Noonan syndrome (NS) is a genetic disorder caused by germline mutations in RAS/MAPK pathway. Predisposition to juvenile myelomonocytic leukemia-like myeloproliferative disorder is well known. Only rare cases of isolated neonatal thrombocytopenia in patients with NS have been described. We report an unusual case of patient with NS and neonatal presentation of severe thrombocytopenia, consistent with diagnosis of congenital amegakaryocytic thrombocytopenia (CAMT) but without MPL (thrombopoietin receptor) gene mutation, who developed bone marrow failure at the age of 4 months. A review of the literature identified four patients with NS and isolated thrombocytopenia but no other case of infant bone marrow failure was previously described. We think that CAMT like clinical picture is rare but nonrandom observation in NS patients with risk of development of bone marrow failure
noonan syndrome, congenital amegakaryocytic thrombocytopenia, bone marrow failure
Abbreviations
NS: Noonan syndrome; CAMT: congenital amegakaryocytic thrombocytopenia; JMML: juvenile myelomonocytic leukemia; MPD: myeloproliferative disorder; HSCT: hematopoetic stem cell transplantation; GCSF: granulocyte colony stimulating factor; ATRUS: amegakaryocytic thrombocytopenia with radioulnar synostosis; TAR: thrombocytopenia with absent radii
Noonan syndrome (NS) is a genetic disorder with multiple organ defects most commonly transferred in an autosomal dominant pattern. It is caused by a heterozygous germline mutation in genes of the RAS/MAPK pathway in the majority of cases. Among the most common features of NS are a distinct face, pulmonary stenosis, various skeletal malformation, cryptorchidism and hematological problems. Typical hematological abnormalities in NS include clotting factor deficiencies, von Willebrand disease, thrombocytopenia and abnormal platelet function [1]. Infants with NS are predisposed to develop juvenile myelomonocytic leukemia (JMML) or a JMML-like myeloproliferative disorder (MPD). PTPN11 is the most commonly involved gene in NS; about 44% of sporadic JMML cases display acquired somatic PTPN11 mutations [2]. About 5.6 % (34 out of 641) of PTPN11 mutated NS patients developed MPD or JMML [3]. Predisposition to JMML and JMML-like MPD is well-known in the literature. However, only rare cases of isolated severe neonatal thrombocytopenia have been described some of which were classified as congenital amegakaryocytic thrombocytopenia (CAMT) [4-6]. CAMT is an extremely rare inherited bone marrow failure syndrome characterized by severe congenital thrombocytopenia due to ineffective megakaryopoiesis, and by exclusion of other congenital syndromes associated with neonatal thrombocytopenia. Most cases of CAMT are caused by defective expression or function of the thrombopoietin receptor due to homozygous or compound heterozygous mutations in the MPL gene [7]. However, a small number of patients have been described in literature [8-10] who fulfill the clinical and laboratory criteria of CAMT without an identified mutation in the MPL gene. The majority of CAMT patients eventually develop bone marrow failure; isolated thrombocytopenia persisting over years without development of pancytopenia is extremely rare [7].
Case description
We report an unusual case of patient with NS manifesting as severe neonatal thrombocytopenia with progression to bone marrow failure over several months. The patient was a girl of nonconsanguineous young parents delivered at the 36th week of gestation with Apgar scores of 7, 7 and 8 (at 1, 5 and 10 minutes). Birthweight was 2450 g (10th percentile), length 43 cm (3rd percentile) and head circumferences 32 cm (10th percentile). She was noted to have prominent petechiae, ecchymoses and dysmorphic facies. Echocardiography revealed obstructive hypertrophic cardiomyopathy, pulmonary stenosis, and atrial and ventricular septal defects. She had no apparent skeletal abnormality, but no extremity X-ray was performed, her karyotype was normal. She also had marked thrombocytopenia (platelet count of 18x10E9/l), leukocytosis (white blood cell count of 39,2x10E9/l, differential count with lymphocytes 62%, neutrophils 31%, eosinophils 2%, monocytes 4% and plasmatic cells 1%) and anemia (erythrocyte count of 2,0x10E12/l and hemoglobin of 79 g/l). The cause of anemia was thoroughly evaluated and it was concluded that anemia was purely due to significant skin and mucosal hemorrhagic diathesis and enterorhagia. She had no hepatomegaly or splenomegaly. The prothrombin time (PT), activated partial thromboplastin time (APTT) and fibrinogen remained within normal limits. Mother's platelet count was normal, evaluation for neonatal alloimmune thrombocytopenia (NAIT) was performed with negative result. The patient had no signs of infection.
Bone marrow aspiration revealed slightly reduced cellularity with normal myeloid and erythroid maturation and a substantial reduction in megakaryocytes. Bone marrow biopsy demonstrated cellularity of 90%, conspicuous hemosiderosis, and only mild reticulin fibrosis. In trilinear hematopoiesis immunohistochemistry indicated a large proportion of B-cell precursors, constituting nearly 40% of total cellularity. There were maturing chloroacetate esterase positive elements of the granulocytic series without an increase in CD34-positive blasts. Erythroid cells formed irregular clusters, but a refractory cytopenia (RC) pattern was not present. There were only a few small megakaryocytes with hypolobated to oval nuclei, and CD61 immunohistochemistry did not reveal any micro-megakaryocytes (Figure 1a). Flow cytometric analysis of hematopoietic progenitors revealed a slightly decreased number of CD34+ cells (1.7%) and normal expression of MPL on early CD34+CD38lo progenitors, excluding an MPL defect as the cause of the thrombocytopenia. Thrombopoetin (THPO) level was elevated (507 +/-6 pg/ml).
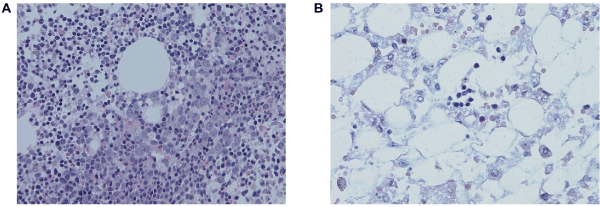
Figure 1. (A) Bone marrow biopsy performed at 3 months of age with trilinear hematopoiesis and megakaryocytes reduction, (B) severely hypocellular bone marrow sample from autopsy
She was referred to our institution at 3 months of age, at which time she was dependent on weekly platelet transfusions and intermittent erythrocyte transfusions, she had normal number of granulocytes and reticulocyte count 3.58%. The facial phenotype as well as the cardiac findings were typical for NS. Subsequently, mutation analysis of NS candidate genes (PTPN11 exons 1-15; SOS1 exons 3-14, 16; RAF1 exons 7, 12, 14, 17; RIT1 exons 2-6) was conducted and revealed a heterozygous missense variant c.1504T > G in PTPN11, resulting in the p.Ser502Ala amino acid substitution, previously described as causative for NS [11].
At the age of 4 months, she developed neutropenia with an absolute neutrophil count <0.2x10E9/l, her anemia progressed with reticulocyte count 0.36%. Repeat bone marrow aspiration was consistent with a diagnosis of bone marrow failure with low cellularity, dominant population of lymphocytes (66.4%), no megakaryocytes and reduced myeloid (16.4%) and erythroid precursors (10.0%). Extensive microbiological screening excluded infectious etiology. Flow cytometry showed significantly reduced CD34+ (0.23%) and CD117+ (0.18%) cells. Conventional cytogenetic confirmed normal karyotype (46, XX) in 20 mitosis with no abnormalities of chromosome 7,8,9 done by fluorescence in situ hybridization (FISH). Hematopoetic stem cell transplantation (HSCT) was considered, but during the next weeks, her cardiac disease progressed and she was no longer a candidate for HSCT. Echocardiography demonstrated pulmonary valvar stenosis, small atrial and ventricular septal defects, severe obstruction of the left ventricular outflow tract, and extreme biventricular hypertrophy. She underwent cardiac surgery at the age of 7.5 months (myectomy of the left ventricular outflow tract obstruction and valvotomy of the pulmonary valve). The surgical procedure was palliative because extreme biventricular hypertrophy as in obstructive cardiomyopathy could not be relieved. Her postoperative course was complicated: she developed third-degree atrioventricular block, right ventricular obstruction progressed to severe heart failure.
Unfortunately, during this time, the pancytopenia also progressed and daily administration of granulocyte colony stimulating factor (GCSF) had no effect and she was platelet and red blood cells transfusion-dependent. At this point, her cardiac disease was not surgically correctable. After discussion with her parents, it was decided that palliative care was the only appropriate course. She died 30 days after surgery due to cardiac failure with severe persistent pancytopenia. On autopsy, the bone marrow was severely hypocellular with stromal edema, hemosiderosis, and focal mild reticulin fibrosis, and dominated by reactive mononuclear cells. No megakaryocytes were found, but immunohistochemistry was not performed (Figure 1b).
Except our patient a review of the literature identified only four other cases of severe congenital thrombocytopenia in NS patients [1,4-6]. All patients including our are summarized in Table 1. The diagnosis of amegakaryocytic thrombocytopenia in Patient No. 1 is controversial, since he experienced early platelet recovery without any treatment [4]. Patient No. 2 was diagnosed in neonatal period and underwent HSCT with diagnosis of CAMT from his HLA-identical mother at the age of 2.8 years [6]. He was diagnosed with Noonan syndrome later and he is described as part of a larger cohort of patients transplanted for CAMT [12]. Patient No. 3 suffered from bleeding diathesis with severe thrombocytopenia from birth (platelet count of 5x10E9/l). Cytogenetic analysis identified PTPN11 gene mutation. The diagnosis of NAIT was excluded, he had no signs of infection. Bone marrow biopsy and aspiration, performed in the neonatal period, revealed normal haematopoiesis not consistent with CAMT and various other diseases were also excluded (Fanconi anemia, thrombocytopenia absent radius syndrome, familiar thrombocytopenia). He was platelet transfusion-dependent until 6 months of age. Administration of immunoglobulin had no effect on platelet count. By the age of 12 months, a mild spontaneous increase in the platelet count had been observed (20-30x10E9/l). No later bone marrow aspiration was reported and the mechanism of thrombocytopenia was not clarified [1]. Patient No. 4 also presented with bleeding diathesis from birth (platelet count of 8x10E9/l). Bone marrow biopsy and aspiration revealed normal myeloid and erythroid maturation with scant megakaryocytes. The plasma TPO concentration was very high (> 1000 pg/ml) and no MPL mutation was revealed. PTPN11 mutation confirmed diagnosis of NS. He was platelet transfusion-dependent until 14 months of age; by 20 months, his platelet count had stabilized around 20x10E9/l. Administration of eltrombopag (an oral agonist of the TPO receptor) had no effect [5].
Table 1. Characteristics of Noonan syndrome patients with severe congenital thrombocytopenia
Patient No. |
Sex/age |
Platelets
at diagnosis |
THPO level
(pg/ml) |
Mpl gene
mutational analysis |
Noonan sy gene analysis |
Outcome |
|
Evans, et al. [4]
|
Boy/neonatal manifestation |
31x10E9/l |
Not done |
Not done |
Not done |
Absence of megakaryocytes in bone marrow, thrombocytes platelets normalized at 7 weeks of age. |
|
Ballmaier, et al. [6,7] |
Boy/neonatal manifestation |
unknown |
2436 |
Normal |
PTPN11 (c.218C>T in exon 3) |
Alive with good marrow function after HSCT. |
Nunes, et al. [1] |
Boy/neonatal manifestation |
5x10E9/l |
Not done |
Not done |
PTPN11 (c.218C>T in exon 3)
|
At 12 months of age stable thrombocytopenia.
No effect of IVIG. |
Christensen, et al. [5] |
Boy/neonatal manifestation |
8x10E9/l |
> 1000 |
Normal |
PTPN11 (c.218C>T in exon 3) |
At 20 months of age stable thrombocytopenia.
No effect of eltrombopag. |
5
[Described in the paper) |
Girl/neonatal manifestation |
18x10E9/l |
507 (at age 3 months)
|
Normal |
PTPN11 (c.1504T>G in exon 13) |
Bone marrow failure at 4 months of age.
No effect of G-CSF. |
THPO: thrombopoietin, IVIG: intravenous immunoglobulins, HSCT: haematopoetic stem cells transplantation, G-CSF: granulocyte colony stimulating factor
In summary 3 of 4 published NS patients had a severe isolated thrombocytopenia lasting for many months and 1 of them was transplanted for CAMT. Thrombopoetin levels were examined in 2 of 4 patients, both were high and none of them had MPL mutation.
The mechanism of severe congenital thrombocytopenia without JMML or JMML like MPD in NS patients is unknown, in our opinion it is very rare, but nonrandom observation. Our patient appears to be unique because of the early development of irreversible bone marrow failure after an initial CAMT-like period. Marrow findings and significantly elevated plasma thrombopoietin (THPO) level were consistent with diagnosis of CAMT in some patients (No. 2, 4, 5) but no mutation in the MPL gene was identified for these patients. In this cohort, four of the five NS patients had PTPN11 mutations and one patient did not undergo genetic diagnosis. There is no evidence of genotype-phenotype correlation, further genetic testing is necessary to reveal potential additional genetic abnormalities.
Although NS patients are usually considered to be predisposed to JMML or JMML-like myeloproliferative disorder, they may also be at risk for CAMT-like congenital thrombocytopenia, followed by bone marrow failure, as demonstrated by our patient’s clinical course. In this situation, early haematopoietic stem cell transplantation is likely to be the only curative therapy. The mechanism of congenital thrombocytopenia and bone marrow failure in NS is not understood and requires further investigation.
The authors declare that there is no conflict of interest.
The authors thank Amy Brown for critical reading of manuscript.
- Nunes P, Aguilar S, Prado SN (2012) Severe congenital thrombocytopaenia-first clinical manifestation of Noonan syndrome. BMJ Case Reports pp: 2-5.
- Pérez B, Kosmider O, Cassinat B (2010) Genetic typing of CBL, ASXL1, RUNX1, TET2 and JAK2 in juvenile myelomonocytic leukaemia reveals a genetic profile distinct from chronic myelomonocytic leukaemia. Br J of Haematol 151: 460-468.
- Strullu M, Caye A, Lachenaud J, Cassinat B, Gazal S, et al. (2014) Juvenile myelomonocytic leukaemia and Noonan syndrome. J Med Genet 51: 689-697. [Crossref]
- Evans DGR, Lonsdale RN, Patton MA (1991) Cutaneous lymphangioma and amegakaryocytic thrombocytopenia in Noonan syndrome. Clinical Genetics 39: 228-232.
- Christensen RD, Yaish HM, Leon EL (2013) De novo T73I mutation in PTPN11 in a neonate with severe and prolonged congenital thrombocytopenia and Noonan syndrome. Neonatology 104: 1-5.
- Ballmaier M, Germeshausen M, Schulze H (2001) C-Mpl mutations are the cause of congenital amegakaryocytic thrombocytopenia. Blood 97: 139-146.
- Ballmaier M, Germeshausen M (2009) Advances in the understanding of congenital amegakaryocytic thrombocytopenia. Br J Haematol 146: 3-16.
- Van den Oudenrijn S, Bruin M, Folman CC (2000) Mutations in the thrombopoietin receptor, mpl, in children with congenital amegakaryocytic thrombocytopenia. Br J Haematol 110: 441-448.
- Bastila Eizaguirre M, Perreda Vicandi A, Pujana Zaldegui I (2008) Congenital amegakaryocytic thrombocytopenia in a 12 years old boy with no signs of pancytopenia: molecular analysis. Anales de Pediatria 68: 353-356.
- Kanaji S, Kanaji T, Migita M (2008) Characterization of a patient with atypical amegakaryocytic thrombocytopenia. European Journal of Hematology 80: 361-364.
- Kratz CP, Niemeyer CM, Castleberry RP (2005) The mutational spectrum of PTPN11 in juvenile myelomonocytic leukemia and Noonan syndrome/myeloproliferative disease. Hematology 106: 2183-2185.
- Lackner A, Basu O, Bierings M (2000) Haematopoietic stem cell transplantation for amegakaryocytic thrombocytopenia. Br J Haematol 109: 773-775.
- Stoddart MT, Connor P, Germeshausen M (2013) Congenital amegakaryocytic thrombocytopenia (CAMT) presenting as severe pancytopenia in the first month of life. Pediatric Blood and Cancer 60: 94-96.