collet TH, Virgini V, Bauer DC, Gussekloo J, et al. (2012) Subclinical thyroid dysfunction and the risk of heart failure events: an individual participant data analysis from 6 prospective cohorts. Circulation 126: 1040-1049. [Crossref]
- Biondi B, Palmieri EA, Lombardi G, Fazio S (2002) Effects of thyroid hormone on cardiac function: the relative importance of heart rate, loading conditions, and myocardial contractility in the regulation of cardiac performance in human hyperthyroidism. J Clin Endocrinol Metab 87: 968-974. [Crossref]
- Kahaly GJ, Dillmann WH (2005) Thyroid hormone action in the heart. Endocr Rev 26: 704-728. [Crossref]
- Fazio S, Palmieri EA, Lombardi G, Biondi B (2004) Effects of thyroid hormone on the cardiovascular system. Recent Prog Horm Res 59: 31-50. [Crossref]
- Biondi B, Palmieri EA, Lombardi G, Fazio S (2001) Subclinical hypothyroidism and cardiac function. Thyroid 12: 505-510. [Crossref]
- Liu Y, Redetzke RA, Said S, Pottala JV, de Escobar GM et al. (2008) Serum thyroid hormone levels may not accurately reflect thyroid tissue levels and cardiac function in mild hypothyroidism. Am J Physiol Heart Circ Physiol 294: H2137-H2143. [Crossref]
- Tang YD, Kuzman JA, Said S, Anderson BE, Wang X, Gerdes AM (2005) Low thyroid function leads to cardiac atrophy with chamber dilatation, impaired myocardial blood flow, loss of arterioles, and severe systolic dysfunction. Circulation 112: 3122-3130. [Crossref]
- Rodondi N, Bauer DC, Cappola AR, Cornuz J, Robbins J et al. (2008) Subclinical thyroid dysfunction, cardiac function, and the risk of heart failure. The Cardiovascular Health Study. J Am Coll Cardiol 52 1152-1159. [Crossref]
- Nanchen D, Gussekloo J, Westendorp RG, Stott DJ, Jukema JW et al. (2012) Subclinical thyroid dysfunction and the risk of heart failure in older persons at high cardiovascular risk. J Clin Endocrinol Metab 97: 852-861. [Crossref]
- Iervasi G, Molinaro S, Landi P, Taddei MC, Galli E et al. (2007) Association between increased mortality mortality and mild thyroid dysfunction in cardiac patients. Arch Intern Med 167: 1526-1532. [Crossref]
- Danzi S, Klein I (2014) Thyroid disease and the cardiovascular system. Endocrinol Metab Clin North Am 43: 517-528. [Crossref]
- Katzeff HL, Powell SR, Ojamaa K (1997) Alterations in cardiac contractility and gene expression during low-T3 syndrome: prevention with T3. Am J Physiol 273(pt 1):E951-E956. [Crossref]
- Gerdes AM, Iervasi G (2010) Thyroid replacement therapy and heart failure. Circulation 122: 385-393. [Crossref]
- Ochs N, Auer R, Bauer DC, Nanchen D, Gussekloo J et al. (2008) Meta-analysis: subclinical thyroid dysfunction and the risk for coronary heart disease and mortality. Ann Intern Med 148: 832-845. [Crossref]
- Razvi S, Shakoor A, Vanderpump M, Weaver JU, Pearce SH (2008) The influence of age on the relationship between subclinical hypothyroidism and ischemic heart disease: a meta-analysis. J Clin Endocrinol Metab 93: 2998-3007. [Crossref]
- Udovcic M, Pena RH, Patham B, Tabatabai L, Kansara A (2017) Hypothyroidism and the heart. Methodist Debakey Cardiovasc J 13: 55. [Crossref]
- Biondi B (2012) Mechanisms in endocrinology: Heart failure and thyroid dysfunction. Eur J Endocrinol 167: 609-618. [Crossref]
- Seol MD, Lee YS, Kim DK, Choi YH, Kim DJ et al. (2014) Dilated cardiomyopathy secondary to hypothyroidism: case report with a review of literatures. J Cardiovasc Ultrasound 22: 32-35. [Crossref]
- Rastogi P, Dua A, Attri S, Sharma H (2018) Hypothyroidism-induced reversible dilated cardiomyopathy. J Postgrad Med 64: 177-179. [Crossref]
- Khochtali I, Hamza N, Harzallah O, Hamdi S, Saad J et al. (2011) Reversible dilated cardiomyopathy caused by hypothyroidism. Int Arch Med 4: 20. [Crossref]
- Kahaly GJ, Dillmann WH (2005) Thyroid hormone action in the heart. Endocr Rev 26: 704-728. [Crossref]
- Rodondi N, Bauer DC, Cappola AR, Cornuz J, Robbins J et al. (2008) Subclinical thyroid dysfunction, cardiac function, and the risk of heart failure. The Cardiovascular Health study. J Am Coll Cardiol 52: 1152-1159. [Crossref]
- Tribulova N, Knezl V, Shainberg A, Seki S, Soukup T (2010) Thyroid hormones and cardiac arrhythmias. Vascul Pharmacol 52: 102-112. [Crossref]
- Wiersinga WM (2001) Thyroid hormone replacement therapy. Horm Res 56: 74-81. [Crossref]
- Wieshammer S, Keck FS, Waitzinger J, Henze E, Loos U et al. (1989) Acute hypothyroidism slows the rate of left ventricular diastolic relaxation. Can J Physiol Pharmacol 67: 1007-1010. [Crossref]
- Crowley WF Jr, Ridgway EC, Bough EW, Francis GS, Daniels GH et al. (1977) Noninvasive evaluation of cardiac function in hypothyroidism: response to gradual thyroxine replacement. N Engl J Med 296: 1-6. [Crossref]
- Osuna PM, Udovcic M, Sharma MD (2017) Hyperthyroidism and the Heart. Methodist Debakey Cardiovasc J 13: 60. [Crossref]
- Dahl P, Danzi S, Klein I (2008) Thyrotoxic cardiac disease. Curr Heart Fail Rep 5: 170-176. [Crossref]
- Vargas-Uricoechea H, Bonelo-Perdomo A, Sierra-Torres CH (2014) Effects of thyroid hormones on the heart. Clin Investig Arterioscler 26: 296-309. [Crossref]
- Forfar JC, Muir AL, Sawers SA, Toft AD (1982) Abnormal left ventricular function in hyperthyroidism: evidence for a possible reversible cardiomyopathy. N Engl J Med 307: 1165-1170. [Crossref]
- Biondi B, Fazio S, Carella C, Sabatini D, Amato G et al. (1994) Control of adrenergic overactivity by beta-blockade improves the quality of life in patients receiving long term suppressive therapy with levothyroxine. J Clin Endocrinol Metab 78: 1028-1033. [Crossref]
- Saccà L, Cittadini A, Fazio S (1994) Growth hormone and the heart. Endocr Rev 15: 555-573. [Crossref]
- Saccà L, Fazio S (1996) Cardiac performance: growth hormone enters the race. Nat Med 2: 29-31. [Crossref]
- Duerr RL, Huang S, Miraliakbar HR, Clark R, Chien KR et al. (1995) Insulin-like growth factor-1 enhances ventricular hypertrophy and function during the onset of experimental cardiac failure. J Clin Invest 95: 619-627. [Crossref]
- Yang R, Bunting S, Gillett N, Clark R, Jin H (1995) Growth hormone improves cardiac performance in experimental heart failure. Circulation 92: 262-267. [Crossref]
- Merola B, Cittadini A, Colao A, Longobardi S, Fazio S et al. (1993) Cardiac structural and functional abnormalities in adult patients with growth hormone deficiency. J Clin Endocrinol Metab 77: 1658-1661. [Crossref]
- Amato G, Carella C, Fazio S, La Montagna G, Cittadini A et al. (1993) Body composition, bone metabolism, and heart structure and function in growth hormone (GH)-deficient adults before and after GH replacement therapy at low doses. J Clin Endocrinol Metab 77: 1671-1676. [Crossref]
- Thuesen L, Christensen SE, Weeke J, Orskov H, Henningsen P (1988) A hyperkinetic heart in uncomplicated active acromegaly: explanation of hypertension in acromegalic patients? Acta Med Scand 223: 337-343. [Crossref]
- Chanson P, Timsit J, Masquet C, Warnet A, Guillausseau PJ et al. (1990) Cardiovascular effects of the somatostatin analog octreotide in acromegaly. Ann Intern Med 113: 921-925. [Crossref]
- Dargad RR, Parekh JD, Dargad RR (2016) Acromegaly with Dilated Cardiomyopathy. J Assoc Physicians India 64: 96-97. [Crossref]
- Sharma MD, Nguyen AV, Brown S, Robbins RJ (2017) Cardiovascular disease in acromegaly. Methodist Debakey Cardiovasc J 13: 64-67. [Crossref]
- Clayton RN (2003) Cardiovascular function in acromegaly. Endocrine Reviews 24: 272-277. [Crossref]
- Lugo G, Pena L, Cordido F (2012) Clinical manifestations and diagnosis of acromegaly. Int J Endocrinol 2012: 1-10. [Crossref]
- Holdaway IM, Rajasoorya C (1999) Epidemiology of acromegaly. Pituitary 2: 29-41. [Crossref]
- Lopes MBS (2010) Growth hormone-secreting adenomas: pathology and cell biology. Neurosurg Focus 29:E2. [Crossref]
- Grey A, Chen Q, Xu X, et al. (2003) Parallel phosphatidylinositol-3 kinase and p42/44 mitogen-activated protein kinase signaling pathways subserve the mitogenic and antiapoptotic actions of insulin-like growth factor I in osteoblastic cells. Endocrinology 144: 4886-4893. [Crossref]
- Sharma AN, Tan M, Amsterdam EA, Singh GD (2018) Acromegalic cardiomyopathy: Epidemiology, diagnosis, and management. Clin Cardiol 41: 419-425. [Crossref]
- McMullen JR, Shioi T, Huang W-Y, et al. (2004) The insulin-like growth factor 1 receptor induces physiological heart growth via the phosphoinositide 3-kinase (p110alpha) pathway. J Biol Chem 279: 4782-4793. [Crossref]
- Kamenicky P, Blanchard A, Frank M, et al. (2011) Body fluid expansion in acromegaly is related to enhanced epithelial sodium channel (ENaC) activity. J Clin Endocrinol Metab 96: 2127-2135. [Crossref]
- Saccà L, Napoli R, Cittadini A (2003) Growth hormone, acromegaly, and heart failure: an intricate triangulation. Clin Endocrinol (Oxf) 59: 660-671. [Crossref]
- Colao A, Marzullo P, Di Somma C, Lombardi G (2001) Growth hormone and the heart. Clin Endocrinol (Oxf) 54: 137-154. [Crossref]
- Zada G, Kelly DF, Cohan P, et al. (2003) Endonasal transsphenoidal approach to treat pituitary adenomas and other sellar lesions: an assessment of efficacy, safety, and patient impressions of the surgery. J Neurosurg 98: 350-358. [Crossref]
- Holdaway IM, Rajasoorya RC, Gamble GD (2004) Factors Influencing Mortality in Acromegaly. J Clin Endocrinol Metab 89: 667-674. [Crossref]
- Lim MJ, Barkan AL, Buda AJ (1992) Rapid reduction of left ventricular hypertrophy in acromegaly after suppression of growth hormone hypersecretion. Ann Intern Med 117: 719-726. [Crossref]
- Thuesen L, Christensen SE, Weeke J, et al. (1989) The Cardiovascular Effects of Otreotide Treatment in Acromegaly: An Echocardiographic Study. Clin Endocrinol (Oxf) 30: 619-625. [Crossref]
- Anumah F, Danbauchi S, Garko S (2008) Acromegaly presenting as cardiac failure. Ethn Dis 18: 104-106. [Crossref]
- Thomas J, Dattani A, Zemrak F, et al. (2017) Renin-Angiotensin System Blockade Improves Cardiac Indices in Acromegaly Patients. Exp Clin Endocrinol Diabetes 125: 365-367. [Crossref]
- Lombardi G, Colao A, Ferone D, Marzullo P, Orio F et al. (1997) Effect of growth hormone on cardiac function. Horm Res 48: 38-42. [Crossref]
- De Boer H, Blok GJ, Van Der Veen E (1995) Clinical aspects of growth hormone deficiency in adults. Endocrine Reviews 16: 63-86. [Crossref]
- Carrol PV, Christ ER (1998) Growth hormone deficiency in adulthood and the effects of growth hormone replacement: a review. Journal of Clinical Endocrinology and Metabolism 83: 382-395. [Crossref]
- Vance ML, Mauras N (1999) Growth hormone therapy in adults and children. New Eng J Med 341: 1206-1216. [Crossref]
- Rosen T, Bengtsson BA (1990) Premature cardiovascular mortality in hypopituitarism-a study of 333 consecutive patients. Lancet 336: 285-288. [Crossref]
- Beshyah SA, Johnston DG (1999) Cardiovascular disease and risk factors in adults with hypopituitarism. Clin Endocrinol 50: 1-15. [Crossref]
- Capalbo D, Lo Vecchio A, Farina V, Spinelli L, Palladino A et al. (2009) Subtle alterations of cardiac performance in children with growth hormone deficiency: results of a two-year prospective, case-control study. J Clin Endocrinol Metab 94: 3347-3355. [Crossref]
- Cuneo RC, Salomon F, Wilmshurst P, Byrne C, Wiles CM et al. (1991) Cardiovascular effects of growth hormone treatment in growth-hormone-deficient adults: stimulation of the reninaldosterone system. Clin Sci (Lond) 81: 587-592. [Crossref]
- Cuneo RC, Salomon F, Wiles CM, Hesp R, Sönksen PH (1991) Growth hormone treatment in growth hormone-deficient adults, II: effects on exercise performance. J Appl Physiol 70: 695-700. [Crossref]
- Isgaard J, Arcopinto M, Karason K, Cittadini A (2015) GH and the cardiovascular system: an update on a topic at heart. Endocrine 48: 25-35. [Crossref]
- Thuesen L, Jørgensen JO, Müller JR, Kristensen BO, Skakkebaek NE et al. (1994) Short and long-term cardiovascular effects of growth hormone therapy in growth hormone deficient adults. Clin Endocrinol (Oxf) 41: 615-620. [Crossref]
- Saccà L (2009) Heart failure as a multiple hormonal deficiency syndrome. Circ Heart Fail 2: 151-156. [Crossref]
- Jankowska EA, Biel B, Majda J, Kristensen BO, Skakkebaek NE et al. (2006) Anabolic deficiency in men with chronic heart failure: prevalence and detrimental impact on survival. Circulation 114: 1829-1837. [Crossref]
- Le Corvoisier P, Hittinger L, Chanson P, Montagne O, MacquinMavier I et al. (2007) Cardiac effects of growth hormone treatment in chronic heart failure: a meta-analysis. J Clin Endocrinol Metab 92: 180-185. [Crossref]
- Cittadini A, Marra AM, Arcopinto M, Bobbio E, Salzano A et al. (2013) Growth hormone replacement delays the progression of chronic heart failure combined with growth hormone deficiency: an extension of a randomized controlled single-blind study. JACC Heart Fail 1: 325-330. [Crossref]
- Jia G, Hill MA, Sowers JR (2018) Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res 122: 624-638. [Crossref]
- Boudina S, Abel ED (2010) Diabetic cardiomyopathy, causes and effects. Rev Endocr Metab Disord 11: 31-39. [Crossref]
- Ryden L, Armstrong PW, Cleland JG, Horowitz JD, Massie BM et al. (2000) Efficacy and safety of high-dose lisinopril in chronic heart failure patients at high cardiovascular risk, including those with diabetes mellitus. Results from the ATLAS trial. Eur Heart J 21: 1967-1978. [Crossref]
- Shindler DM, Kostis JB, Yusuf S, Quinones MA, Pitt B et al. (1996) Diabetes mellitus, a predictor of morbidity and mortality in the Studies of Left Ventricular Dysfunction (SOLVD) Trials and Registry. Am J Cardiol 77: 1017-1020. [Crossref]
- Thrainsdottir IS, Aspelund T, Thorgeirsson G, Gudnason V, Hardarson T et al. (2005) The association between glucose abnormalities and heart failure in the population-based Reykjavik study. Diabetes Care 28: 612-616. [Crossref]
- Kannel WB, Hjortland M, Castelli WP (1974) Role of diabetes in congestive heart failure: the Framingham study. Am J Cardiol 34: 29-34. [Crossref]
- Lorenzo-Almoros A, Tunon J, Orejas M, Cortés M, Egido J, Lorenzo Ó (2017) Diagnostic approaches for diabetic cardiomyopathy. Cardiovasc Diabeto16: 28. [Crossref]
- Lee WS, Kim J (2017) Diabetic cardiomyopathy: where we are and where we are going. Korean J Intern Med.32: 404. [Crossref]
- Cai L, Li W, Wang G, Guo L, Jiang Y, Kang YJ (2002) Hyperglycemia-induced apoptosis in mouse myocardium: mitochondrial cytochrome C-mediated caspase-3 activation pathway. Diabetes 51: 1938-1948. [Crossref]
- Aragno M, Mastrocola R, Medana C, et al. (2006) Oxidative stress-dependent impairment of cardiac-specific transcription factors in experimental diabetes. Endocrinology 147: 5967-5974. [Crossref]
- Gawlowski T, Stratmann B, Stork I, et al. (2009) Heat shock protein 27 modification is increased in the human diabetic failing heart. Horm Metab Res: 41: 594-599. [Crossref]
- Feng B, Chen S, Chiu J, George B, Chakrabarti S (2008) Regulation of cardiomyocyte hypertrophy in diabetes at the transcriptional level. Am J Physiol Endocrinol Metab 294: E1119-E1126. [Crossref]
- Factor SM, Minase T, Cho S, Fein F, Capasso JM et al. (1984) Coronary microvascular abnormalities in the hypertensive-diabetic rat: a primary cause of cardiomyopathy? Am J Pathol 116: 9-20. [Crossref]
- Adameova A, Dhalla NS (2014) Role of microangiopathy in diabetic cardiomyopathy. Heart Fail Rev 19: 25-33. [Crossref]
- Zhou X, Ma L, Habibi J, et al. (2010) Nebivolol improves diastolic dysfunction and myocardial remodelling through reductions in oxidative stress in the Zucker obese rat. Hypertension 55: 880-888. [Crossref]
- Hayden MR, Habibi J, Joginpally T, Karuparthi PR, Sowers JR (2012) Ultrastructure study of transgenic Ren2 rat aorta. Part 1: rndothelium and intima. Cardiorenal Med 2: 66-82 [Crossref]
- Blaha MJ, DeFilippis AP, Rivera JJ, et al. (2011) The relationship between insulin resistance and incidence and progression of coronary artery calcification: the Multi-Ethnic Study of Atherosclerosis (MESA). Diabetes Care 34: 749-751. [Crossref]
- Olesen P, Nguyen K, Wogensen L, Ledet T, Rasmussen LM (2007) Calcification of human vascular smooth muscle cells: associations with osteoprotegerin expression and acceleration by high-dose insulin. Am J Physiol Heart Circ Physiol 292: H1058-H1064. [Crossref]
- Yuan LQ, Zhu JH, Wang HW, et al. (2011) RANKL is a downstream mediator for insulin-induced osteoblastic differentiation of vascular smooth muscle cells. PLoS One 6: e29037. [Crossref]
- Voulgari C, Papadogiannis D, Tentolouris N (2010) Diabetic cardiomyopathy: from the pathophysiology of the cardiac myocytes to current diagnosis and management strategies. Vasc Health Risk Manag 6: 883-903. [Crossref]
- Nunes S, Soares E, Fernandes J, et al. (2013) Early cardiac changes in a rat model of prediabetes: brain natriuretic peptide overexpression seems to be the best marker. Cardiovasc Diabetol 12: 44. [Crossref]
- Quilliot D, Alla F, Bohme P, et al. (2005) Myocardial collagen turnover in normotensive obese patients: relation to insulin resistance. Int J Obes (Lond) 29: 1321-1328. [Crossref]
- Russell NE, Higgins MF, Amaruso M, Foley M, McAuliffe FM (2009) Troponin T and pro-B-type natriuretic peptide in fetuses of type 1 diabetic mothers. Diabetes Care 32: 2050-2055. [Crossref]
- Whalen RK, Althausen AF, Daniels GH (1992) Extra-adrenal pheochromocytoma. J Urol 147: 1-10. [Crossref]
- Bravo EL, Gifford Jr RW (1984) Pheochromocytoma: diagnosis, localization and management. New Eng J Med 311: 1298-303. [Crossref]
- Vilcant V (2017) Pheochromocytoma-Induced Cardiomyopathy Mimicking Acute Coronary Syndrome. J Am Osteopath Assoc 117: 537-540. [Crossref]
- Otusanya O, Goraya H, Iyer P, Landi K, Tibb A, Msaouel P (2015) A vicious cycle of acute catecholamine cardiomyopathy and circulatory collapse secondary to pheochromocytoma. Oxf Med Case Reports 2015: 343-345. [Crossref]
- Chiang YL, Chen PC, Lee CC, Chua SK (2016) Adrenal pheochromocytoma presenting with takotsubo-pattern cardiomyopathy and acute heart failure: a case report and literature review. Medicine 95: e4846 [Crossref]
- Varghese RT, John AM, Paul TV (2013) Catecholamine induced cardiomyopathy in pheochromocytoma. Indian J Endocrinol Metab 17: 733-735. [Crossref]
- Batisse-Lignier M, Pereira B, Motreff P, Pierrard R, Burnot C et al. (2015) Acute and chronic pheochromocytoma-induced cardiomyopathies: different prognoses?: a systematic analytical review. Medicine 94: e2198. [Crossref]
- Mobine HR, Baker AB, Wang L, Wakimoto H, Jacobsen KC et al. (2009) Pheochromocytoma-induced cardiomyopathy is modulated by the synergistic effects of cell-secreted factors. Circ Heart Fail 2:121-128. [Crossref]
- Sanabria C, Vendrell M (2016) Severe cardiomyopathy secondary to pheochromocytoma: Usefulness of magnesium sulfate. Case report. Colombian Journal of Anesthesiology 44: 58-62.
- Jiang JP, Downing SE (1990) Catecholamine cardiomyopathy: review and analysis of pathogenetic mechanisms. Yale J Biol Med 63: 581-591. [Crossref]
- Sadowski D, Cujec B, McMeekin JD, Wilson TW (1989) Reversibility of catecholamine-induced cardiomyopathy in a woman with pheochromocytoma. CMAJ 141: 923-924. [Crossref]
- Salathe M, Weiss P, Ritz R (1992) Rapid reversal of heart failure in a patient with phaeochromocytoma and catecholamine-induced cardiomyopathy who was treated with captopril. Br Heart J 68: 527-528. [Crossref]
- Orth DN (1995) Cushing's syndrome. New Eng J Med 332:791-803. [Crossref]
- Newell-Price J, Bertagna X, Grossman AB, Nieman LK (2016) Cushing's syndrome. Lancet 367: 1605-1617. [Crossref]
- Aydoğan Bİ, Gerede DM, Canpolat AG, Erdoğan MF (2015) Cushing’s Disease Presented by Reversible Dilated Cardiomyopathy. Case Rep Cardiol 2015: 2015:980897. [Crossref]
- Yong TY, Li JY (2010) Reversible dilated cardiomyopathy in a patient with Cushing’s syndrome. Congestive Heart Fail 16: 77-79. [Crossref]
- Frustaci A, Letizia C, Verardo R, Grande C, Petramala L et al. (2016) Cushing syndrome cardiomyopathy: clinicopathologic impact of cortisol normalization. Circ Cardiovasc Imaging 9: e004569. [Crossref]
- Marchand L, Segrestin B, Lapoirie M, Favrel V, Dementhon J et al. (2015) Dilated cardiomyopathy revealing Cushing disease: a case report and literature review. Medicine 94: e2011. [Crossref]
- Kamenický P, Redheuil A, Roux C, Salenave S, Kachenoura N et al. (2014) Cardiac structure and function in Cushing's syndrome: a cardiac magnetic resonance imaging study. J Clin Endocrinol Metab 99: E2144-E2153. [Crossref]
- Sbardella E, Minnetti M, D’Aluisio D, Rizza L, Di Giorgio MR et al. (2018) Cardiovascular features of possible autonomous cortisol secretion in patients with adrenal incidentalomas. Eur J Endocrinol 178: 501-511. [Crossref]
- Opie LH, Commerford PJ, Gersh BJ, et al. (2006) Controversies in ventricular remodelling. Lancet 367: 356-367. [Crossref]
- Fedak PWM, Verma S, Weisel RD, et al.(2005) Cardiac remodeling and failure – From molecules to man. Cardiovasc Pathol 14: 1-11. [Crossref]
- Ullian ME (1999) The role of corticosteroids in the regulation of vascular tone. Cardiovasc Res 41: 55-64. [Crossref]
- Whitworth JA, Mangos GJ, Kelly JJ (2000) Cushing, cortisol and cardiovascular disease. Hypertension 6: 912-916. [Crossref]
- Saruta T, Suzuki H, Handa M, et al. (1986) Multiple factors contribute to the pathogenesis of hypertension in Cushing’s syndrome. J Clin Endocrinol Metab 62: 275-279. [Crossref]
- Mercuro G, Zoncu S, Colonna P, Cherchi P, Mariotti S et al. (2000) Cardiac dysfunction in acromegaly: evidence by pulsed wave tissue Doppler imaging. Eur J Endocrinol 2000 Sep 1;143(3):363-369. [Crossref]
- Damjanovic SS, Neskovic AN, Petakov MS, Popovic V, Vujisic B et al. (2002) High output heart failure in patients with newly diagnosed acromegaly. Am J Med 112: 610-616. [Crossref]
- Herrmann BL, Bruch C, Saller B, Bartel T, Ferdin S et al. (2002) Acromegaly: evidence for a direct relation between disease activity and cardiac dysfunction in patients without ventricular hypertrophy. Clin Endocrinol (Oxf) 56: 595-602. [Crossref]
- Van der Klaauw AA, Bax JJ, Smit JW, Holman ER, Delgado V et al. (2008) Increased aortic root diameters in patients with acromegaly. Eur J Endocrinol 159: 97-103. [Crossref]
- Guo X, Gao L, Zhang S, Li Y, Wu Y et al. (2015) Cardiovascular system changes and related risk factors in acromegaly patients: a case-control study. Int J Endocrinol 2015:573643 [Crossref]
- Orosz A, Csajbók É, Czékus C, Gavallér H, Magony S et al. (2015) Increased short-term beat-to-beat variability of QT interval in patients with acromegaly. PloS one 10: e0125639. [Crossref]
- Hoffman I, Lowrey RD (1960). The electrocardiogram in thyrotoxicosis. Am J Cardiol 6: 893-904. [Crossref]
- Osman F, Franklyn JA, Holder RL, Sheppard MC, Gammage MD (2007) Cardiovascular manifestations of hyperthyroidism before and after antithyroid therapy: a matched case-control study. J Am Coll Cardiol 49: 71-81. [Crossref]
- Satpathy PK, Diggikar PM, Sachdeva V, Laddha M, Agarwal A et al. (2013) Lipid profile and electrocardiographic changes in thyroid dysfunction. Medical Journal of Dr. DY Patil University 6: 250-253. [Crossref]
- Anakwue RC, Onwubere BJ, Ikeh V, Anisiuba B, Ike S et al. (2015) Echocardiographic assessment of left ventricular function in thyrotoxicosis and implications for the therapeutics of thyrotoxic cardiac disease. Ther Clin Risk Manag 11: 189-200. [Crossref]
- Goyal S, Goyal V (2016). A study of electrocardiographic changes in thyroid disorders. International Journal of Medical Research and Review, 4: 486-490. [Crossref]
- Widya RL, Van Der Meer RW, Smit JW, Rijzewijk LJ, Diamant M et al. (2013) Right ventricular involvement in diabetic cardiomyopathy. Diabetes Care 36: 457-462. [Crossref]
- Garg P, Jindal S, Gangajalia C (2015) A study of prevalence of cardiomyopathy in diabetes mellitus as evidenced by electrocardiography and 2-D echo. Int J Adv Med 2: 218-222.
- Pham I, Cosson E, Nguyen MT, Banu I, Genevois I et al. (2015) Evidence for a specific diabetic cardiomyopathy: an observational retrospective echocardiographic study in 656 asymptomatic type 2 diabetic patients. Int J Endocrinol 2015: 743503. [Crossref]
- Jørgensen PG, Jensen MT, Mogelvang R, von Scholten BJ, Bech J et al. (2016) Abnormal echocardiography in patients with type 2 diabetes and relation to symptoms and clinical characteristics. Diab Vasc Dis Res 2016 13: 321-330. [Crossref]
- Zakria E, Elsayed NM, Ghanem NS, Youssif E, Kalaf MM et al. (2017) Assessment of left ventricular functions in patients with type 2 diabetes mellitus using tissue Doppler imaging and its correlation with a novel cardiac biomarker. Egyptian Journal of Internal Medicine 29: 181.
- Park JH, Kim KS, Sul JY, Shin SK, Kim JH et al. (2011) Prevalence and patterns of left ventricular dysfunction in patients with pheochromocytoma. J Cardiovasc Ultrasound 19: 76-82. [Crossref]