The role of High Performance Computing (HPC) in Medicine is greatly increase in these last years, moving from basic research to the clinics. With the advent of Next Generation Sequencing (NGS) technologies, diverse areas of human health have been investigated through different omics techniques. The extensive use of these NGS platforms to high throughput profile human health issues in a cost-efficient manner, is generating huge amount of sequencing data pushing bioinformatic research in the big-data field. Speed, accuracy and reproducibility of massively sequencing analysis have allowed to transfer molecular biology knowledge into precision medicine. Furthermore, Molecular Dynamics (MD) earned a great importance in aiding genome research. Sequencing studies of cancer have allowed to detect and characterize mutated genes that drive tumorigenesis. As a complementary approach, from a biophysical perspective, MD simulations, executed on HPC architectures, have permitted to investigate the role played by pathological mutations on the molecular mechanism of activation.
The goal of genomics research is to identify genetic variants associated with disease, response to treatment, or future patient prognosis. Whole Genome Sequencing (WGS) is a genomics technique that allows to detect all types of genetic variations (single nucleotide and deletion/insertion polymorphism) across the entire genome. This powerful feature joined to the maps of genetic variation in populations is a very robust and effective tool for identifying pathogenic variants thus enabling the integration of diagnosis, genetic counselling into treatment decision-making. In 2015 Taylor et al. extensively applied whole-genome sequencing as tool for diagnosis of genetic disorders in routine clinical practice on 500 patients (including 156 independent cases) [1]. They identified of at least one variant with a high level of evidence of pathogenicity in 21% of cases (33/156) using several analysis strategies that improved the accuracy of variant calling and detection rates. More in general WGS provides a picture of the whole landscape of driver mutation and mutational signature in diseases. Several HPC bioinformatic pipelines have been developed to characterize and prioritize genetics variant [2-3].
Whole-exome sequencing (WES) is a genomic technique for sequencing all of the protein-coding genes in a genome (also known as the exome) [4-5]. It has been applied to cancer and rare diseases to identify both the actionable somatic variants in the coding regions and efficiently detect the Mendelian disorder variants; WES has been used extensively to diagnose novel diseases and find novel causative mutations for known disease phenotypes [6]. WES has been also applied for diagnosis of young patients without all spectrum of symptoms [7] and prenatal diagnosis [8]. Furthermore, detecting the causative mutation can suggest how to modify the treatment and prevent more invasive tests, confirming diagnoses and open the access to clinical trials.
Targeted-exome sequencing (TES) is a genomic technique for which a subset of genes or regions of the genome are isolated and sequenced. This technique allows researchers to focus data analysis on specific genomic ranges of interest and enables sequencing at much higher coverage levels. In this way, specific gene panels [9-10] become valuable tools to detect mutations in genes or genomic regions that are known or suspected to be associated to the disease of interest; the panel can be custom designed to amplify the regions of interest. TES offers a more sensitive approach for the analysis of the cancer genome. It eliminates in short time much of the background noise generated by WES, since it provides higher coverage at a lower cost. This feature makes TES an ideal tool for translational medicine and clinical settings.
RNA sequencing (RNA-Seq) is a sequencing technique able to reveal the presence and quantity of RNA in a biological sample at a given moment and specific experimental condition. RNA-Seq is used to analyse the continuously changing cellular transcriptome. It has been extensively applied to patients to identify the molecular bases of many biological processes and diseases, including cancer [11-12]. In particular, transcriptome-wide gene expression profiling has provided a better comprehension of the molecular mechanisms underlying prognosis and drug sensitivity. It addresses several aspects of the expression process (e.g. identification and quantification of expressed genes and transcripts, alternative splicing and polyadenylation, fusion genes and trans-splicing, post-transcriptional events, etc.) [13-17].
The Cancer Genome Atlas consortium [18] provides access to a big-data secure repository for storing, cataloguing and querying cancer genome ‘omics data. Through the TCGA Data Portal (https://tcga-data.nci.nih.gov) cancer genome sequences, alignments, mutation information and molecular changes in cancer genome datasets, such as new aberrations in several cancer types, are now available to scientific community. Another two available big-data resources on cancer are the Cancer Cell Line Encyclopedia (CCLE) [19] and the Genomics of Drug Sensitivity in Cancer [20]. As translational immediate impact on precision medicine, link among genomic biomarkers and drug sensitivity in hundreds of cancer cell lines are available for patients. With particular reference to CCLE, a big-data HPC analysis has been extensively performed on 935 paired-end RNA-seq experiments downloaded from CCLE repository, aiming at addressing novel putative cell-line specific gene fusion events in human malignancies [21]. Several gene fusion detection algorithms have been applied to the CCLE dataset in order to provide in silico a reliable consensus result set of about 1,700 predicted novel fusion gene candidates in all the human malignant cell lines. Such results, querieble on gene fusion database web portal (Ligea - http://hpc-bioinformatics.cineca.it/fusion) could represent the starting point for detecting in wet lab novel cancer biomarkers and specific drug targets. In (Figure 1) a composition of screenshots of the Ligea portal are shown.
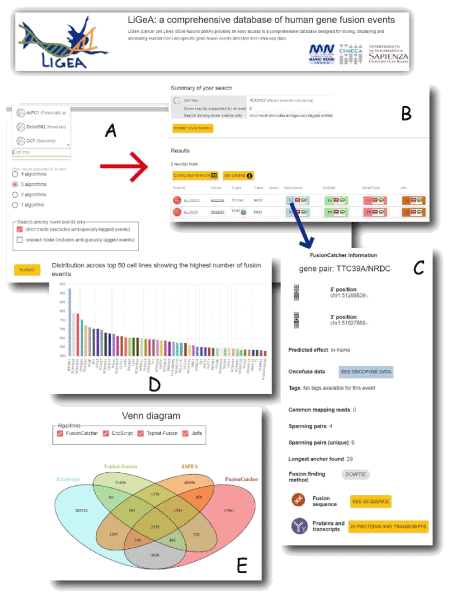
Figure 1. Screenshots of Ligea Portal: a) A ’Search by Cell line’ form allows to navigate the database by indicating a specific cell line name; b) the corresponding results table reporting the gene fusion prediction for each used algorithm (Fusioncatcher, EricScript, Tophat Fusion, Jaffa); c) by clicking on the light blue button (corresponding to fusioncatcher algorithm result), a popup window opens showing details about the putative detected gene fusion event; d) the Venn diagram shows the intersection of the putative gene fusion events identified by the four algorithms; e) the number of the different cell lines derived from the same diseases. Both d) and e) panels can be visualized in the Ligea home page.
The availability of human gene expression profiles for normal (GTEx), tumor (TCGA) and cancer cell line (CCLE) tissues provide a first picture of the structure of global gene expression. However, the complexity functional tumour molecular profiles poses great challenges to translate information contained in the big-data bioinformatics repositories into new cancer drugs and molecular diagnostics. The role of HPC in bioinformatics and computational biology is essential to reach these goals in a reasonable time.
ChIP-seq is a sequencing technique that combines chromatin immunoprecipitation (ChIP) with massively parallel DNA sequencing. It is a powerful method to identify genome-wide DNA binding sites for transcription factors and other proteins. Furthermore, it can be used to precisely map global binding sites for any protein of interest [22-24].
Epigenetic alterations are modifications in gene expression that are independent of the DNA gene sequence. They are considered to be very influential in both the normal and disease states of an organism. In particular they may influence the epigenetic inheritance and epigenetic carcinogenesis, or any other disease related to alterations in an organism. Main epigenetic mechanisms modificating gene expression are: DNA methylation [25], histone modifications [26], chromatin remodeling and microRNAs that act as regulatory molecules [27]. Epigenetics changes provides a molecular profiling of interactions between genomic and environmental conditions [28]; they are responsible for the regulation of specific gene expression networks that differ in behaviour between normal and diseased phenotype. In case of pancreatic ductal adenocarcinoma (PDAC) subtypes the study of epigenomic landscapes integrated with data of Chip-seq and RNAseq has allowed to predict aggressiveness and survival in some subtype of PADC [29], thus providing potential new markers and therapeutic targets.
Metagenomics is a sequencing technique that allows to study the genetic material recovered directly from environmental samples. It has been extensively applied to characterize virus genome heterogeneity, without in vitro replication biases, in the microbial community present in the clinical samples. High-throughput pyrosequencing has been used to detect and characterize 2009 pandemic influenza A (H1N1) virus directly in nasopharyngeal swabs in the context of the microbial community [30-33].
Nowadays another translational clinical field is growing in metagenomic research area: the study of human microbiome, responsible for influencing individuals in both health and disease. It is a major player in the immune system, since researchers believes that immune reactions are closely linked to the distribution of microbial communities throughout a person’s life [34].
Historically, the HPC role in Medicine is even precedent to the NGS revolution, starting in the ‘90 with the availability of accurate in silico models for the simulation of biological macromolecules (first proteins in aqueous environment and then nucleic acids and membrane proteins).
HPC, in particular, has been widely applied in cancer research with Molecular Dynamics simulations characterizing cancer related proteins [35-38]; evaluating the impact of somatic mutations or the activity of anticancer drugs [39-42]. MD has been also applied for the characterization of viral proteins [43-44].
The growing availability of genomic information, and in particular non-synonymous SNPs obtained by NGS and microarray-based platforms, has increased the need for in silico methods capable to provide information at atomic level on the structural and dynamic alterations produced in mutated proteins. MD simulation is routinely complemented by other complementary methods such as Homology modelling, Molecular docking, and Drug Design. Application of these methods has become a standard tool in human genome research, since they proved to be able to rationalize the impact of pathogenic mutations [45-47].
MD simulations, in particular, allows one to address specific questions about structural properties and long-range dynamics of protein and nucleic acids, thus allowing the formulation of rational hypothesis of clinical data [48-51]. In (Figure 2) location of clinically relevant tubulin cofactor D (TBCD) variants and MD simulations results showing the structural perturbation induced by the Ala586Val clinically observed substitution.
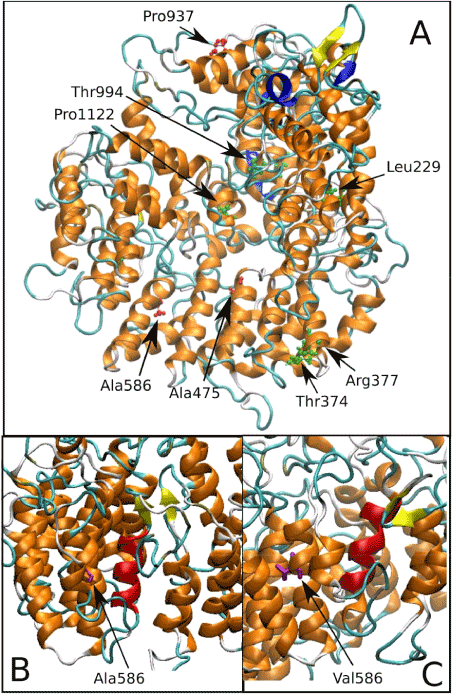
Figure 2. (a) Location of disease-associated amino acid substitutions in tubulin cofactor D (TBCD). The three variants described in 45-46 have the lateral chain highlighted in pink. (b) Ala586 is a buried residue located in a region of α helices. (c) MD simulations performed to investigate the structural perturbation induced by the Ala586Val substitution identified a local rearrangement of these helices, resulting in a substantial rearrangement of their relative orientation.
This research was supported by:The “Departments of Excellence-2018” Program (Dipartimenti di Eccellenza) of the Italian Ministry of Education, University and Research-MIUR, DIBAF-Department Project “Landscape 4.0 – food, wellbeing and environment; The “Fondi di Ateneo per la ricerca 2017”, University of Rome, Sapienza.
- Taylor, et al. (2015) Factors influencing success of clinical genome sequencing across a broad spectrum of disorders. Nat Genet 47: 717-726. [Crossref]
- Causey JL, Ashby C, Walker K, et al. (2018) DNAp: A Pipeline for DNA-seq Data Analysis. Scientific Reports, volume 8, Article number 6793.
- Chiara M, Gioiosa S, Chillemi G, D'Antonio M, Flati T, et al. (2018) CoVaCS: a Consensus Variant Calling System. BMC Genomics 19: 120.
- D'Antonio M, D'Onorio De Meo P, Paoletti D, Elmi B, Pallocca M, et al. (2013) WEP: a high-performance analysis pipeline for whole-exome data. BMC Bioinformatics 14 Suppl 7: S11. [Crossref]
- Neri M, Bovolenta M, Scotton C, De Grandis D, Castrignanò T, et al. (2012) Whole exome sequencing filtered by novel candidate genes as tool for gene discovery in a recessive family with Parkinson and ataxia. Neuromuscular Disorders 22: 810.
- de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, et al. (2012) Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med 367: 1921-1929. [Crossref]
- Iglesias A, Anyane-Yeboa K, Wynn J, Wilson A, Truitt Cho M, et al. (2014). The usefulness of whole-exome sequencing in routine clinical practice. Genet Med 16: 922-931.
- Xu Y, Xiao B, Jiang WT, Wang L, Gen HQ, et al. (2014). A novel mutation identified in PKHD1 by targeted exome sequencing: guiding prenatal diagnosis for an ARPKD family. Gene 551: 33-38.
- D’Antonio M, D’Onorio De Meo P, Castrignanò T, Erbacci G, Pallocca M, et al. (2014) ODESSA: a High Performance Analysis Pipeline for Ultra Deep Targeted Exome Sequencing Data. International Conference on High Performance Computing & Simulation 608-615.
- Miller EM, Patterson NE, Zechmeister JM, et al. (2017). Development and validation of a targeted next generation DNA sequencing panel outperforming whole exome sequencing for the identification of clinically relevant genetic variants. Oncotarget 8:102033-102045. [Crossref]
- Byron SA, Van Keuren-Jensen KR, Engelthaler DM, Carpten JD, Craig DW (2016) Translating RNA sequencing into clinical diagnostics: opportunities and challenges. Nat Rev Genet 17: 257-271. [Crossref]
- Cieślik M, Chinnaiyan AM, et al. (2018) Cancer transcriptome profiling at the juncture of clinical translation. Nat Rev Genet 19: 93-109. [Crossref]
- D'Antonio M, D'Onorio De Meo P, Pallocca M, Picardi E, D'Erchia AM, et al. (2015) RAP: RNA-Seq Analysis Pipeline, a new cloud-based NGS web application. BMC Genomics 16: S3. [Crossref]
- Picardi E1, D'Antonio M, Carrabino D, Castrignanò T, Pesole G (2011) ExpEdit: a webserver to explore human RNA editing in RNA-Seq experiments. Bioinformatics 27: 1311-1312. [Crossref]
- Bolis M, Garattini E, Paroni G, Zanetti A, Kurosaki M, et al. (2017). Network-guided modelling allows tumor-type independent prediction of sensitivity to all-trans retinoic acid. Ann Oncol 28: 611-621.
- Scotton C, Bovolenta M, Schwartz E, et al. (2016). Deep RNA profiling identified Clock and molecular clock genes as pathophysiological signatures in collagen VI myopathy. J Cell Sci. 129: 1671-84.
- Silvestri V, Zelli V, Valentini V, et al. (2017) Whole-exome sequencing and targeted gene sequencing provide insights into the role of PALB2 as a male breast cancer susceptibility gene. Cancer 123: 210-218.
- Cancer Genome Atlas Research Network, Weinstein JN, et al. (2013) The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet 45: 1113-1120. [Crossref]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, et al. (2012) The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483: 603-607. [Crossref]
- Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, et al. (2012) Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 483: 570-575. [Crossref]
- Gioiosa S, Bolis M, Flati T, Massini A, Garattini E, et al. (2018) Massive NGS Data Analysis Reveals Hundreds of Potential Novel Gene Fusions in Human Cell Lines. Gigascience
- Desantis A, Bruno T, Catena V, De Nicola F, Goeman F, et al. (2015) Che-1-induced inhibition of mTOR pathway enables stress-induced autophagy. EMBO J 34: 1214-1230. [Crossref]
- Goeman F, De Nicola F, D'Onorio De Meo P, Pallocca M, Elmi B, et al. (2014) VDR primary targets by genome-wide transcriptional profiling. J Steroid Biochem Mol Biol 143: 348-356. [Crossref]
- Botti E, Spallone G, Moretti F, Marinari B, Pinetti V, et al. (2011) Developmental factor IRF6 exhibits tumor suppressor activity in squamous cell carcinomas. Proc Natl Acad Sci U S A 108: 13710-13715. [Crossref]
- Sadikovic B, Al-Romaih K, Squire JA, Zielenska M (2008) Cause and consequences of genetic and epigenetic alterations in human cancer. Curr Genomics 9: 394-408. [Crossref]
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, et al. (2007) Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448: 553-560. [Crossref]
- Choi SW, Friso S (2010) Epigenetics: A New Bridge between Nutrition and Health. Adv Nutr 1: 8-16. [Crossref]
- Nebbioso A, Tambaro FP, Dell'Aversana C, Altucci L (2018) Cancer epigenetics: Moving forward. PLoS Genet 14: e1007362. [Crossref]
- Lomberk G (2018) Distinct epigenetic landscapes underlie the pathobiology of pancreatic cancer subtypes. Nature Communications 9: 1978.
- Bartolini B, Chillemi G, Abbate I, Bruselles A, Rozera G, et al. (2011) Assembly and characterization of pandemic influenza A H1N1 genome in nasopharyngeal swabs using high-throughput pyrosequencing. New Microbiol 34: 391-377.
- Rozera G, (2009) Archived HIV-1 minority variants detected by ultra-deep pyrosequencing in provirus may be fully replication competent AIDS 23: 2541-2543.
- Castilletti C, Carletti F Gruber CE, Bordi L, Lalle E (2015) Molecular Characterization of the First Ebola Virus Isolated in Italy, from a Health Care Worker Repatriated from Sierra Leone. Genome Announcements 18: 3. [Crossref]
- Capobianchi MR, Gruber CE, Carletti F, Meschi S, Castilletti C, et al. (2015) Molecular Signature of the Ebola Virus Associated with the Fishermen Community Outbreak in Aberdeen, Sierra Leone, in February 2015. Genome Announcements 3. [Crossref]
- Palm N.W., de Zoete M. R., and A Flavell (2015). Immune-microbiota interactions in health and disease. Clinical Immunology 159: 122-127. [Crossref]
- Chillemi G, Castrignanò T, Desideri A (2001) Structure and hydration of the DNA-human topoisomerase I covalent complex. Biophys J 81: 490-500. [Crossref]
- Chillemi G, Davidovich P, D'Abramo M, Mametnabiev T, Garabadzhiu AV, et al. (2013) Molecular dynamics of the full-length p53 monomer. Cell Cycle 12: 3098-3108. [Crossref]
- D'Annessa I, Coletta A, Sutthibutpong T, Mitchell J, Chillemi G, et al. (2014) Simulations of DNA topoisomerase 1B bound to supercoiled DNA reveal changes in the flexibility pattern of the enzyme and a secondary protein-DNA binding site. Nucleic Acids Res 42: 9304-9312. [Crossref]
- Capranico G, Marinello J, Chillemi G (2017) Type I DNA Topoisomerases. J Med Chem 60: 2169-2192. [Crossref]
- Tesauro C, Fiorani P, D'annessa I, Chillemi G, Turchi G, et al. (2010) Erybraedin C, a natural compound from the plant Bituminaria bituminosa, inhibits both the cleavage and religation activities of human topoisomerase I. Biochemical J 425: 531-539.
- Mancini G, D'Annessa I, Coletta A, Sanna N, Chillemi G, et al. (2010) Structural and dynamical effects induced by the anticancer drug topotecan on the human topoisomerase I - DNA complex. PLoS One 5: e10934. [Crossref]
- Mancini G, D'Annessa I, Coletta A, Chillemi G, Pommier Y, et al. (2012) Binding of an Indenoisoquinoline to the topoisomerase-DNA complex induces reduction of linker mobility and strengthening of protein-DNA interaction. PLoS One 7: e51354 [Crossref]
- Sanna N, Chillemi G, Gontrani L, Grandi A, Mancini G, et al. (2009) UV-vis spectra of the anticancer campothecin family drugs in aqueous solution: Specific spectroscopic signatures unraveled by a combined computational and experimental study. J Phys Chem B 113: 5369-5375.
- Chandramouli B, Chillemi G, Desideri A (2014) Structural dynamics of V3 loop in a trimeric ambiance, a molecular dynamics study on gp120-CD4 trimeric mimic. J Struct Biol 186: 132-140. [Crossref]
- Chandramouli B, Chillemi G, Abbate I, Capobianchi MR, Rozera G, et al. (2012) Importance of V3 loop flexibility and net charge in the context of co-receptor recognition. A molecular dynamics study on HIV gp120. J Biomol Struct Dyn 29: 879-891.
- D'Annessa I, Tesauro C, Fiorani P, Chillemi G, Castelli S, et al. (2012) Role of Flexibility in Protein-DNA-Drug Recognition: The Case of Asp677Gly-Val703Ile Topoisomerase Mutant Hypersensitive to Camptothecin. J Amino Acids 2012: 206083. [Crossref]
- Fiorani P, Tesauro C, Mancini G, Chillemi G, D'Annessa I, et al. (2009) Evidence of the crucial role of the linker domain on the catalytic activity of human topoisomerase I by experimental and simulative characterization of the Lys681Ala mutant. Nucleic Acids Res 37: 6849-6858.
- Biagini T, Chillemi G, Mazzoccoli G, Grottesi A, Fusilli C, et al. ( 2017) Molecular Dynamics Recipes for Genome Research. Brief Bioinform 18. [Crossref]
- Pode-Shakked B, Barash H, Ziv L, Gripp KW, Flex E, et al. (2017) Microcephaly, intractable seizures and developmental delay caused by biallelic variants in TBCD: Further delineation of a new chaperone-mediated tubulinopathy. Clin Genet. 91(5):725-738. [Crossref]
- Flex E, Niceta M, Cecchetti S, Thiffault I, Au MG, et al. (2016) Biallelic mutations in TBCD, encoding the tubulin folding cofactor D, perturb microtubule dynamics and cause early-onset encephalopathy. Am J Hum Genet 99: 962-973. [Crossref]
- Motta M, Chillemi G, Fodale V, Cecchetti S, Coppola S. (2016) SHOC2 subcellular shuttling requires the KEKE motif-rich region and N-terminal leucine-rich repeat domain and impacts on ERK signalling. Human Molecular Genetics 25: 3824-3835 [Crossref]
- Dionisi-Vici C, Shteyer E, Niceta M, Rizzo C, Pode-Shakked B, et al. (2016). Expanding the molecular diversity and associated phenotype of Glycerol-3 Phosphate Dehydrogenase 1 deficiency. J Inherit Metab Dis 39689-39695.