Pulmonary arterial hypertension (PAH) is characterized by vascular cell growth and proliferation leading to increased pulmonary vascular resistance, increased pulmonary arterial pressure, right ventricular failure, and death. The main objective of the study was to investigate the prophylactic and therapeutic benefits of over expression of EC-SOD in an animal model with chronic PAH. Chronic PAH was induced by exposing adult male C57BL6 mice to hypoxia (FiO2 10%) for 3 weeks and Sugen[E Miller1] administration, once per week (3 doses). Hemodynamic, histologic, immunohistochemistry, NO Pathway, angiogenesis markers and inflammatory marker studies were performed at 3, 7, 11- and 14-weeks post-exposure. Transgenic mice which overexpress EC-SOD showed less evidence of PAH, associated with mild inflammation which was resolved by 11-14 weeks post exposure compared with wildtype mice under the same conditions. Overexpression of EC-SOD post-establishment of chronic progressive PAH showed significant therapeutic benefits. There was a significant decrease in pulmonary pressures associated with resolution of the chronic changes at the level of the parenchyma in transfected hypoxic wild type animals compared to wild type hypoxic non-transfected. The data suggest that EC-SOD has a marked and a significant role in ameliorating and reversing the chronic, progressive form of PAH. in murine mo[E Miller2]del. [E Miller3]
pulmonary hypertension, chronic hypoxia, sugen, EC-SOD, prophylaxis, therapy
Pulmonary arterial hypertension (PAH), occurs when pulmonary vascular resistance (PVR) remains abnormally elevated. This in turn leads to severe hypoxemia that may not respond to conventional respiratory support and can lead to right heart failure and death [1]. PAH encompasses a multifactorial group of pulmonary vascular disorders [2]. Major factors contributing to the complicated pathogenesis include sustained vasoconstriction and progressive fixed vascular remodeling [3]. Despite huge advances in the understanding of the pathogenesis of PAH, the complexity of its origins, and the pathway interactions involved require further elucidation. In particular, the equilibrium between pulmonary smooth cells and endothelial cells, the complexity of hemodynamic changes i, the prevention and reversion of pulmonary hypertension remain unexplained [4].
One of the major contributory factors in the pathophysiology of PAH is hypoxia, which plays a pivotal role [5], leading to pulmonary vessel constriction, proliferation of endothelial cells, smooth muscle cells and adventitial fibroblasts resulting in vessel narrowing [5-8].
Exposure of most animals to low levels of environmental oxygen results in alveolar hypoxia and reliably causes chronic pulmonary hypertension and morphological alterations of the precapillary pulmonary vessels [9-11]. The major limitation of the frequently studied small animal models of pulmonary hypertension (induced by chronic hypoxia), is that they do not always develop the chronic and irreversible changes associated with high mortality and morbidity in humans [12,13]. Thus, hypoxia (acute or chronic) alone is not sufficient to cause severe pulmonary hypertension and/or chronic changes. Two hits models, as described a decade ago, continue to be investigated and provide insight into the nature of the lung vascular lesions and mechanisms of cardiac adaptation to an altered lung circulation. In thes animal models, a combination of VEGF [E Miller4]receptor blockade with Su5416[E Miller5] and chronic hypoxia has been shown to induce a pulmonary hypertension and severe pulmonary arteriopathy, including concentric neointimal and complex plexiform-like lesions; [13-15].
EC-SOD overexpression has been shown to attenuate, prevent, and reverse [16-19] PAH in previous studies in animals with PAH induced by hypoxia alone. However, these studies, have not examined EC-SOD in animals with chronic progressive PAH associated with advanced stage of vasculopathy. In our previous studies, we showed that EC-SOD is uniquely positioned to oppose the development of PAH because of its ability to catalyze the dismutation of superoxide radicals [20,21], increase NO bioavailability [22], and decrease the inflammatory response associated with oxidative stress [20,22], and increase VGEF expression [23]. In the current study, our overall goal was to evaluate the role of EC-SOD overexpression in preventing the progression of the pulmonary hypertension to its chronic and irreversible stages.
All animal experiments were approved by the Institutional Animal Care and Use Committee of the Feinstein Institute for Medical Research.
Animal model
We established the chronic progressive form of PAH by exposing male C57BL/6 mice (9-11wks old) to hypoxia 10% and intraperitoneal administration (20 mg/kg body weight per dose) of the vascular endothelial receptor (VEGF) blocker SU5416 (Sugen) at days 0, 7 and 14. Sugen was suspended within a mixture of 0.5% carboxy methyl cellulose sodium, 0.9% sodium chloride, 0.4% polysorbate 80, and 0.9% benzyl alcohol in deionized water.
Study design
Prophylactic study: Wild type C57B6 mice (WT) and transgenics (TG) expressing an additional copy of human EC-SOD, were studied as follows:
Group 1 (n=5): WT housed in room air
Group 2 (n= 5): TG housed in room air
Group 3 (n= 20): WT housed in hypoxia+ sugen.
Group 4 (n= 20): TG housed in hypoxia chamber + sugen.[E Miller6]
After 3 weeks of exposure, all animals were housed in room air. Five animals were euthanized and studied from each group at each study time point (3,7,11&14 weeks post exposure).
Therapeutic study: Chronic pulmonary hypertension was developed over 3 weeks as described above. After the exposure period, half of the animals were transfected with hEC-SOD/in vivo-jetPEI [E Miller7]complexes 50 µL/dose via intratracheal instillation under general anesthesia as described previously [24,25]. The remaining animals were not transfected. Both non transfected & transfected were then housed in room air and five animals/group were studied and euthanized at 7, 11 weeks post exposure.
Hemodynamic studies
RT Ventricular Pressure Assessment (N=5 mice/group): was evaluated at 3, 7, 11, 14 weeks post hypoxia exposure as described before [18]. Briefly, animals were anesthetized with ketamine 20 mg/kg/dose, a 26-gauge needle connected to a transducer was introduced into the right ventricle of the heart trans-diaphragmatically. Right ventricular systolic pressure (RVSP) was measured and recorded using a computerized hemodynamic recording system (HAEMODYN, Harvard Apparatus, MA, USA). The mice were then euthanized by exsanguination.
RV/S + LV ratio (N=5 mice/group): Hearts were removed, dissected and weighed for calculation of different of cardiac indices. The heart weights were be expressed as a ratio of the weight of the right ventricle to that of the septum plus left ventricle (RV/S + LV), or the right ventricle mass (Right ventricle / weight - RV/W), was expressed the ratio of the weight of the right ventricle to the weight of the animal [20].
Quantification of microvessel density and vascular wall thickness (N=5 mice/group): The left lung was perfused with 4% paraformaldehyde (PFA), inflated by infusion of 4% PFA at a constant pressure of 25 cm H2O through a cannula inserted in the trachea, fixed in 4% PFA overnight at 4°C and then embedded in OCT,[E Miller8] Subsequently 5-μm-thick sections were taken and stained with hematoxylin and eosin (H&E) Images of individual pulmonary arteries were captured using a digital camera, mounted on a light microscope, and linked to a computer. Microvessel density was quantified by counting the percentage area of positive pixels per image with at least 21 images per sample (5 animals, 3 samples per animal, and 7 sections per sample as described previously) [26].
Vascular wall thickness: was measured using a Zeiss Axiovert 200M light microscope – CCD camera AxioCam (mRm) color camera and expressed as the percentage of total vessel size. Percent wall thickness was calculated as (2 × wall thickness)/external diameter × 100% (8,10). External diameter and internal diameter of 50 alveolar vessels (with an external diameter of 100–200 μm) per animal were determined and recorded by an independent investigator blinded to the treatment regimen. The ratio of vessel wall area to total area (WA%) and the ratio of pulmonary arteriole wall thickness to vascular external diameter (WT%) were measured using Zeiss axial program of 3 random wall sections.
Histological studies (N=5 mice/group)
Lung morphometric was assessed on lungs from each group. Lung tissues were embedded in paraffin wax. Serial sections were stained with H&E The stained sections were analyzed using a Nikon Eclipse E400 microscope and images were obtained using a Nikon DS-Fi1 camera with 5-megapixel CCD.
Immunohistochemistry Studies (N=5 mice/group)
Immunohistochemical staining was performed with the Vectastain Universal Quick kit (Vector Laboratories, Burlingame, CA) (13), using antibodies to detect von Willebrand factor (Dako, Glostrup, Denmark), smooth muscle actin (Abcam, Cambridge, Mass), and VEGF, (Santa Cruz Biotechnology, CA).
Evaluations of nitric oxide synthase pathway, angiogenesis markers and inflammatory (N=5 mice/group)
Lung tissues were perfused and rinsed with phosphate-buffered saline (pH 7.4) containing heparin (2 unit/ml) to remove any red blood cells and clots and were then homogenized at 48oC. NO pathway analysis was performed in lung homogenates by Western blot analysis using i) EC-SOD antibody at 1:1,000 (Santa Cruz, Biotechnology, CA); ii) mouse monoclonal endothelial nitric oxide synthase (eNOS) at 1:1,000 (BD Transduction, San Jose, CA); iii) rabbit polyclonal phosphonitric oxide synthase (pNOS) at 1:1,000 (Abcam, Cambridge, Mass); iv)rabbit polyclonal sGC [E Miller9]at 1:1,000 (Cayman Chemical, Ann Arbor, Michigan). Assay of the angiogenesis markers sFlt-1 (sVEGFR-1), was performed using the Quantikine mouse soluble VEGFR1/Flt-1 Immunoassay Kit (R&D Systems, Minneapolis, MN), and cGMP measurements by enzyme immunoassay kit Cayman Chemical.
Western blot experiments (N=5 mice/group)
Control and experimental tissues were lysed in RIPA buffer containing 50 mM Tris·Cl (pH 7.5), 150 mM NaCl, 1 mM EDTA[E Miller10], 1% Nonidet P-40, 0.25% deoxycholate, 0.1% SDS, 1 protease inhibitor cocktail (Cocktail Set I, Calbiochem, San Diego, CA), 1 mM PMSF[E Miller11], and 0.2 mM sodium orthovanadate. The protein concentration was measured using the Bio-Rad Protein Assay (Pierce, Rockford, IL). Total protein lysates (20 g/lane)[E Miller12] were loaded on a 10% polyacrylamide gel (Bio-Rad, Hercules, CA) and transferred onto a nitrocellulose membrane using a Bio-Rad miniblot apparatus. Nitrocellulose membranes were processed for immunostaining with primary antibodies and subsequently with the appropriate horseradish peroxidase-labeled secondary antibodies. Blots were developed using a chemiluminescence detection kit (Pierce, Rockford, IL) and exposed to X-ray film (Eastman Kodak, Rochester, NY). Equal protein loading and protein transfer were confirmed by immunoblot analysis of B-actin protein (Santa Cruz Biotechnology, CA) on the same Western blots.
Statistics
Values in the figures are reported as mean ± SD of samples taken at least in triplicates. A one-way ANOVA with ‘Bonferroni's Multiple Comparison Test’, posttest was performed using GraphPad Prism version 5 for Windows, (GraphPad Software, La Jolla, CA, USA), and P values <0.05 were considered significant.
Prophylactic study
Our data showed that TG mice had significantly lower RVSP , RV/S + LV ratio and RV/W ratio at 3, 7 11 and 14 weeks compared with WTmice (P< 0.05) which showed chronic progressive pulmonary hypertension as shown by significantly elevated RVSP , RV/S + LV ratio and RV/W ratio (Figure 1).
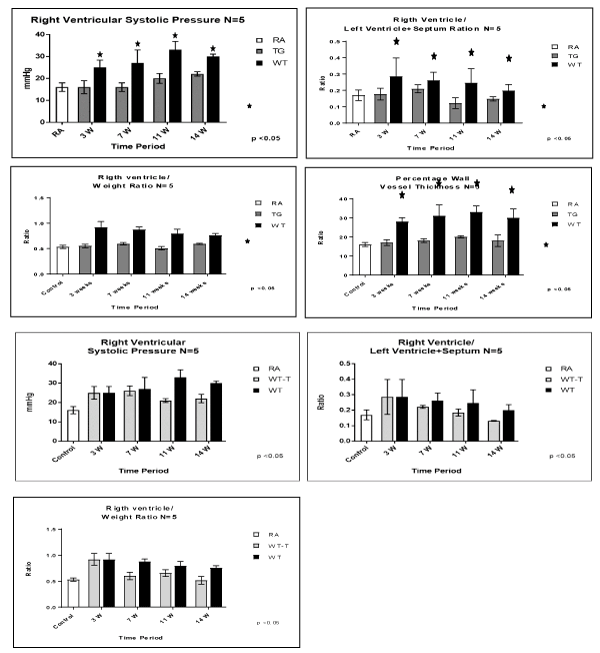
Figure 1. Evaluation of PAH model and Cardiac hypertrophy after expose mice to a fraction on inspirited oxygen 10% for 3 weeks, and injection of vascular endothelial growth factor (VEGF) receptor bloker Sugen (SU) 5416, once a 1 week x3 doses. Animals studies were done at 3, 7, 11 and 14 weeks post-exposure, among RA control, TG hypoxic and WT hypoxic groups. (A) right ventricular systolic pressure (RVSP), (B) right ventricular/Left ventricle + septum ratio, (C) right ventricle/body weight, (D) percentage wall vessel thickness (PWVT). Fig. 1B: Evaluation of PAH model and Cardiac hypertrophy after expose mice to a fraction on inspirited oxygen 10% for 3 weeks, and injection of vascular endothelial growth factor (VEGF) receptor bloker Sugen (SU) 5416, once a 1 week 3 doses. After Hypoxia haft of the animals were transfected and other haft not transfected. Animals studies were done at 3, 7, 11, 14 weeks post exposure, in. (A) right ventricular systolic pressure (RVSP), (B) rightventricular/left ventricule + septum ratio, (C) rigth ventricle/body weight. N/group=5 animals. *P<0.05
At the level of the lung parenchyma (Figure 2), TG mice showed mild inflammatory changes (increased number of interstitial cells) at 3 and 7 weeks, with improvement comparable to control mice at 11 and 14 weeks. Wild type animals showed progressive interstitial and peri-lymphocytic infiltrates and abnormal alveolarization. At the level of the pulmonary arteries (Figure 3), TG mice showed normal wall thickness, endothelial and adventitia (comparable to control mice) after 3 weeks and maintained its normal morphology at 11 weeks post exposure. However wild type mice showed significant, progressive muscular wall thickness over the time without regression at the end 11 and 14 weeks. Angiogenesis markers, (including alpha-smooth muscle cell, VEGF , VWF and soluble family member like tyrosine kinase type 1 (sFlt-1)) (Figures 4-7) and Inflammatory markers (Figure 8) (IL1, IL 6 and TNF-alpha) were increased in WT hypoxic mice and remained increased over the 14 weeks compared with TG mice and control groups (P<0.05). Overexpression of EC-SOD and increased bioavailability of NO was studied by assessment of both cyclic Guanosine Monophosphate (cGMP) and soluble Guanylate Cyclase (sGC), which were decreased at 3 and 14 weeks among WT hypoxic animals compared to those in room air (<0.05) (Figure 9). In hypoxic TG animals, overexpression of EC-SOD had a significant protective action on the NO pathway. All hypoxic TG animals showed similarlevels of both cGMP and sGC compared to the RA control group. But was significantly higher than WT hypoxic animals (P<0.05).
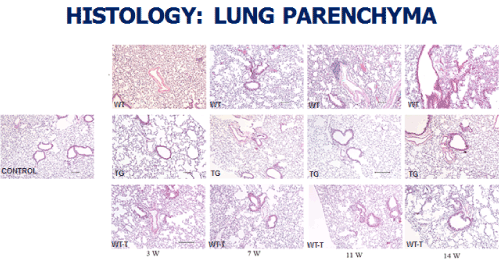
Figure 2. Histopathologycal studies of Lung Parenchyma in all four studied groups: RA control, Hypoxic WT, Hypoxic Transgenic, and Hypoxic transfected groups
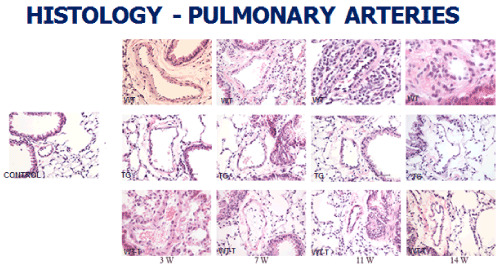
Figure 3. Histopathologycal studies of lung pulmonary arteries in all four studied groups: RA control, Hypoxic WT, Hypoxic Transgenic, and Hypoxic transfected groups
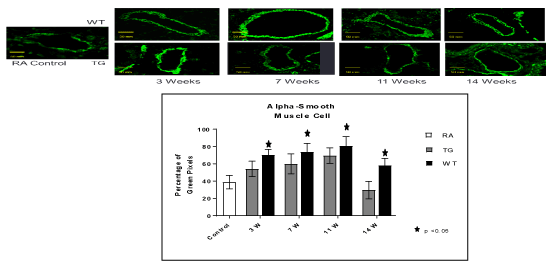
Figure 4. Histo-chemistry studies of pulmonary arteries smooth muscles in RA, Hypoxic WT and hypoxic TG groups. A: Immunostaining for alpha smooth muscle antibody (α-SMA). B: Table shows a quantification of α-SMA pixels percentage. N/group=5 animals. *P<0.05
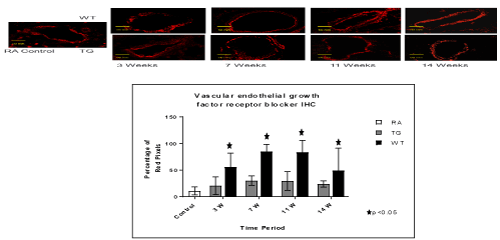
Figure 5. Histo-chemistry studies of pulmonary arteries smooth muscles in RA, Hypoxic WT and hypoxic TG groups. A: Immunostaining for vascular endothelial growth factor (VEGF). B: Table shows a quantification of VGEF pixels percentage. N/group=5 animals. *P<0.05
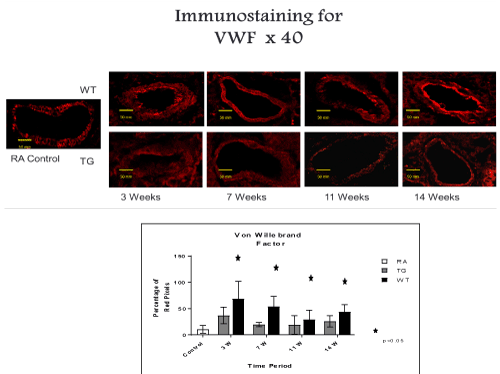
Figure 6. Histo-chemistry studies of pulmonary arteries smooth muscles in RA, Hypoxic WT and hypoxic TG groups. A: Immunostaining for Von Willebrand factor (vWF). B: Table shows a quantification of vWF pixels percentage. N/group=5 animals. *P<0.05
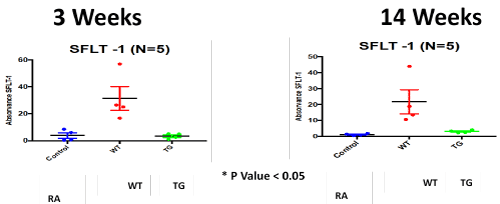
Figure 7. ELISA Assay of angiogenesis marker; soluble family member like tyrosine kinase type 1 (sFlt-1); in RA, Hypoxic WT and hypoxic TG groups at 3 and 14 weeks post-exposure. N/group=5 animals. *P<0.05
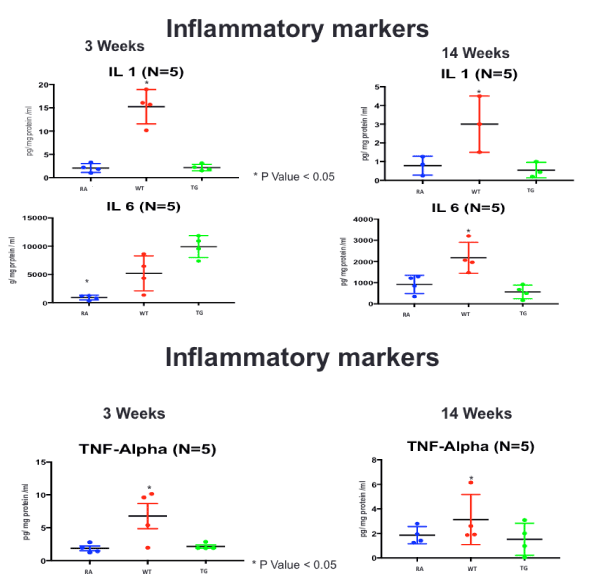
Figure 8. ELISA Assay of inflammatory markers (IL-1, IL-6 and TNF-a), at 3 and 14 weeks post-exposure, in RA, Hypoxic WT and hypoxic TG groups N/group=5 animals. *P<0.05
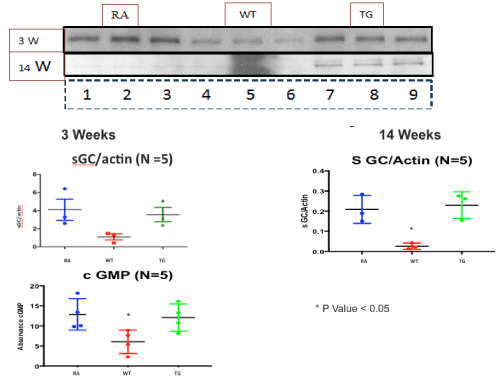
Figure 9. Western blot assay of NO pathway at 3 and 14 weeks post-exposure, in RA, Hypoxic WT and hypoxic TG groups. A: Assay of PeNOS/eNOS ratio. B: Assay of Soluble guanylate cyclase (sGC). N/group=5 animals. *P<0.05
Therapeutic effect study
Animal transfection using EC-SOD plasmid showed a significant therapeutic role by preventing the progression to chronic pulmonary changes and improving the pulmonary hypertension status compared to WT hypoxic animals (P<0.05). Transfected mice (WT-T) showed decreased right ventricular systolic pressure, right ventricle mass RV/W ratio and right ventricle/weight ratio (cardiac mass index) (RV/W) compared with WT mice at 7 weeks with significant reductions in these parameters at 11 and 14 weeks post-exposure,compared to the RA control group (Figure 1). At the level of the pulmonary parenchyma, transfected mice showed attenuation of the inflammatory changes at 7 weeks with continuous improvement at 11 and 14 weeks without the development of chronic pulmonary changes, which was obvious and significant among the non-treated WT mice (Figure 2). At the level of the pulmonary arteries, post-transfected, treated WT hypoxic mice group, showed normal pulmonary artery morphology at 7, 11, 14 weeks post-exposure, compared with the control RA group. WT hypoxic non-treated group showed a progressive increase in wall thickness, muscularization and endothelial proliferation (Figure 3).
In this study, we report an intervention that can prevent and treat pathological changes induced by chronic hypoxia and SU5416[E Miller13] in an adult mouse model with PAH. Both transgenic mice (with overexpression of EC-SOD), and transfected mice (with hEC-SOD/in vivo-jetPEI complex via intra-tracheal instillation[E Miller14]), were protected and experienced both normalization of pulmonary pressures and attenuation of chronic pulmonary changes in both pulmonary vasculature and lung parenchyma.
PAH is associated with a hig[E Miller15]h morbidity and mortality in all age groups from neonates to the elder population. Although, the prophylactic role of EC-SOD to prevent PAH induced by hypoxia has previously been shown (16–20), this is the first study to show the ability of EC-SOD to prevent and reverse the progression of PAH and chronic pulmonary changes in the more severe Su5416/chronic hypoxia murine model.
Abe, et al. (2010) showed that Su5416/chronic hypoxia is a particularly robust model of pulmonary hypertension [14]. Plexiform lesion is a characteristic structure of the pulmonary arteriopathy in severe PAH and was found in the SuHx model at 14 weeks (11 weeks on normoxia) in addition to the other stages of vasculopathy (1-3) [14]. Vitali, et al. (2014) reported that weekly SU5416 combined with 3 weeks of hypoxia followed for an additional 10 weeks in normoxia yielded an enhancement of pulmonary hypertensive and structural responses causes a more profound PAH phenotype in mice than hypoxia alone, but with very few angio-obliterative lesions were found at the 10-week follow-up time point in SuHx mouse lungs [27]. Recently, enhanced glycolysis was found to be associated with the pulmonary vasculature and tissue matrix in the SuHx mouse model [28]. In our mouse model with SU5416 combined with 3 weeks of hypoxia, a progressive and profound form of PAH phenotype was noted in comparison to hypoxia alone and similar to that observed in human severe PAH [13]. In our model, there were significant arteriopathy changes including concentric laminar neointimal proliferation and irreversible changes, but patterns of complex plexiform lesions were not seen at 11, 14 weeks follow-up time points, in agreement with other studies [27]. Most of the studies agree that the SuHx mouse model can be an important tool in unravelling the multifactorial pathogenic mechanisms associated with PAH [29].
TG animals that were treated with Su5416 and exposed to hypoxia showed normal alveolar and parenchymal architecture, in comparison to WT treated ganimals and controls housed in room air (Figures 2 and 3). Immunostaining for α-SMA (Figure 4), VEGF (Figure 5) and Von Willebrand factor(vWF) (Figure 6), showed minimal medial wall proliferation, with no progression to chronic changes (Figures 2 and 3). Interestingly, among WT mice that were initially exposed to Sug5416/hypoxia x3 weeks (to establish the disease), and were then transfected with EC-SOD (Transfected group), showed significant evidence of reversial of their established parenchymal pathological changes, with marked improvement and resolution of the inflammation at the 11 and 14 weeks’ time points (Figure 2). At the pulmonary arterial level, the marked muscular wall thickness, muscularization and endothelial proliferation occurred after exposure (3 weeks) and started to regress markedly at 11, 14 weeks follow-up time points (Figure 3).
In parallel to the inflammation and vasculopathy findings, arterial pulmonary pressure measurements, right ventricular hypertrophy and vascular wall thickness showed similar trends among studied groups. TG hypoxic mice showed normal limits of those parameters, post exposure compared to hypoxic and control groups (Figure 1). While the WT hypoxic group, showed a significant increase of RVP and right ventricle hypertrophy post-exposure, transfection of EC-SOD, started to show progressive reduction of RVP and right cardiac hypertrophy at 7, 11 & 14 weeks post-exposure time points upon transfection with EC-SOD in comparison to non-transfected group which showed progressive increase of RVP (Figure 1). These data support both the prophylactic and therapeutic role of EC-SOD in preventing both the initiation, and the progression of the pathological changes including vascular remodeling triggered.
In our model, we showed significant elevation of inflammatory markers mainly; IL-B1, IL-6 and TNF-α; in accordance with previously described findings in humans with idiopathic and familial severe pulmonary hypertension, and which correlate with the worst outcome [30]. Experimental evidence in hypoxia-induced pulmonary hypertension has shown diverse evidence with increase IL-1B in monocrotaline animal model, hypoxic sugen rat model, but no such findings were reported in hypoxic mouse models [30,31].
We have demonstrated that sFlt-1 expression in wild type mice was up-regulated under hypoxia + sugen model and was normalized in hypoxic transgenic mice and control, in accordance with what demonstrated by Xiong, et al. [32]. VEGF-A overexpression has been shown to protect against pulmonary hypertension in monocrotaline and hypoxia animal models [33,34]. sFlt-1-mediated VEGF-A inhibition has previously been proposed to contribute to PAH pathology [35]. sFlt-1 infusions have furthermore been shown to elevate blood pressure in mice by augmenting endothelin-1-mediated vasoconstriction [36] Our results do not agree with other studies that showed down regulation of sFlt-1 or normal levels [37-39]. The discrepancy between the conclusions of previous studies and the current study, may be due to the difference between different types of microvasculature, acute vs chronic different stressors and different hypoxia models [40].
Despite no change in total lung EC-SOD content, the selective depletion of SMC-derived pulmonary vascular EC-SOD worsened chronic hypoxic PAH [34]. The augmented PAH was shown by measures of hemodynamics as well as analysis of pulmonary vascular remodeling. In this model, enhanced PAH in the setting of low vascular EC-SOD was due to impaired cGMP-dependent signaling, indicating that impaired eNOS activity could account for the exaggerated PAH in the mice with insufficient pulmonary vascular EC-SOD [41]. Our studies demonstrate the necessity to consider the compartmentalization of oxidant production and antioxidant activity and the importance of extracellular oxidant/antioxidant imbalance on specific redox-sensitive signaling pathways [42]. In our model, studying, nitric oxide synthase pathway showed significant reduction of p-eNOS in WT hypoxic mice, with elevated values in hypoxic transgenic mice that become equivalent to control animals at 7 weeks post-exposure (Figure 9).
We recognize a few limitations with this study. The mouse model of chronic hypoxia-induced PH, while reproducible and well-characterized, does not reflect the marked severe degree of pulmonary vascular remodeling or inflammation that is observed in human disease. Although we demonstrated worsened medial and adventitial wall remodeling, this model does not enable us to assess the impact of insufficient vascular EC-SOD on intimal changes, even though this is an important feature of pulmonary artery remodeling in the clinical setting. Another limitation is that, since we used lung tissue to evaluate the sGC-cGMP-PDE5 axis, we may not have detected more subtle changes in pulmonary artery expression or activity. The lung changes may not fully reflect the changes observed in the pulmonary artery, as has been shown in large animal models or human patients [10,11]. In the future, we plan to test more robust models of PAH associated with remodeling in all three layers of the vessel wall as well as inflammation.
In conclusion, we were able to establish a chronic PAH in adult mice. EC-SOD has a marked and a significant role in ameliorating and attenuating pulmonary vasculopathy and subsequently the progression of chronic PAH induced by hypoxia. Overexpression of EC-SOD leads to regression of the inflammatory process associated with vascular remodel in chronic model of pulmonary hypertension. [E Miller16]
- Steinhorn RH (2010) Neonatal pulmonary hypertension. Pediatr Crit Care Med 11: 79-84.
- Walsh-Sukys MC, Tyson JE, Wright LL, Bauer CR, Korones SB, et al. (2000) Persistent pulmonary hypertension of the newborn in the era before nitric oxide: practice variation and outcomes. Pediatrics 105: 14-20. [Crossref]
- Aikio O, Metsola J, Vuolteenaho R (2012) Transient defect in nitric oxide generation after rupture of fetal membranes and responsiveness to inhaled nitric oxide in very preterm infants with hypoxic respiratory failure. J Pediatr 161: 397-403. [Crossref]
- Khemani E, McElhinney DB, Rhein L (2007) Pulmonary artery hypertension in formerly premature infants with bronchopulmonary dysplasia: clinical features and outcomes in the surfactant era. Pediatrics 120: 1260-1269.
- Farquhar M, Fitzgerald DA (2010) Pulmonary hypertension in chronic neonatal lung disease. Paediatr Respir Rev 11: 149-153.
- Stenmark KR, Fagan KA, Frid MG (2006) Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res 99: 675-691. [Crossref]
- Humbert M, Sitbon O, Simonneau G (2004) Treatment of pulmonary arterial hypertension. N Engl J Med 351: 1425-1436. [Crossref]
- Bland RD (2005) Neonatal chronic lung disease in the post-surfactant era. Biol Neonate 88: 181-191. [Crossref]
- Henderson-Smart DJ (2006) Prenatal predictors of chronic lung disease in very preterm infants. Arch Dis Child Fetal Neonatal Ed 91: 40-54.
- Tuder RM, Yun JH, Bhunia A, Fijalkowska I (2007) Hypoxia and chronic lung disease. J Mol Med (Berl) 85: 1317-1324. [Crossref]
- Deruelle P, Grover TR, Abman SH (2005) Pulmonary vascular effects of nitric oxide-cGMP augmentation in a model of chronic pulmonary hypertension in fetal and neonatal sheep. Am J Physiol Lung Cell Mol Physiol 289: 798–806.
- Li FH, Xia W, Li AW, Zhao CF, Sun RP (2007) Inhibition of rho kinase attenuates high flow induced pulmonary hypertension in rats. Chin Med J (Engl) 120: 22-29. [Crossref]
- Voelkel NF, Tuder RM (2000) Hypoxia-induced pulmonary vascular remodeling: a model for what human disease? J Clin Invest 106: 733-738.
- Abe K, Toba M, Alzoubi A (2010) Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation 121: 2747-2754.
- Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, et al. (2001) Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. Faseb J 15: 427-438.
- Nicolls MR, Mizuno S, Taraseviciene-Stewart L (2012) New models of pulmonary hypertension based on VEGF receptor blockade-induced endothelial cell apoptosis. Pulm Circ 2: 434-442.
- Mizuno S, Farkas L, Al Husseini A, Farkas D, Gomez-Arroyo J, et al. (2012) Severe pulmonary arterial hypertension induced by SU5416 and ovalbumin immunization. Am J Respir Cell Mol Biol 47: 679-687. [Crossref]
- Nozik-Grayck E (2008) Lung EC-SOD overexpression attenuates hypoxic induction of Egr-1and chronic hypoxic pulmonary vascular remodeling. Am J Physiol Lung Cell Mol Physiol 295: 422-430.
- Ahmed MN, Zhang Y, Codipilly C, Zaghloul N, Patel D, et al. (2012) Extracellular superoxide dismutase overexpression can reverse the course of hypoxia-induced pulmonary hypertension. Mol Med 18: 38-46.
- Elmedal B, de Dam MY, Mulvany MJ, Simonsen U (2004) The superoxide dismutase mimetic, tempol, blunts right ventricular hypertrophy in chronic hypoxic rats. Br J Pharmacol 141: 105-113. [Crossref]
- Ahmed MN, Codipilly C, Hogg A, Auten RL (2011) The protective effects of overexpression of extracellular superoxide dismutase on nitric oxide bioavailability in the lung after exposure to hyperoxia stress. Exp Lung Res 37: 10-17.
- Perveen S, Patel H, Arif A, Younis S, Codipilly CN, et al. (2012) Role of EC-SOD overexpression in preserving pulmonary angiogenesis inhibited by oxidative stress. Plos One 7: e51945.
- Kamezaki F, Tasaki H, Yamashita K, Tsutsui M, Koide S, et al. (2008) Gene transfer of extracellular superoxide dismutase ameliorates pulmonary hypertension in rats. Am J Respir Crit Care Med 177: 219-226.
- Ahmed MN, Masson SN, Whorton MA, Auten RL (2002) Gene transfer of human extracellular superoxide dismutase (hEC-SOD) cDNA using polyethylenimine (PEI) carrier express hEC-SOD in lung epithelium of adult mice after 7 days after treatment. Pediatr Res 1: 1-10.
- Morishita T, Tsutsui M, Shimokawa H, Sabanai K, Tasaki H, et al. (2005) Nephrogenic diabetes insipidus in mice lacking all nitric oxide synthase isoforms. Proc Natl Acad Sci USA 102: 10616-10621. [Crossref]
- Vitali SH, Hansmann G, Rose C, Fernandez‐Gonzalez A, Scheid A, et al. (2014) The Sugen 5416/hypoxia mouse model of pulmonary hypertension revisited: long‐term follow‐up. Pulm Circ 4: 619-629.
- Penumatsa KC, Toksoz D, Warburton RR, Kharnaf M, Preston IR, et al. (2017) Transglutaminase 2 in pulmonary and cardiac tissue remodeling in experimental pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 313: 752-762. [Crossref]
- Penumatsa K, Warburton R, Hill N, Fanburg B (2019) CrossTalk proposal: The mouse SuHx model is a good model of pulmonary arterial hypertension. J of Physiol 4: 975-977. [Crossref]
- Soon E, Holmes AM, Treacy CM, Doughty NJ, Southgate L, et al. (2010) Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 122: 920-927.
- Voelkel NF, Tuder RM, Bridges J, Arend WP (1994) Interleukin-1 receptor antagonist treatment reduces pulmonary hypertension generated in rats by monocrotaline. Am J Respir Cell Mol Biol 11: 664-675. [Crossref]
- Xiong Y, Liebermann DA, Tront JS, Holtzman EJ, Huang Y, et al. (2009) Gadd45a stress signaling regulates sFlt-1 expression in pre-eclampsia. J Cell Physiol 220: 632-639.
- Campbell AI, Zhao Y, Sandhu R, Stewart DJ (2001) Cell-based gene transfer of vascular endothelial growth factor attenuates monocrotaline-induced pulmonary hypertension. Circulation 104: 2242-2248.
- Partovian C, Adnot S, Raffestin B, Louzier V, Levame M, et al. (2000) Adenovirus-mediated lung vascular endothelial growth factor overexpression protects against hypoxic pulmonary hypertension in rats. Am J Resp Cell Mol Biol 23: 762-771. [Crossref]
- Voelkel NF, Gomez AJ (2014) The role of vascular endothelial growth factor in pulmonary arterial hypertension. The angiogenesis paradox. Am J Resp Cell Mol Biol 51: 474-484.
- Amraoui F, Spijkers L, Hassani LH, Vogt L, Van der Post J, et al. (2014) SFlt-1 elevates blood pressure by augmenting endothelin-1-mediated vasoconstriction in mice. PloS One 9: e91897.
- Ikeda T (2011) Hypoxia down-regulates sFlt-1 (sVEGFR-1) expression in human microvascular endothelial cells by a mechanism involving mRNA alternative processing. Biochem J 436: 399-407.
- Kubo T, Wada T, Yamaguchi Y, Shimizu A, Handa H (2006) Knock-down of kDa subunit of cleavage factor Im in Hela cells alters alternative polyadenylation within-UTRs. Nucleic Acids Res 34: 6264-6271.
- Wu FTH, Stefanini MO, Gabhann FM, Kontos CD, Annex BH, et al. (2010) A systems biology perspective on sVEGFR1: its biological function, pathogenic role and therapeutic use. J Cell Mol Med 14: 528-552.
- Xiong Y, Liebermann DA, Tront JS, Holtzman EJ, Huang Y, et al. (2009) Stress signaling regulates sFlt-1 expression in preeclampsia. J Cell Physiol 220: 632-639.
- Nozik-Grayck E, Woods C, Taylor JM, Benninger RK, Johnson RD, et al. (2014) Selective depletion of vascular EC-SOD augments chronic hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 307: L868-876. [Crossref]
- Tuder RM, Cool CD, Geraci MW, Wang J, Abman SH, et al. (1999) Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med 159: 1925-1932.
- Wen FQ, Watanabe K, Tanaka H, Yoshida M (1997) Cytokines and lipopolysaccharide enhance basal and thrombin-stimulated production of PGI2 by cultured human pulmonary artery smooth muscle cells. Prostaglandins Leukot Essent Fatty Acids 56: 185-192. [Crossref]